
Abstract
Background
Sepsis-associated encephalopathy (SAE) patients often experience changes in intracranial pressure and impaired cerebral autoregulation. Mean arterial pressure (MAP) plays a crucial role in cerebral perfusion pressure, but its relationship with mortality in SAE patients remains unclear. This study aims to investigate the relationship between MAP and the risk of 28-day and in-hospital mortality in SAE patients, providing clinicians with the optimal MAP target.
Methods
We retrospectively collected clinical data of patients diagnosed with SAE on the first day of ICU admission from the MIMIC-IV (v2.2) database. Patients were divided into four groups based on MAP quartiles. Kruskal-Wallis H test and Chi-square test were used to compare clinical characteristics among the groups. Restricted cubic spline and segmented Cox regression models, both unadjusted and adjusted for multiple variables, were employed to elucidate the relationship between MAP and the risk of 28-day and in-hospital mortality in SAE patients and to identify the optimal MAP. Subgroup analyses were conducted to assess the stability of the results.
Results
A total of 3,816 SAE patients were included. The Q1 group had higher rates of acute kidney injury and vasoactive drug use on the first day of ICU admission compared to other groups (P < 0.01). The Q1 and Q4 groups had longer ICU and hospital stays (P < 0.01). The 28-day and in-hospital mortality rates were highest in the Q1 group and lowest in the Q3 group. Multivariable adjustment restricted cubic spline curves indicated a nonlinear relationship between MAP and mortality risk (P for nonlinearity < 0.05). The MAP ranges associated with HRs below 1 for 28-day and in-hospital mortality were 74.6–90.2 mmHg and 74.6–89.3 mmHg, respectively.The inflection point for mortality risk, determined by the minimum hazard ratio (HR), was identified at a MAP of 81.5 mmHg. The multivariable adjusted segmented Cox regression models showed that for MAP < 81.5 mmHg, an increase in MAP was associated with a decreased risk of 28-day and in-hospital mortality (P < 0.05). In Model 4, each 5 mmHg increase in MAP was associated with a 15% decrease in 28-day mortality risk (HR: 0.85, 95% CI: 0.79–0.91, p < 0.05) and a 14% decrease in in-hospital mortality risk (HR: 0.86, 95% CI: 0.80–0.93, p < 0.05). However, for MAP ≥ 81.5 mmHg, there was no significant association between MAP and mortality risk (P > 0.05). Subgroup analyses based on age, congestive heart failure, use of vasoactive drugs, and acute kidney injury showed consistent results across different subgroups.Subsequent analysis of SAE patients with septic shock also showed results similar to those of the original cohort.However, for comatose SAE patients (GCS ≤ 8), there was a negative correlation between MAP and the risk of 28-day and in-hospital mortality when MAP was < 81.5 mmHg, but a positive correlation when MAP was ≥ 81.5 mmHg in adjusted models 2 and 4.
Conclusion
There is a nonlinear relationship between MAP and the risk of 28-day and in-hospital mortality in SAE patients. The optimal MAP target for SAE patients in clinical practice appears to be 81.5 mmHg.
Similar content being viewed by others

Background
Sepsis-associated encephalopathy (SAE) is a brain dysfunction caused by infection without direct central nervous system infection, excluding other potential causes of brain dysfunction. The clinical manifestations of SAE can range from somnolence or delirium to severe cognitive impairment and deep coma. In the intensive care unit (ICU), the incidence of SAE in sepsis patients could be as high as 43.6% [1]. This significantly affects patient outcomes, including long-term neurological dysfunction and high mortality rates.The pathophysiological mechanisms of SAE are complex and not fully elucidated. It is currently believed to involve multiple potential mechanisms, primarily including inflammatory responses, reduced cerebral blood flow (CBF), glial cell activation, blood-brain barrier dysfunction, and other factors [2,3,4,5]. Studies have shown that up to 50% of septic patients had impaired cerebral autoregulation [6], which reduces the brain’s ability to adapt to changes in systemic blood pressure, subsequently affecting intracranial perfusion pressure and reducing CBF. Given the critical role of reduced CBF in the pathogenesis of SAE, maintaining adequate intracranial perfusion pressure to improve CBF is crucial for managing SAE patients.
Optimal mean arterial pressure (MAP) is crucial for tissue and organ perfusion and is essential in managing sepsis, especially septic shock. Sepsis and septic shock management guidelines recommended maintaining MAP ≥ 65 mmHg [7] to reduce organ damage and mortality risk. However, because organs have different anatomical and physiological characteristics and varying tolerance to ischemia and hypoxia, the optimal MAP should be adjusted based on the disease.MAP, a key parameter in determining intracranial perfusion pressure, influences CBF, oxygenation, and metabolism of brain cells. Although some studies have explored the impact of MAP on neurological outcomes in critically ill patients, the results remain controversial. Some research indicated that lower MAP was associated with more neurological sequelae and higher mortality in critically ill patients, likely due to brain hypoxia and damage caused by inadequate cerebral perfusion [8, 9]. Conversely, other studies suggested that excessively high MAP could lead to hyperperfusion, increased intracranial pressure, and potentially brain edema and further brain injury [10].
To date, there are no specific studies on the optimal MAP for SAE patients. Determining an appropriate MAP target is crucial for improving the prognosis of SAE patients. This study aims to investigate the relationship between different MAP levels and 28-day and in-hospital mortality risks in SAE patients. It provides clinical practitioners with the optimal MAP for managing SAE.
Methods
Data sources
This retrospective study extracted all data from the MIMIC-IV (v2.2) database.The MIMIC-IV database is a publicly available critical care database that contains clinical data from over 70,000 ICU admissions at the Beth Israel Deaconess Medical Center and the Massachusetts Institute of Technology, spanning from 2008 to 2019. This database includes comprehensive patient information such as demographics, vital signs, laboratory results, imaging studies, and treatments. One of researchers has complied with the data usage agreement (certificate number: 53211641). The MIMIC-IV database used in this study was approved by the Institutional Review Boards (IRB) of the institution, with a waiver of informed consent for patients and guardians.
Study population and important definitions
The definition of sepsis patients in this study was based on the third edition of the sepsis diagnostic criteria (sepsis 3.0), which combined Sequential Organ Failure Assessment (SOFA ≥ 2) and suspected or confirmed infection [7]. SAE was characterized by altered mental status due to sepsis, excluding other possible causes of altered mental status. In this study, altered mental status referred to a Glasgow Coma Scale (GCS) score < 15 or a delirium diagnosis (based on ICD-9 codes 2930 and 2931) [11, 12]. Coma was defined as a GCS score ≤ 8 points.Specifically, the inclusion and exclusion criteria for this study were as follows [13, 14]. The inclusion criteria were: (1) diagnosed with sepsis on the first day of ICU admission; (2) admitted to the ICU for the first time; (3) had a GCS score < 15 or a diagnosis of delirium. The exclusion criteria were: (1) ICU stays less than 24 h; (2)primary hypertension; (3)patients with brain injuries not caused by sepsis, such as encephalitis, intracranial abscess, epilepsy, brain trauma, cerebral hemorrhage, cerebral infarction, other cerebrovascular diseases, mental disorders, alcohol or drug dependence, hypoglycemic coma, ischemic-hypoxic encephalopathy, metabolic encephalopathy, hypertensive encephalopathy, hepatic encephalopathy, or other encephalopathies.
Data extraction and variables
We used Navicat 16 and SQL (Structured Query Language) to extract data from the MIMIC-IV (v2.2) database. The extracted variables included demographic characteristics (gender, age, weight, and race), comorbidities (congestive heart failure, chronic pulmonary disease, renal disease, severe liver disease, diabetes, malignant cancer, etc.), average vital signs within the first 24 h of ICU admission (heart rate, respiratory rate, temperature, mean arterial pressure), treatments (mechanical ventilation, vasoactive drugs, renal replacement therapy), laboratory results (pH, PaO2, PaCO2, white blood cell count, hemoglobin, platelets, glucose, anion gap, calcium, sodium, potassium, creatinine, international normalized ratio, prothrombin time), and severity of illness scores (GCS, SOFA), and acute kidney injury(AKI) stage.
Endpoints
The primary endpoints were defined as the 28-day mortality following ICU admission and the in-hospital mortality.
Statistical analyses
For continuous variables, the Shapiro-Wilk test was used to assess normality. Normally distributed data were presented as mean ± standard deviation, while non-normally distributed data were presented as median (interquartile range). Categorical variables were expressed as frequencies or percentages. Based on the interquartile range of the MAP in the included population, subjects were divided into four groups. One-way ANOVA (for normally distributed variables), Kruskal-Wallis H test (for non-normally distributed variables), and Chi-square test (for categorical variables) were used to determine statistical differences between groups.
To investigate the relationship between MAP and 28-day and in-hospital mortality in SAE patients, we used unadjusted and multivariable adjusted (variables in Table 1) restricted cubic splines(RCS) to fit MAP and incorporated it into the Cox proportional hazards model. By fitting the spline model, we captured the nonlinear relationship in the data. Based on our research objectives and clinical applicability, the inflection point was determined by identifying the minimum hazard ratio (HR) [15, 16]. After identifying the inflection point, we divided the data into two segments and fitted four Cox proportional hazards models for each segment. First, MAP divided by 5 was entered as a continuous variable into the model. Then, MAP was categorized into six groups based on 10 mmHg increments or decrements around the inflection point and entered as a categorical variable into the models. The four Cox regression models included: Model 1 (unadjusted); Model 2 adjusted for gender, age, weight, and race; Model 3 adjusted for Model 2 plus Charlson Comorbidity Index, AKI Stage, SOFA Score, and GCS; Model 4 adjusted for Model 3 plus number of vasoactive drugs, ventilation status, and renal replacement therapy.
To explore the stability of the results, segmented Cox regression analyses were conducted in SAE patients with shock and severe SAE (coma, GCS ≤ 8) to examine the relationship between MAP and 28-day and in-hospital mortality. Additionally, given that age, congestive heart failure, use of vasoactive drugs, and acute kidney injury were important confounders for 28-day and in-hospital mortality in SAE patients, we performed subgroup and interaction analyses using the adjusted Cox regression model (Model 4) to further evaluate the stability of the relationship between MAP and 28-day and in-hospital mortality. We selected confounders in the model based on clinical relevance and used variance inflation factors to control for multicollinearity.To avoid bias due to missing data, robust linear regression was used to impute multiple missing columns, and variables with more than 30% missing values were excluded. Statistical analyses were performed using R software (R version 4.3.3). A two-tailed P value < 0.05 was considered statistically significant.
Results
Baseline characteristics of patients
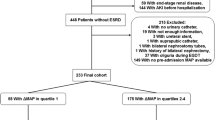
According to the inclusion and exclusion criteria (Fig. 1), 3,816 patients were included in our study.Among these patients, 39.1% were male, and 68.8% were white. The median age was 70.04 years (IQR: 59.66, 79.85), and the median weight was 80.00 kg (IQR: 67.20, 95.00). The median Charlson Comorbidity Index was 5.00 (IQR: 3.00, 7.00), the median GCS score was 14.00 (IQR: 10.00, 14.00), and the median SOFA score was 6.00 (IQR: 4.00, 8.00). Across the cohort, the MAP had a median of 74.72 mmHg (IQR: 69.60, 80.89).In terms of laboratory findings, patients in the Q1 group had significantly lower pH, PO2, glucose, sodium, calcium, and hemoglobin levels (p < 0.01), and significantly higher anion gap, potassium, creatinine, INR, and PT levels (p < 0.01). Among all patients included, 57.7% (n = 2,200) experienced acute kidney injury (AKI) on the first day of ICU admission, with a higher incidence in the Q1 group. Patients in the Q1 group also had higher usage of vasoactive drugs (p < 0.01). The Q1 and Q4 groups had longer ICU and hospital stays (p < 0.01). The 28-day mortality rate was 15.5% (n = 590) in all SAE patients, with rates of 23.8%, 13.9%, 11.4%, and 12.7% across the MAP quartiles, respectively. The 28-day mortality and in-hospital mortality rates were highest in the Q1 group and lowest in the Q3 group (p < 0.01).Baseline demographic, clinical characteristics and outcome of the study patients stratified by MAP quartiles, were shown in Table 1.

Flow chart of study patients
Nonlinear relationship between MAP and mortality risk in all patients
Using unadjusted and multivariable-adjusted (variables in Table 1,except outcome) restricted cubic splines (RCS) to model the relationship between MAP and the risk of 28-day and in-hospital mortality, we found a nonlinear association (P < 0.05). After multivariable adjustment, the inflection point, determined by the minimum hazard ratio (HR) for mortality risk, was identified at a MAP of 81.5 mmHg. The MAP ranges associated with HRs below 1 for 28-day and in-hospital mortality were 74.6–90.2 mmHg and 74.6–89.3 mmHg, respectively (Fig. 2).Based on this inflection point, we divided the data into two segments (MAP < 81.5 mmHg and MAP ≥ 81.5 mmHg) and fitted four Cox proportional hazards models for each segment. When MAP was entered as a continuous variable, results indicated that for MAP < 81.5 mmHg, an increase in MAP was associated with a decreased risk of 28-day ICU and in-hospital mortality (P < 0.05). In Model 4, for each 5 mmHg increase in MAP, the risk of 28-day ICU mortality decreased by 15% (HR: 0.85, 95% CI: 0.79–0.91, P < 0.01), and the risk of in-hospital mortality decreased by 14% (HR: 0.86, 95% CI: 0.80–0.93, P < 0.01). However, for MAP ≥ 81.5 mmHg, there was no significant association between MAP and the risk of 28-day ICU and in-hospital mortality (P > 0.05). Additionally, using 81.5 mmHg as the inflection point, we categorized MAP into six groups based on 10 mmHg increments or decrements and entered MAP as a categorical variable in the model (with 71.5–81.5 mmHg as the reference). Except in Model 3, where the MAP level of 91.5–101.5 mmHg was associated with a higher risk of 28-day ICU and in-hospital mortality compared to 71.5–81.5 mmHg (P < 0.05), the results were consistent with those when MAP was treated as a continuous variable (Table 2).

Unadjusted and multivariable-adjusted restricted cubic splines between mean arterial pressure(MAP) and mortality. (A): RCS curve of MAP and unadjusted HR (95% CI) for 28-day mortality; (B): RCS curve of MAP and unadjusted HR (95% CI) for hospital mortality; (C): RCS curve of MAP and multivariable-adjusted HR (95% CI) for 28-day mortality; (D):RCS curve of MAP and multivariable-adjusted HR (95% CI) for hospital mortality
Sensitivity analysis of MAP and mortality risk in SAE patients with septic shock or coma
To verify the stability of our results, we conducted further segmented Cox regression analyses on SAE patients with septic shock (n = 2328) and coma (n = 793). In patients with septic shock, when MAP was < 81.5 mmHg, both unadjusted and adjusted models showed that the risk of 28-day ICU mortality and in-hospital mortality decreased as MAP increased (p < 0.05). In Model 4, for every 5 mmHg increase in MAP, the 28-day mortality risk in SAE patients with septic shock decreased by 15% (HR: 0.85, 95% CI: 0.78–0.93, p < 0.01), and the in-hospital mortality risk decreased by 14% (HR: 0.86, 95% CI: 0.79–0.94, p < 0.05). However, when MAP was ≥ 81.5 mmHg, no significant association was observed between MAP and mortality risk in both unadjusted and adjusted models (p > 0.05). In patients with coma (GCS ≤ 8), when MAP was < 81.5 mmHg, the risk of 28-day ICU and in-hospital mortality decreased as MAP increased (p < 0.05). However, when MAP was ≥ 81.5 mmHg, in the adjusted Models 2 and 4, every 5 mmHg increase in MAP was associated with a 22% (HR: 1.22, 95% CI: 1.03–1.44, p < 0.05) and 28% (HR: 1.28, 95% CI: 1.05–1.56, p < 0.05) increase in 28-day mortality, respectively, and a 23% (HR: 1.23, 95% CI: 1.04–1.45, p < 0.05) and 32% (HR: 1.32, 95% CI: 1.08–1.60, p < 0.05) increase in in-hospital mortality, respectively. In unadjusted Model 1 and adjusted Model 3, no significant association was found between MAP and mortality risk in comatose patients (p > 0.05), as shown in Table 3.
Subgroup analyses of the association between MAP and mortality risk in all patients
Using the adjusted Model 4 Cox regression model, we performed subgroup and interaction analyses based on age, congestive heart failure, use of vasoactive drugs, and acute kidney injury to further assess the stability of the relationship between MAP and 28-day ICU and in-hospital mortality. The study showed that when MAP was < 81.5 mmHg, an increase in MAP was associated with a decrease in 28-day ICU and in-hospital mortality risks across all subgroups (P < 0.05), except for patients with stage 0–1 acute kidney injury, where the association with in-hospital mortality did not reach statistical significance (HR: 0.90, 95% CI: 0.8–1.01, p = 0.07). When MAP was ≥ 81.5 mmHg, no significant association was found between MAP and 28-day ICU or in-hospital mortality in any subgroup (P > 0.05). Interaction analysis revealed a significant interaction between MAP and the presence of heart failure on in-hospital mortality risk (P = 0.03), as shown in Table 4.
Discussion
This study analyzed 3,816 ICU patients with SAE and found a significant nonlinear association between MAP and the risk of 28-day and in-hospital mortality, even after multivariable adjustment. The lowest mortality risk was identified at a MAP of 81.5 mmHg. The MAP ranges associated with HRs below 1 for 28-day and in-hospital mortality were 74.6–90.2 mmHg and 74.6–89.3 mmHg, respectively. Segmented Cox regression analysis revealed that increasing MAP below 81.5 mmHg reduced both 28-day and in-hospital mortality risks. However, at MAP levels of 81.5 mmHg or higher, there was no statistically significant relationship between MAP and mortality risk. Subgroup analyses by age, heart failure, acute kidney injury, and vasopressor use confirmed the stability of these findings. In comatose SAE patients, increasing MAP below 81.5 mmHg significantly reduced mortality risk, whereas MAP at or above 81.5 mmHg increased 28-day and in-hospital mortality risks.
The optimal MAP for sepsis and septic shock patients has been a major focus in medical research [17, 18]. Adequate MAP is essential for tissue and organ perfusion, with guidelines recommending a MAP of ≥ 65 mmHg [7]. Zhong et al. [19] found that among 14,031 sepsis patients, those with MAP over 65 mmHg had lower 30-day, 60-day, and 100-day mortality rates and shorter ICU stays compared to those with MAP of 60–65 mmHg. Cao et al. [20]identified a nonlinear relationship between MAP and 30-day mortality, with a turning point at 68.6 mmHg. Below this point, MAP was negatively correlated with 30-day mortality; above this point, there was no significant association. Conversely, Lamontagne et al. [21] found that permissive hypotension (MAP 60–65 mmHg) reduced vasopressor exposure and arrhythmia incidence without increasing mortality. The appropriate MAP for different diseases remains controversial [22], and due to physiological and anatomical differences among organs, the optimal MAP may vary [23]. Maiwall et al. [24]found no survival benefit in the higher MAP group compared to the lower MAP group in septic shock patients with cirrhosis, although the higher MAP group improved dialysis tolerance, lactate clearance, and renal recovery, and were associated with improved endothelial function markers. Zhu et al. [25] found that in hypertensive patients with septic shock, a lower MAP target (65–70 mmHg) compared to a higher MAP target (75–80 mmHg) reduced inflammatory cytokine levels, fluid volume, and vasopressor duration, while improving mesenteric blood flow and protecting gastrointestinal function. Khanna et al. [26] reported that in 2,833 postoperative surgical ICU patients, those with a MAP at the 25th percentile (78 mmHg) had a 23% higher risk of myocardial injury or death compared to those with a median MAP of 87 mmHg. These studies suggested that optimal MAP varied across diseases and significantly impacted outcomes in critically ill patients, necessitating research into disease-specific optimal MAPs.
There had been few studies on the optimal MAP for SAE. Animal studies suggested that correcting hypotension with vasopressors during septic shock might improve cerebral oxygenation but not reverse cerebral microcirculation or metabolic changes [27]; other studies showed reduced small vessel perfusion during sepsis-induced encephalopathy [28]. A preliminary study on six patients with extracranial sepsis using NIRS-derived cerebral oxygen saturation determined an optimal MAP range of 55 to 115 mmHg for SAE patients [29]. These studies, being either animal experiments or small-sample studies, had limited representativeness for determining the optimal MAP range.Our large-sample study used multivariable-adjusted segmented Cox regression models and restricted cubic spline analysis, revealing that in SAE patients with septic shock, MAP was negatively correlated with 28-day and in-hospital mortality risks below the inflection point, with no correlation above it. Our findings can guide the management of target MAP in patients with SAE combined with septic shock, including comprehensive management of fluid resuscitation, use of vasoactive drugs, and other aspects. Notably, our optimal MAP of 81.5 mmHg was slightly higher than 65 mmHg recommended by sepsis management guidelines, possibly due to differences in patient conditions and backgrounds across studies, as our study focused on SAE patients who were more sensitive to ischemia and hypoxia.
The impact of MAP on 28-day and in-hospital mortality risk in SAE patients may be mediated through CBF regulation. Decreased CBF is a mechanism of SAE, varying with cerebral perfusion pressure (CPP), which is determined by the difference between MAP and intracranial pressure (ICP) (i.e., CPP = MAP - ICP) [30, 31]. In normal physiological conditions, CBF has self-regulating ability, meaning it maintains a stable blood flow by altering the diameter of cerebral blood vessels [32].However, in SAE patients, this autoregulation may be impaired, making cerebral blood flow more sensitive to blood pressure changes [29]. Crippa et al. [33] found altered cerebral hemodynamics, particularly CPP, in one-third of critically ill sepsis patients. Thus, appropriate MAP levels are crucial for maintaining cerebral perfusion and preventing brain injury. Our study found that when MAP was below 81.5 mmHg, each 5 mmHg increase in MAP in the adjusted Model 4 reduced 28-day mortality risk by 15% and in-hospital mortality risk by 14%, with similar trends in SAE patients with septic shock or coma. This suggests that SAE patients may rely on higher MAP to maintain stable CBF due to impaired autoregulation. Low MAP may reduce CBF, allowing inflammatory mediators to accumulate in brain tissue, directly causing cellular damage and dysfunction; impaired clearance of metabolic byproducts (e.g., lactate) may exacerbate brain cell metabolic burden and damage; hypoxia in brain tissue increases oxidative stress, generating more free radicals, leading to neuron death and worsening brain function; insufficient cerebral microvascular perfusion affects oxygen and nutrient supply, increases blood viscosity and microthrombosis, further hindering cerebral blood flow, increasing brain injury and mortality risk [34, 35]. This was similar to Erickson’s [9] study, where lower MAP within the first 12 h of PICU admission in children with severe traumatic brain injury was associated with poor discharge outcomes. However, when MAP was 81.5 mmHg or higher, the association between MAP and mortality risk was not significant. Sensitivity analysis revealed that in the adjusted Model 4, every 5 mmHg increase in MAP was associated with a 28% increase in 28-day mortality and a 32% increase in in-hospital mortality, suggesting limited benefits from further increasing MAP for cerebral perfusion. Excessively high MAP might increase cerebrovascular pressure and fluid overload, causing cerebral edema and hemorrhage, further damaging brain function.In comatose SAE patients, analysis showed a positive correlation between high MAP and mortality after the inflection point. This might be due to more severe impairment of cerebral autoregulation in comatose patients [36]. Therefore, it was crucial to maintain an appropriate MAP range in comatose SAE patients to avoid both low and high MAP.
Given that age, heart failure [37], use of vasopressors, and acute kidney injury were important confounders in assessing the relationship between MAP and mortality risk.Lamontagne et al. [38] conducted a multicenter study and found that for critically ill patients with vasodilatory shock, there was no statistical difference in hospital mortality between lower (60–65 mmHg) and higher (75–80 mmHg) MAP targets, with arrhythmia risks of 20% and 36% and mortality rates of 30% and 33%, respectively. However, in patients aged 75 or older, a lower MAP target was associated with reduced hospital mortality (13% vs. 60%, p = 0.03). Zhao et al. [39]found that AKI incidence might be related to MAP levels in sepsis patients. To ensure renal perfusion, hypertensive AKI patients might have required higher MAPs than non-hypertensive patients, at 70–80 mmHg and 65–73 mmHg, respectively.Subgroup analysis of our study population yielded results similar to the original cohort. Although the overall results supported our conclusions, in patients with stage 0–1 AKI, the association between increased MAP and in-hospital mortality risk did not reach statistical significance (HR: 0.90, 95% CI: 0.8–1.01, p = 0.07), possibly due to reduced sample size after subgrouping. Additionally, interaction analysis between significant confounders and MAP revealed a significant interaction only for heart failure in the assessment of in-hospital mortality risk. This indicated that heart failure patients required special attention in blood pressure management. Heart failure affected cardiac pumping function, impacting systemic and cerebral perfusion, warranting further research.
Despite thoroughly examining the relationship between MAP and 28-day and in-hospital mortality rates, this study had limitations. Firstly, as a retrospective analysis, and in the absence of a gold standard for diagnosing SAE, we defined SAE based on previous literature as sepsis with a GCS score less than 15 or the presence of delirium, while excluding other causes of brain injury. Although this approach might have introduced selection bias, our study was conducted using a large public database and aligned with established SAE research criteria [13, 14]. Secondly, this study had some missing data for certain variables, and some covariates had too much missing data to be included in the analysis. However, we used robust linear regression to fill in multiple missing columns, minimizing bias from missing data. We adjusted for many covariates to reduce confounding bias, and conducted sensitivity and subgroup analyses to enhance the reliability and generalizability of our results. Furthermore, the study lacked data on cranial TCD, EEG, cerebral oxygen monitoring, and cranial imaging. This made it hard to objectively assess how different blood pressure levels affected cerebral autoregulation and blood flow. Additionally, although our retrospective study had a large sample size, statistical significance may not fully reflect clinical relevance. Future research should focus on prospective randomized controlled trials aimed at optimal MAP management strategies, incorporating multimodal brain function monitoring, to further improve clinical outcomes.
In conclusion
Taken together, this study demonstrated a nonlinear relationship between MAP and 28-day and in-hospital mortality rates, suggesting that different MAP levels required different blood pressure management strategies. Clinically, aiming for an optimal MAP of 81.5 mmHg was crucial for improving survival rates in SAE. Further research on the pathophysiological mechanisms of SAE and bedside cerebral hemodynamic monitoring is needed to better understand the relationship between MAP and SAE prognosis, thereby providing more effective treatment strategies for clinical practice.
Data availability
The data supporting this study can be obtained from the MIMIC-IV database, but its use requires a license. Therefore, it is not publicly accessible. However, the authors can provide the data upon reasonable request and with permission from the database holders.
Abbreviations
- MAP:
-
Mean arterial pressure
- SAE:
-
Sepsis-associated encephalopathy
- ICU:
-
Intensive care unit
- SOFA:
-
Sequential organ failure assessment
- GCS:
-
Glasgow Coma Scale
- CBF:
-
Cerebral blood flow
- AKI:
-
Acute kidney injury
- IQR:
-
Interquartile range
- HR:
-
Hazard ratio
- CI:
-
Confidence Interval
References
Chen J, Shi X, Diao M, et al. A retrospective study of sepsis-associated encephalopathy: epidemiology, clinical features and adverse outcomes. BMC Emerg Med. 2020;20(1):77. https://doi.org/10.1186/s12873-020-00374-3.
Griton M, Dhaya I, Nicolas R, et al. Experimental sepsis-associated encephalopathy is accompanied by altered cerebral blood perfusion and water diffusion and related to changes in cyclooxygenase-2 expression and glial cell morphology but not to blood-brain barrier breakdown[J]. Brain Behav Immun. 2020;83:200–13. https://doi.org/10.1016/j.bbi.2019.10.012.
Gao Q, Hernandes MS. Sepsis-associated encephalopathy and blood-brain barrier dysfunction[J]. Inflammation. 2021;44(6):2143–50. https://doi.org/10.1007/s10753-021-01501-3.
Qin N, Miao Y, Xie L, et al. Sepsis-associated encephalopathy: autophagy and mirnas regulate microglial activation[J]. Physiol Rep. 2024;12(5):e15964. https://doi.org/10.14814/phy2.15964.
Algebaly H, Elsherbini S, Galal A, et al. Transcranial doppler can predict development and outcome of sepsis-associated encephalopathy in pediatrics with severe sepsis or septic shock[J]. Front Pediatr. 2020;8:450. https://doi.org/10.3389/fped.2020.00450.
Crippa IA, Subira C, Vincent JL, et al. Impaired cerebral autoregulation is associated with brain dysfunction in patients with sepsis[J]. Crit Care. 2018;22(1):327. https://doi.org/10.1186/s13054-018-2258-8.
Evans L, Rhodes A, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021[J]. Intensive Care Med. 2021;47(11):1181–247. https://doi.org/10.1007/s00134-021-06506-y.
Jouan Y, Seegers V, Meziani F, et al. Effects of mean arterial pressure on arousal in sedated ventilated patients with septic shock: a sepsispam post hoc exploratory study[J]. Ann Intensive Care. 2019;9(1):54. https://doi.org/10.1186/s13613-019-0528-5.
Erickson SL, Killien EY, Wainwright M, et al. Mean arterial pressure and discharge outcomes in severe pediatric traumatic brain injury[J]. Neurocrit Care. 2021;34(3):1017–25. https://doi.org/10.1007/s12028-020-01121-z.
Lin YH, Liu HM. Update on cerebral hyperperfusion syndrome[J]. J Neurointerv Surg. 2020;12(8):788–93. https://doi.org/10.1136/neurintsurg-2019-015621.
Ge C, Deng F, Chen W, et al. Machine learning for early prediction of sepsis-associated acute brain injury[J]. Front Med (Lausanne). 2022;9:962027. https://doi.org/10.3389/fmed.2022.962027.
Yang Y, Liang S, Geng J, et al. Development of a nomogram to predict 30-day mortality of patients with sepsis-associated encephalopathy: a retrospective cohort study[J]. J Intensive Care. 2020;8:45. https://doi.org/10.1186/s40560-020-00459-y.
Zhao L, Wang Y, Ge Z, et al. Mechanical learning for prediction of sepsis-associated encephalopathy[J]. Front Comput Neurosci. 2021;15:739265. https://doi.org/10.3389/fncom.2021.739265.
Lu X, Qin M, Walline JH, et al. Clinical phenotypes of sepsis-associated encephalopathy: a retrospective cohort study[J]. Shock. 2023;59(4):583–90. https://doi.org/10.1097/SHK.0000000000002092.
Pei J, Wang X, Xing Z, et al. Association between admission systolic blood pressure and major adverse cardiovascular events in patients with acute myocardial infarction[J]. PLoS ONE. 2020;15(6):e234935. https://doi.org/10.1371/journal.pone.0234935.
Zhang H, Xu Y, Xu Y. The association of the platelet/high-density lipoprotein cholesterol ratio with self-reported stroke and cardiovascular mortality: a population-based observational study[J]. Lipids Health Dis. 2024;23(1):121. https://doi.org/10.1186/s12944-024-02115-y.
Beloncle F, Radermacher P, Guerin C, et al. Mean arterial pressure target in patients with septic shock[J]. Minerva Anestesiol. 2016;82(7):777–84.
Dari MA, Fayaz A, Sharif S, et al. Comparison of high-normal versus low-normal mean arterial pressure at target on outcomes in sepsis or shock patients: a meta-analysis of randomized control trials[J]. Cureus. 2024;16(1):e52258. https://doi.org/10.7759/cureus.52258.
Zhong X, Li H, Chen Q, et al. Association between different map levels and 30-day mortality in sepsis patients: a propensity-score-matched, retrospective cohort study[J]. BMC Anesthesiol. 2023;23(1):116. https://doi.org/10.1186/s12871-023-02047-7.
Cao B, Chen Q, Tang T, et al. Non-linear relationship between baseline mean arterial pressure and 30-day mortality in patients with sepsis: a retrospective cohort study based on the mimic-iii database[J]. Ann Transl Med. 2022;10(16):872. https://doi.org/10.21037/atm-22-3457.
Lamontagne F, Richards-Belle A, Thomas K, et al. Effect of reduced exposure to vasopressors on 90-day mortality in older critically ill patients with vasodilatory hypotension: a randomized clinical trial[J]. JAMA. 2020;323(10):938–49. https://doi.org/10.1001/jama.2020.0930.
Marshall JC. Choosing the best blood pressure target for vasopressor therapy[J]. JAMA. 2020;323(10):931–3. https://doi.org/10.1001/jama.2019.22526.
Meng L, Wang Y, Zhang L, et al. Heterogeneity and variability in pressure autoregulation of organ blood flow: lessons learned over 100 + years[J]. Crit Care Med. 2019;47(3):436–48. https://doi.org/10.1097/CCM.0000000000003569.
Maiwall R, Rao PS, Hidam AK, et al. A randomised-controlled trial (target-c) of high vs. low target mean arterial pressure in patients with cirrhosis and septic shock[J]. J Hepatol. 2023;79(2):349–61. https://doi.org/10.1016/j.jhep.2023.04.006.
Zhu X, Hou J, Zhang Q, et al. [Effects of treatment based on different target mean arterial pressure on gastrointestinal function in septic shock patients with hypertension][J]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2021;33(5):517–22. https://doi.org/10.3760/cma.j.cn121430-20200713-00515.
Khanna AK, Maheshwari K, Mao G, et al. Association between mean arterial pressure and acute kidney injury and a composite of myocardial injury and mortality in postoperative critically ill patients: a retrospective cohort analysis[J]. Crit Care Med. 2019;47(7):910–7. https://doi.org/10.1097/CCM.0000000000003763.
Taccone FS, Su F, He X, et al. Effects of reversal of hypotension on cerebral microcirculation and metabolism in experimental sepsis[J]. Biomedicines. 2022;10(4). https://doi.org/10.3390/biomedicines10040923.
Taccone FS, Su F, Pierrakos C, et al. Cerebral microcirculation is impaired during sepsis: an experimental study[J]. Crit Care. 2010;14(4):R140. https://doi.org/10.1186/cc9205.
Rosenblatt K, Walker KA, Goodson C, et al. Cerebral autoregulation-guided optimal blood pressure in sepsis-associated encephalopathy: a case series[J]. J Intensive Care Med. 2020;35(12):1453–64. https://doi.org/10.1177/0885066619828293.
Kow CY, Harley B, Li C, et al. Escalating mean arterial pressure in severe traumatic brain injury: a prospective, observational study[J]. J Neurotrauma. 2021;38(14):1995–2002. https://doi.org/10.1089/neu.2020.7289.
Dietvorst S, Depreitere B, Meyfroidt G. Beyond intracranial pressure: monitoring cerebral perfusion and autoregulation in severe traumatic brain injury[J]. Curr Opin Crit Care. 2023;29(2):85–8. https://doi.org/10.1097/MCC.0000000000001026.
Claassen J, Thijssen D, Panerai RB, et al. Regulation of cerebral blood flow in humans: physiology and clinical implications of autoregulation[J]. Physiol Rev. 2021;101(4):1487–559. https://doi.org/10.1152/physrev.00022.2020.
Crippa IA, Vincent JL, Zama CF, et al. Estimated cerebral perfusion pressure and intracranial pressure in septic patients[J]. Neurocrit Care. 2024;40(2):577–86. https://doi.org/10.1007/s12028-023-01783-5.
Taccone FS, Scolletta S, Franchi F, et al. Brain perfusion in sepsis[J]. Curr Vasc Pharmacol. 2013;11(2):170–86. https://doi.org/10.2174/1570161111311020007.
Pan S, Lv Z, Wang R et al. Sepsis-induced brain dysfunction: pathogenesis, diagnosis, and treatment[J]. Oxid Med Cell Longev, 2022,2022: 1328729. https://doi.org/10.1155/2022/1328729
Figaji AA, Zwane E, Fieggen AG, et al. Pressure autoregulation, intracranial pressure, and brain tissue oxygenation in children with severe traumatic brain injury[J]. J Neurosurg Pediatr. 2009;4(5):420–8. https://doi.org/10.3171/2009.6.PEDS096.
Osteresch R, Diehl K, Dierks P, et al. Influence of the ratio of mean arterial pressure to right atrial pressure on outcome after successful percutaneous edge-to-edge repair for severe mitral valve regurgitation[J]. Int J Cardiol Heart Vasc. 2021;37:100903. https://doi.org/10.1016/j.ijcha.2021.100903.
Lamontagne F, Day AG, Meade MO, et al. Pooled analysis of higher versus lower blood pressure targets for vasopressor therapy septic and vasodilatory shock[J]. Intensive Care Med. 2018;44(1):12–21. https://doi.org/10.1007/s00134-017-5016-5.
Zhao L, Fan Y, Wang Z, et al. The blood pressure targets in sepsis patients with acute kidney injury: an observational cohort study of multiple icus[J]. Front Immunol. 2022;13:1060612. https://doi.org/10.3389/fimmu.2022.1060612.
Acknowledgements
Not applicable.
Funding
This study was funded by Guangzhou Health Science and Technology Project (20231A011031).
Author information
Authors and Affiliations
Contributions
YYY and HYP conceived and designed the study. HYP, ZXL and SXZ Collected and analyzed data. HYPand ZXL wrote the manuscript.All authors had reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study involved a retrospective analysis of anonymized data. One of the researchers in this study, Hongyan Peng, who passed the “Protecting Human Research Participants” exam, accessed the database and was responsible for data extraction (Record ID: 53211641). The MIMIC-IV database used in this study was approved by the Institutional Review Boards (IRB) of Beth Israel Deaconess Medical Center and the Massachusetts Institute of Technology, with a waiver of informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Peng, H., Liang, Z., Zhang, S. et al. Optimal target mean arterial pressure for patients with sepsis-associated encephalopathy: a retrospective cohort study. BMC Infect Dis 24, 902 (2024). https://doi.org/10.1186/s12879-024-09789-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-024-09789-w