
Abstract
HIV-1 remains a global health concern and to date, nearly 38 million people are living with HIV. The complexity of HIV-1 pathogenesis and its subsequent prevalence is influenced by several factors including the HIV-1 subtype. HIV-1 subtype variation extends to sequence variation in the amino acids of the HIV-1 viral proteins. Of particular interest is the transactivation of transcription (Tat) protein due to its key function in viral transcription. The Tat protein predominantly functions by binding to the transactivation response (TAR) RNA element to activate HIV-1 transcriptional elongation. Subtype-specific Tat protein sequence variation influences Tat-TAR binding affinity. Despite several studies investigating Tat-TAR binding, it is not clear which regions of the Tat protein and/or individual Tat amino acid residues may contribute to TAR binding affinity. We, therefore, conducted a scoping review on studies investigating Tat-TAR binding. We aimed to synthesize the published data to determine (1) the regions of the Tat protein that may be involved in TAR binding, (2) key Tat amino acids involved in TAR binding and (3) if Tat subtype-specific variation influences TAR binding. A total of thirteen studies met our inclusion criteria and the key findings were that (1) both N-terminal and C-terminal amino acids outside the basic domain (47–59) may be important in increasing Tat-TAR binding affinity, (2) substitution of the amino acids Lysine and Arginine (47–59) resulted in a reduction in binding affinity to TAR, and (3) none of the included studies have investigated Tat subtype-specific substitutions and therefore no commentary could be made regarding which subtype may have a higher Tat-TAR binding affinity. Future studies investigating Tat-TAR binding should therefore use full-length Tat proteins and compare subtype-specific variations. Studies of such a nature may help explain why we see differential pathogenesis and prevalence when comparing HIV-1 subtypes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Human immunodeficiency virus (HIV) is a retrovirus that functions by integrating a copy of its retroviral deoxyribonucleic acid (DNA) genome into the DNA of the infected human cell for replication [1]. HIV was discovered in the early 1980s when the virus had already established a pandemic [2]. HIV-1 is responsible for the development of Acquired Immune Deficiency Syndrome (AIDS) [3]. Since the 1980s to date, about 74.9 million people have been infected by HIV, with 32 million people succumbing to AIDS (the stage defined as a decrease in CD4+ cells below 200 cells/µl) [4]. In 2021, an estimated 38.4 million people were recorded to be living with HIV worldwide, with 650,000 deaths and 1.5 million new infections [5]. Adding to this, the coronavirus disease 2019 (COVID-19) pandemic had negatively influenced the progression of HIV. In South Africa, HIV-1 testing, and anti-retroviral therapy (ART) initiations were heavily impacted [6]. Further, the shortage of anti-retroviral drugs (ARVs) due to the shutdowns of certain drug manufacturers [7] and the disruption in the delivery of HIV care due to the COVID-19 pandemic may have increased morbidity and mortality among people living with HIV (PLWH) [8]. The diversion of more healthcare workers to care for COVID-19 patients may also have contributed to an increase in the prevalence of HIV infections and disease progression [9,10,11]. In both severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) and HIV, evolutionary strategies are at play [8] which influences the change in the genetic makeup of these viruses.
Two main types of HIV exist, namely HIV-1 and HIV-2, of which HIV-1 predominates worldwide [9]. HIV-1 is classified into three genetic groups, M, O and N [10]. Most HIV-1 infections worldwide are caused by the group M subtypes which are designated by letters A, B, C, D, F, G, H, J and K [10]. HIV-1 subtypes, also known as clades, are linked geographically or epidemiologically [11]. Subtype C is the most prevalent HIV strain and is the predominant subtype in India and Southern Africa [12]. Subtype B is prevalent in almost all parts of Europe and the Americas, while a diverse variety of subtypes are found in West and Central Africa [13]. Statistically, subtype C represents approximately 50% of the world’s HIV infected while the second most prevalent subtype B accounts for about 12% of PLWH [12, 14]. Subtype D is prevalent in East and Central Africa, with sporadic cases observed in Southern and Western Africa [15]. Collectively, the subtypes F, H, J and K account for 0.94% of all global infections [16]. Subtypes H, J and K are found in Central, Southern and West Africa [17], with subtype K, in particular, being identified in the Democratic Republic of Congo and Cameroon [18]. Subtypes G and A viruses have been identified in western and eastern Africa and also in central Europe [19, 20]. Subtype F is endemic in South and South-East Asia [21].
The HIV virion has a spherical structure with a diameter of approximately 100–130 nm. Its lipid membrane envelope is derived from the host cell and cellular proteins [22]. The HIV genome contains the retroviral genes gag, pol, and env. In addition, HIV has six regulatory genes (tat, rev, nef, vif, vpr, and vpu) and is therefore considered a “complex” retrovirus [22]. HIV encodes for 15 distinct proteins [23, 24], and these include: structural (matrix, capsid, nucleocapsid, p6, surface and transmembrane), Pol enzymes (protease, reverse transcriptase, integrase), regulatory proteins (Transactivation of transcription (Tat) and regulator of expression of virion proteins (Rev)) and accessory proteins (Negative regulatory factor (Nef), viral infectivity factor (Vif), viral protein R (Vpr) and virus protein U (Vpu)). Of particular interest is the Tat protein due to its multifunctional activity within HIV pathogenesis.
HIV-1 Tat is a regulatory protein encoded by the tat gene. The Tat protein has a variable weight of 14–16 kilodaltons (kDa), has an amino acid composition varying from 86 to 104 amino acids [25, 26] and is an intrinsically unstructured protein [27] (Figs. 1 and 2). Tat is divided into six different regions and contains various conserved areas within its overall sequence, which are crucial for its function [28]. Region I has a conserved tryptophan (Trp)-11 while the cysteine-rich region II contains seven well-conserved cysteines at positions 22, 25, 27, 30, 31, 34 and 37 (22–37) [29]. In region III (38–48), a conserved Phenylalanine-38 motif Arginine-Lysine-Leucine-Glycine-Isoleucine at 43–48 was observed in HIV-1 subtypes [30]. Region IV, the basic domain (49–59) is the key factor in Tat-TAR binding and is highly conserved among all Tat variants [31, 32]. Region V (60–72) is a glutamine-rich region [33]. This region shows the highest rate of sequence variation [29]. Region VI contains the C-terminus of Tat encoded by the second exon and it shows similarities among Tat variants [34] (Fig. 2).

3D predicted structure of the HIV-1 subtype B Tat protein (subtype B, Isolate MN) (1–86) using Swiss-model webserver. The alpha-helical structure is coloured red and the N and C- terminals are yellow coloured

Multiple sequence alignment of various HIV-1 Tat subtypes. From top to bottom; Tat subtype H (isolate 90CF056), subtype D (isolate ELI), subtype G (isolate SE6165), subtype B (isolate MN), subtype K (isolate 96CM-MP535), subtype A (isolate U455), subtype J (isolate SE9280) and subtype C (isolate 92BR025). Tat protein is encoded by two exons, exon one spans the region of amino acids 1–72 and exon two spans the region of 73–101. The Tat protein is made up of six function regions including the proline-rich region (1–21), the cysteine-rich region (22–37), the core region (38–48), the basic, arginine-rich domain (49–59), the glutamine-rich domain (60–72) and the RGD domain (73–101). The black arrows indicate the two exons
The various regions/domains of the Tat protein serve specific functions. In particular, the N-terminal (1–48) is considered crucial for the activation of transcription. This is due to the cysteine-rich domain (21–37) being multifunctional and required for dimerization, metal binding, and stabilization of the protein structure [35]. In addition, the hydrophobic core motif (38–48) is critical for transactivation activity and also binds to the RNA TAR element of nascent RNA [36]. The arginine-rich domain (49–59) was shown to be important for Tat localization in the nucleus, binding to the HIV long terminal repeat (LTR) TAR RNAs and Tat internalization into cells by binding to cellular heparan sulphate proteoglycans [37,38,39,40]. The glutamine-rich region (60–72) has also been shown to function in TAR interaction and is important for the Tat-apoptosis function [41, 42]. The second exon (73–86) was shown to be less important for transactivation activity, however, studies suggest having a role in virus replication in lymphocytes and macrophages [43,44,45]. Finally, the C-terminal domain, containing the arginylglycylaspartic acid (RGD) motif is essential for binding and signalling through the same integrin receptors α5β1, αvβ3 and αvβ5, which recognize the RGD region of extracellular matrix proteins [46, 47].
Mutations in the tat gene result in variations in its protein amino acid sequence and this may affect Tat function [48]. This can be seen with subtype-specific variation which results in changes to the amino acid sequences within the HIV Tat protein (Fig. 2). Several subtype-specific mutations, specifically the basic Arginine (Arg)-rich region (49–59) exist when comparing the HIV-1 subtypes A, B, C, D, G, H, J and K (Fig. 2). These naturally occurring sequence variations in the Tat protein have been linked to differential pathogenesis [49] and neuropathogenesis [50]. In particular, mutations within the glutamine-rich region (60–72) have been involved in the induction of apoptosis in T-cells [51]. Another study showed that the two-point mutations, L43V and S46F increased the transcriptional activity of Tat and increased its apoptosis induction potential [52]. The naturally occurring glutamate substitution at amino acid 63 that is largely present in subtype C [53] leads to greater transcriptional activity in human CD4 T-cells which are the target of HIV, thus allowing HIV to achieve a high-level of transcription [54]. Further, the Tat substitutions (R57S) have been linked to differential levels of inflammation in cell culture and human studies [40, 55]. Mutations in the Tat protein have also been associated with differential levels of neurotoxicity. An in vivo study has shown that the mutation C22G resulted in significantly less neurotoxicity due to reduced levels of apoptosis [56]. The C22G Tat mutant cannot interact with cyclin-dependent kinase 9 (CDK9) which is critical for RNA Polymerase II (Pol II) transcription initiation and elongation. It is, therefore, transactivation negative [57].
The HIV-1 transactivation response element (TAR) binds to Tat, facilitating viral replication in its latent state [58]. The primary role of Tat is to recognize the 5’-TAR element in the HIV-1 RNA, and its flexible and disorderly structure promotes high-affinity complexes with the RNA [59,60,61]. Tat recruits the positive transcriptional elongation factor (P-TEFb) onto the nascent viral TAR RNA in order to overcome the elongation pause for activation transcription of the entire viral genome [62]. This elongation factor consists of CDK9 and Cyclin T1. In the absence of Tat, P-TEFb exists in the cell as a large inactive complex composed of 7SK snRNA and MAQ1/HEXIM1 proteins [63, 64]. Once the elongation factor is recruited by Tat to TAR RNA, CDK9 phosphorylates the carboxyl-terminal domain of RNA Polymerase II (RNAP II) and thereby activates elongation [61]. The result of these post-translational modifications is the synthesis of high levels of full-length viral transcripts. Tat is known to interact with multiple host factors that ensure the binding affinity of Tat to TAR, however, for the purpose of this review, we were particularly interested in the major interacting partner which is TAR.
As highlighted above, mutations in the Tat protein influence the pathogenesis and neuropathogenesis of HIV-1. Tat mutations may also influence Tat-TAR binding and subsequent viral transcription. Of the two important functional domains of HIV-1 Tat, mutational analysis has shown that the Arg-rich basic region (47–59) is required for binding to TAR RNA [65]. The basic domain located in regions 47–59 of the Tat forms an alpha helix during Tat-TAR binding [51]. Modifications in amino acid sequence on the functional groups of Tat proteins have also been shown to affect hydrogen bonding to TAR RNA, lowering the binding affinity by up to 20-fold [66]. The amino acid substitution S46F in the Tat core region could lead to a conformational change to Tat resulting in more hydrogen bond interactions than in the wild-type making it a highly potent transactivator [67]. In addition, the K51R mutation was shown to make Tat more flexible in this location, giving it a direct hydrogen interaction which is more non-rigid than in the wild-type [68]. Thus, from this knowledge, Tat mutations may affect Tat-TAR interaction and the rate of transcription and ultimately the rate of viral replication.
Although many studies have investigated Tat mutations in Tat-TAR interactions [69,70,71,72], to our best knowledge, no study has summarised findings for which Tat amino acids and/or regions are the most important in Tat-TAR binding affinity and if subtype-specific Tat sequence variation influences Tat-TAR binding. Therefore, the primary aims of this scoping review were to determine; (1) the regions of the Tat protein that may be involved in TAR binding, (2) the key Tat amino acids involved in TAR binding; and (3) if Tat subtype-specific variation influences TAR binding. The secondary aims were to determine (1) the value of undertaking a full systematic review and meta-analysis; and (2) the extent of the available evidence by reviewing all literature on this topic to date. Findings from this study may help further develop our understanding of the subtype-specific Tat function.
Methods
Study design
This is a descriptive and narrative scoping review aimed at synthesising the extant literature of basic/fundamental studies investigating HIV-1 Tat-TAR binding.
Eligibility criteria
For inclusion, studies needed to investigate Tat-TAR interaction/binding to identify key Tat amino acid regions and/or specific amino acids which may influence Tat-TAR binding affinity. Only studies investigating the HIV-1 Tat protein and/or Tat-derived peptides were included. Investigations of all other Tat proteins (HIV-2, bovine Tat etc.) were excluded. Studies that investigated HIV-1 Tat protein amino acid variation in transactivation assays with no direct binding assays were also excluded. Therefore, the studies had to investigate Tat-TAR binding with a relevant binding assay ((e.g., electrophoretic mobility shift assay (EMSA), surface plasmon resonance (SPR) etc.)) to be included. To ensure uniformity in the included studies, only those studies that reported dissociation constants (Kd) as a measure for the binding affinity between Tat-TAR were included. Studies not published in English were excluded and no data was extracted. Review articles, thesis, conference proceedings and book chapters were also excluded.
Data sources
We electronically searched for publications in PubMed, Scopus and Web of Science databases based on all studies published until 28/11/2022. The search strategy was executed without publication date limitations. The full search criteria for each database are included in the Additional file 1: File S1. The following search terms were applied to PubMed: (HIV [mh] OR HIV [tw] OR Acquired Immunodeficiency Syndrome [mh] OR “acquired immunodeficiency syndrome” [tw] OR AIDS [tw]) AND (Gene Products, Tat [mh] OR transactivation of transcription [tw] OR Tat [tw]) AND (transactivating response region [tw] OR TAR [tw] OR Tat-TAR [tw] OR HIV Long Terminal Repeat [mh] OR Tat-TAR binding [tw]).
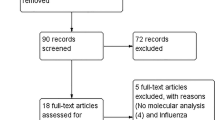
In addition, we also (1) reviewed reference sections of eligible articles and manually searched for relevant publications and (2) consulted with the corresponding authors of the included studies. This search strategy and the retrieved articles are shown in Fig. 3.

Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram for results of the search strategy
Data selection
All articles were retrieved and loaded onto a single database using a reference manager (EndNote X9, Clarivate, PA, USA). Two authors, PTG and MEW independently identified studies meeting the inclusion criteria. Where there was a discrepancy in article inclusion/exclusion, this was discussed amongst all authors, and a decision was made regarding its suitability.
Quality assessment
The quality of the included studies was assessed by PTG and MEW and a kappa statistic was calculated. The quality criterion has been adopted from the CRIS Guidelines (Checklist for Reporting In-vitro Studies) [73]. Here we have amended the CRIS Guidelines by implementing a Likert scale [74] to provide a quantitative measure of study quality. The CRIS guidelines suggest that several areas need to be addressed to promote the quality and transparency of evidence. However, we have selected those areas that may have influenced the findings in the included studies, and these included the reporting of (1) sample size calculation; (2) sample preparation and handling; and (3) statistical analysis. Therefore, these areas were addressed with the following questions (1) were the sample sizes clearly defined; (2) was there a detailed explanation about sample preparation and sample handling to ensure replication of the experimentation; and (3) have appropriate statistical analysis been applied to address the research question. Each question was rated for 0 = no, 1 = partly and 2 = yes. Studies that addressed all the above questions and had a total rating of 6 were classified as high quality. Studies with a rating between 3 and 5 were considered intermediate-quality and less than 3 was low quality.
Results
Study characteristics
Using this criterion and search strategy (section “Eligibility criteria”, “Data sources”), 3708 articles were extracted. Duplicates (n = 1039) were removed, resulting in 2669 studies. Thereafter, abstracts and titles were screened and a total of 2325 studies were excluded which comprised of:
-
Review articles/thesis/book chapters/conference proceedings (n = 618).
-
Studies not investigating HIV-1 Tat-TAR interactions in general (n = 1493).
-
Studies not published in English (n = 10).
-
Studies not investigating HIV-1 in general (n = 175).
-
Studies investigating HIV-2 (n = 23).
-
Studies investigating Tat-TAR interactions, but not investigating Tat amino acids/regions influencing interactions (n = 6).
Full-text articles assessed for eligibility were done for 344 studies, and an additional n = 331 were excluded:
-
Review articles/book chapters/conference proceedings (n = 21).
-
Studies not investigating HIV-1 Tat-TAR interactions in general (n = 14).
-
Studies not investigating HIV-1 in general (n = 6).
-
Studies investigating Tat-TAR interactions, but not investigating Tat amino acids/regions influencing interactions (n = 217).
-
Studies investigating Tat transactivation but not reporting findings for Tat-TAR binding affinities (n = 12).
-
Studies investigating Tat-TAR interactions, but only reporting TAR mutants (n = 5). These studies were excluded because the focus of the manuscript was to identify the possible variation of the Tat amino acids only and not the variation of TAR.
-
Studies investigating Tat-TAR interactions but with the addition of post-translational modifications of Tat amino acids and use of unnatural Tat peptides (e.g., acetylation, methylation etc.) (n = 29). These studies were excluded as we wanted to determine the Tat amino acids involved in TAR binding interaction and affinity without the potential confounding influence of Tat modifications on these interactions.
-
Studies investigating Tat-TAR interaction in the presence of additional interacting partners (e.g., neomycin) (n = 4). These studies were excluded as we wanted to determine the Tat amino acids involved in TAR binding interaction and affinity without the potential confounding influence of additional interacting partners on these interactions.
-
Studies not investigating HIV-1 Tat proteins (e.g., Bovine Tat) (n = 4).
-
Studies not reporting Kd values (n = 14).
-
In silico studies only (n = 5).
Using this criterion (section “Study design”– “Data sources”), a total of 13 fundamental studies were included for data extraction (Fig. 3).
Quality assessment
The quality of the included studies was assessed by PTG and MEW independently and the inter-rater reliability was assessed. The Kappa statistic for inter-rater agreement and reliability was 0.683, indicating substantial agreement [75]. The majority of articles were rated as intermediate (62%) followed by high quality (38%). No study was rated as low quality (Additional file 2: Table S1A and B).
Regions of the Tat protein that influence Tat-TAR binding
Certain regions of the Tat protein may be important for TAR binding. The majority of studies used Tat peptides covering the basic domain (47–58) for investigating Tat-TAR interactions (Table 1). As a baseline, studies investigated binding affinity to TAR using Tat peptides with only the basic domain (47–58). Thereafter, amino acids were added to either domain individually or both the N-terminal and C-terminal domain together and thereafter binding affinity was measured. Of all studies, four studies investigated the addition of amino acids to the Arg-rich domain (47–58) [69, 76,77,78] (Table 1). It is relevant to note that this region has been widely investigated due to its confirmed function in Tat-TAR binding [79], and Tat mutations found in this region have influenced Tat-RNA interaction in vivo [80]. A study found that the addition of only N-terminal domain amino acids resulted in no difference in binding affinity to TAR [69]. This is an interesting finding as others have found the N-terminal domain to be involved in TAR interaction [35, 36]. However, several studies reported that the addition of C-terminal Tat amino acids resulted in increased binding affinity to TAR (> 50% increase in binding affinity) [69, 77, 78]. The addition of both N and C terminal Tat amino acids increased binding affinity to TAR (52–85%) [76] (Table 1). A similar trend was noted when amino acids were removed from full-length Tat proteins (1–86) [36, 81]. Based on these findings, it may be hypothesized that the Tat regions outside the basic domain may be important for Tat-TAR interactions, however, this warrants further investigation. With the removal of N and C terminal amino acids resulting in peptides spanning the region of amino acids 37–72 or 48–72, it was reported that binding affinity was decreased (> 1000%) [36]. Interestingly, one study found that removing the N-terminal amino acids 1–29 resulted in no binding of TAR [81] (Table 1). The significance of these regions/domains of the Tat protein in TAR binding is discussed further in the “Discussion” section.
Multiple and single Tat amino acid substitutions that may influence Tat-TAR binding
Of all included studies, six studies investigated Tat-TAR binding and the influence of multiple amino acid substitutions (Table 2) [69, 70, 72, 82,83,84]. None of the studies investigated naturally occurring Tat subtype-specific mutations. The majority of studies introduced either Lysine (Lys) or Alanine (Ala) to investigate Tat-TAR binding within the cysteine-rich domain (22–38) or basic Arg domain (47–58). One study reported a double substitution in the cysteine-rich domain (22–38), in particular, C34S and C37W which resulted in a lower binding affinity to TAR (19.7%) [70]. All other studies investigated two to eight Tat substitutions within the basic domain (47–58) (Table 2). The substitution of only Ala resulted in the largest percentage decrease in binding affinity across all studies (≥ 1566%) [69, 82]. Because substitution with Ala removes all side-chain atoms past the β-carbon, the effects of individual Ala mutations can be used to infer the roles of wild-type individual side chains and ultimately elucidate the role of particular amino acids in Tat-TAR binding [85]. Studies introducing two to four Lys amino acids only within the basic domain (47–58) resulted in smaller decreases in binding affinity (25%-116%) [69, 83, 84]. Studies reporting five to seven Lys substitutions within the basic domain (47–58) reported much higher decreases in binding affinity (> 1000%) compared to substitutions of two to four Lys amino acids (Table 2). Two studies substituted a combination of multiple Ala, Lys and Arg amino acids within the Tat protein, and this resulted in either no binding [82] or a higher decrease in binding affinity (≥ 250%) compared to wild-type Tat peptides used in the respective studies [82](Table 2). It is also relevant to note that different binding interactions and affinities were recorded by different techniques including Nuclear Magnetic Resonance Spectroscopy (NMR) and Fluorescence spectroscopy [70, 72], Fluorescence resonance energy transfer (FRET), Matrix-assisted laser desorption/ionization-Time of Flight Mass Spectrometry (MALDI-TOFMS) and Fluorescence binding assay [82], EMSA [69, 84], Gel electrophoresis and circular dichroism (CD) [83] and Electron paramagnetic resonance spectroscopy (EPR) [72] (Table 2).
Several studies investigated the influence of single amino acid substitutions in Tat-TAR binding (Table 3). In particular, four studies [69, 71, 82, 86] investigated single amino acid substitutions (Table 3). None of the studies investigated naturally occurring Tat subtype-specific mutations but rather substituted wild-type amino acids with Ala. The majority of studies investigated the basic domain (47–58) as this is the known interacting partner for TAR. The largest decreases in binding affinities (> 1900%) were recorded for the Tat substitutions K49A, K50A, K51A, K53A, K54A and K56A (Table 2). When Lys was mutated to Ala, a significant decrease in binding affinity was observed. In contrast to this, when Arg or Glutamine (Gln) was mutated to Ala, smaller decreases in binding affinity were observed. An example of this is when Arg was mutated (i.e., R5K, R52A, R53A, R55A, R56A), and the binding affinity showed a small decrease (40–70%) compared to when Lys was mutated (i.e., K49A, K50A, K51A, K53A, K54A and K56A) which resulted in a larger decrease in binding affinity (> 1900%) (Table 3). Interestingly, one study reported that the Q54A substitution resulted in an increased binding affinity compared to the wild-type Tat peptide (20% increase) [69] (Table 3). These suggest that the binding affinity of Tat-TAR may be influenced by which amino acids are present in the wild-type Tat protein (Lys, Arg or Gln). Further, this may suggest that, compared to Arg amino acids in the basic domain (47–58), Lys amino acids may be greater contributors to the binding affinity to TAR. It is relevant to note that the binding interaction and affinities were recorded by different techniques SPR [86], Absorption spectroscopy, Gel shift assays, CD Spectroscopy [71], Electrophoretic mobility shift assay (EMSA) [69] and Fluorescence resonance energy transfer (FRET), MALDI-TOFMS, Fluorescence binding assay [82] (Table 3). Even though different techniques were employed across these four studies, a consistent trend was noted that when wild-type amino acids were substituted, a lower binding affinity was observed, confirming the notion that amino acids within the basic domain (47–58) are important for TAR binding. A summary of all key findings is given in Table 4.
Discussion
Several findings were highlighted in this review, and these included (1) both N-terminal and C-terminal amino acids outside the basic domain (47–58) may be important in Tat-TAR binding, (2) substitution of wild-type Tat amino acid Lys and Arg within the basic domain (47–58) results in a reduction in binding to TAR and (3) none of the included studies have investigated Tat subtype-specific substitutions and therefore no commentary could be made regarding which subtype may have a higher Tat-TAR binding affinity. Lastly, a full systematic review/meta-analysis would not be able to be conducted due to the heterogeneity of the available studies.
First, the general consensus is that when studying Tat-TAR interactions, Tat peptides encompassing the basic domain (47–58) may be sufficient for such investigations [31]. However, based on the findings reported in the included studies, we propose that the Tat regions outside the basic domain may be important for Tat-TAR interactions, however, this warrants further investigation. Peptide fragments showed significantly reduced affinities for TAR in comparison to the full-length Tat protein [36, 87]. In line with this, transactivation analysis also revealed that Tat 1–86 is 20-fold more active than Tat 1–57 and it indicates the role played by the sequence 57–86 in post-transactivation [88]. The majority of studies included in this review have used Tat-derived peptides instead of full-length Tat proteins. It may be that researchers opt to use Tat peptides in such experiments due to the difficulty of recombinantly expressing full-length Tat proteins and their inherent toxicity to cells [89], or the difficulty in artificially synthesizing full-length Tat peptides due to the high percentage of cysteine amino acids that may be at risk for oxidation [90]. Here we highlight that full-length Tat protein may be more suited for understanding Tat-TAR binding interactions and its potential downstream effects.
Second, we found that the substitution of wild-type Tat amino acids Lys, Arg and Gln with Ala resulted in decreases in binding affinity. This may be because even though the virus evolves at a high rate [91] certain amino acids may be functionally conserved [31, 32] and therefore any substitution from the wild-type will result in reduced binding. The percentage decrease in binding affinity is influenced by which amino acids were present in the wild-type Tat peptide or protein. In particular, Lys substitutions within the basic Arg domain (47–58) resulted in the largest decrease in TAR binding, and this may suggest that HIV-1 subtypes (e.g., subtype A) with a greater number of Lys amino acids within the basic domain may have higher binding affinities to TAR. Similarly, we found that substituting Arg with Ala, also resulted in a decrease in TAR binding, albeit at a smaller percentage compared to that of Lys. Therefore, it is plausible to suggest that a Lys and Arg-rich basic domain (47–58) may have a higher binding affinity to TAR compared to Tat with a lower percentage of these amino acids in the basic domain (47–58). This may be plausible for subtype B compared to subtype C, whereby subtype B has a greater number of Lys and Arg in the basic domain (47–58). In a previous computational study done within our group, it was found that Tat subtype B had a higher affinity for the TAR RNA element compared to Tat subtype C based on a higher docking score of − 187.37, a higher binding free energy value of − 9834.63 ± 216.17 kJ/mol, and a higher number of proteins–nucleotide interactions of 26 [92]. However, this warrants further molecular investigation.
Third, no study has compared subtype-specific Tat variation and its influence on Tat-TAR binding. All mutations introduced were Ala for the purpose of removing the function of particular wild-type amino acid side chains. Computational studies done by our group [92] and others [67] indicated that subtype-specific variation may influence TAR binding, however molecular validation of such findings remains unstudied. Therefore, it is not clear which subtype variation may influence Tat-TAR binding in the basic domain (47–58) specifically, as well as the domains in the Tat protein. It is therefore important that future studies investigate Tat subtype-specific variation in TAR binding with the use of full-length Tat proteins. Findings from such studies may help explain why we see different levels of HIV-1 pathogenesis and prevalence when comparing HIV-1 subtypes.
Lastly, based on the findings highlighted in this review, several recommendations can be made. Studies were heterogeneous in design and therefore a full systematic review and meta-analysis could not be conducted. Having said this, it may be important to develop a pipeline to allow studies in this line of work to be conducted uniformly. Therefore, studies should clearly define sample sizes used to answer the research question, sample preparation and handling and the relevant statistical analysis. This review also highlights all studies in this particular area of research to date, and it is relevant to note that the majority of studies have been conducted before the year 2000 (53%). Considering the advancement of research techniques in the genome sequencing of HIV-1, more recent investigations may be able to provide clearer reporting of findings. Lastly, no study has investigated subtype-specific mutations. Considering that HIV-1 may be considered a chronic disease, in addition to investigating targets to block Tat-TAR interactions, studies should also investigate which amino acids contribute to the level of transcription, disease phenotypes in PLWH and the prevalence of HIV-1.
Conclusion
The HIV Tat protein binds to TAR which is responsible for the initiation of transcription. Based on the findings reported in this review, we propose that Tat amino acids outside the basic Arg domain (47–58) may be important in Tat-TAR binding and full-length Tat peptides should be used for the investigation of Tat-TAR binding. Further, wild-type Tat containing a higher percentage of Lys and Arg amino acids within the basic domain (47–58) may have a higher binding to TAR. To date, limited studies have investigated subtype-specific variation within the Tat protein and its binding to TAR and therefore, it is not clear which subtype may have the highest binding affinity. Future studies should investigate subtype-specific variation and implement full-length Tat protein in such studies. Findings from such studies will aid in understanding Tat-TAR binding and potential downstream effects. In addition, this may also provide new therapeutic targets to prevent Tat-initiated transcription.
Availability of data and materials
All supporting data (Additional files/tables) is attached to this manuscript.
Abbreviations
- AIDS:
-
Acquired Immune Deficiency Syndrome
- Ala:
-
Alanine
- Arg:
-
Arginine
- ART:
-
Anti-retroviral therapy
- ARV:
-
Anti-retroviral drugs
- CCL2:
-
C–C motif chemokine ligand 2
- CD4:
-
Cluster of differentiation four
- CDK9:
-
Cyclin-dependent kinase 9
- CD:
-
Circular dichroism
- COVID-19:
-
Coronavirus disease 2019
- CRIS:
-
Checklist for Reporting In-vitro Studies
- DNA:
-
Deoxyribonucleic acid
- EMSA:
-
Electrophoretic mobility shift assay
- EPR:
-
Electron Paramagnetic Resonance
- FRET:
-
Fluorescence resonance energy transfer
- Gln:
-
Glutamine
- HAND:
-
HIV associated neurocognitive disorder
- HIV:
-
Human immunodeficiency virus
- IRIS:
-
Immune reconstitution inflammatory syndrome
- kDa:
-
Kilodaltons
- Lys:
-
Lysine
- MALDI-TOFMS:
-
Matrix-assisted laser desorption/ionization-Time of Flight Mass Spectrometry
- Nef:
-
Negative regulatory factor
- NMR:
-
Nuclear magnetic resonance
- PLWH:
-
People living with HIV
- Pol II:
-
RNA Polymerase II
- P-TEFb:
-
Positive transcriptional elongation factor
- RGD:
-
Arginylglycylaspartic acid
- Rev:
-
Regulator of expression of virion proteins
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus type 2
- SPR:
-
Surface plasmon resonance
- TAR:
-
Transactivation response element
- Tat:
-
Transactivation of transcription
- Vif:
-
Viral infectivity factor
- Vpr:
-
Viral protein R
- Vpu:
-
Virus protein U
References
Gelderblom HR, Hausmann EH, Özel M, Pauli G, Koch MA. Fine structure of human immunodeficiency virus (HIV) and immunolocalization of structural proteins. Virology. 1987;156(1):171–6.
Morison L. The global epidemiology of HIV/AIDS. Br Med Bull. 2001;58(1):7–18.
Barré-Sinoussi F, Chermann J-C, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vézinet-Brun F, Rouzioux C. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science. 1983;220(4599):868–71.
Montana JF, Ferreira GRON, Cunha CLF, de Queiroz AAR, Fernandes WAA, Polaro SHI, Gonçalves LHT, Couto DCC, Gir E, Reis RK. The HIV epidemic in Colombia: spatial and temporal trends analysis. BMC Public Health. 2021;21(1):1–14.
UNAIDS. Danger: UNAIDS global AIDS update 2022. In: 2022.
Dorward J, Khubone T, Gate K, Ngobese H, Sookrajh Y, Mkhize S, Jeewa A, Bottomley C, Lewis L, Baisley K, et al. The impact of the COVID-19 lockdown on HIV care in 65 South African primary care clinics: an interrupted time series analysis. Lancet HIV. 2021;8(3):e158–65.
Gatechompol S, Avihingsanon A, Putcharoen O, Ruxrungtham K, Kuritzkes DR. COVID-19 and HIV infection co-pandemics and their impact: a review of the literature. AIDS Res Ther. 2021;18(1):1–9.
Fischer W, Giorgi EE, Chakraborty S, Nguyen K, Bhattacharya T, Theiler J, Goloboff PA, Yoon H, Abfalterer W, Foley BT, et al. HIV-1 and SARS-CoV-2: patterns in the evolution of two pandemic pathogens. Cell Host Microbe. 2021;29(7):1093–110.
Nyamweya S, Hegedus A, Jaye A, Rowland-Jones S, Flanagan KL, Macallan DC. Comparing HIV-1 and HIV-2 infection: lessons for viral immunopathogenesis. Rev Med Virol. 2013;23(4):221–40.
Hemelaar J, Elangovan R, Yun J, Dickson-Tetteh L, Kirtley S, Gouws-Williams E, Ghys PD, Alash’le GA, Agwale S, Archibald C. Global and regional epidemiology of HIV-1 recombinants in 1990–2015: a systematic review and global survey. Lancet HIV. 2020;7(11):e772–81.
Taylor BS, Sobieszczyk ME, McCutchan FE, Hammer SM. The challenge of HIV-1 subtype diversity. N Engl J Med. 2008;358(15):1590–602.
Gartner MJ, Roche M, Churchill MJ, Gorry PR, Flynn JK. Understanding the mechanisms driving the spread of subtype C HIV-1. EBioMedicine. 2020;53: 102682.
Bbosa N, Kaleebu P, Ssemwanga D. HIV subtype diversity worldwide. Curr Opin HIV AIDS. 2019;14(3):153–60.
Hemelaar J, Elangovan R, Yun J, Dickson-Tetteh L, Fleminger I, Kirtley S, Williams B, Gouws-Williams E, Ghys PD. Global and regional molecular epidemiology of HIV-1, 1990–2015: a systematic review, global survey, and trend analysis. Lancet Infect Dis. 2019;19(2):143–55.
Wainberg MA. HIV-1 subtype distribution and the problem of drug resistance. AIDS. 2004;18:S63–8.
Hemelaar J, Gouws E, Ghys PD, Osmanov S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS. 2006;20(16):W13–23.
Hemelaar J, Gouws E, Ghys PD, Osmanov S. Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS. 2011;25(5):679.
Triques K, Bourgeois A, Vidal N, Mpoudi-Ngole E, Mulanga-Kabeya C, Nzilambi N, Torimiro N, Saman E, Delaporte E, Peeters M. Near-full-length genome sequencing of divergent African HIV type 1 subtype F viruses leads to the identification of a new HIV type 1 subtype designated K. AIDS Res Hum Retroviruses. 2000;16(2):139–51.
Piot P, Bartos M. The epidemiology of HIV and AIDS. In: Essex M, Mboup S, Kanki PJ, Marlink RG, Tlou SD, Holme M, editors. AIDS in Africa. Boston: Springer; 2002. p. 200–17.
Buonaguro L, Tornesello M, Buonaguro F. Human immunodeficiency virus type 1 subtype distribution in the worldwide epidemic: pathogenetic and therapeutic implications. J Virol. 2007;81(19):10209–19.
Calvez V, Marcelin A-G, Vingerhoets J, Hill A, Hadacek B, Moecklinghoff C. Systematic review to determine the prevalence of transmitted drug resistance mutations to rilpivirine in HIV-infected treatment-naive persons. Antivir Ther. 2016;21(5):405–12.
Kirchhoff F. HIV life cycle: overview. Encyclopedia AIDS. 2013. https://doi.org/10.1007/978-1-4614-9610-6_60-1.
Doolittle JM, Gomez SM. Structural similarity-based predictions of protein interactions between HIV-1 and Homo sapiens. Virol J. 2010;7(1):1–15.
Frankel AD, Young JA. HIV-1: fifteen proteins and an RNA. Annu Rev Biochem. 1998;67:1–25.
Pugliese A, Vidotto V, Beltramo T, Petrini S, Torre D. A review of HIV-1 Tat protein biological effects. Cell Biochem Funct. 2005;23(4):223–7.
Siddappa NB, Venkatramanan M, Venkatesh P, Janki MV, Jayasuryan N, Desai A, Ravi V, Ranga U. Transactivation and signaling functions of Tat are not correlated: biological and immunological characterization of HIV-1 subtype-C Tat protein. Retrovirology. 2006;3:53.
Shojania S, O’Neil JD. HIV-1 Tat is a natively unfolded protein: the solution conformation and dynamics of reduced HIV-1 Tat-(1–72) by NMR spectroscopy. J Biol Chem. 2006;281(13):8347–56.
Kuppuswamy M, Subramanian T, Srinivasan A, Chinnadurai G. Multiple functional domains of Tat, the trans-activator of HIV-1, defined by mutational analysis. Nucleic Acids Res. 1989;17(9):3551–61.
Campbell GR, Loret EP. What does the structure-function relationship of the HIV-1 Tat protein teach us about developing an AIDS vaccine? Retrovirology. 2009;6(1):1–13.
Jeang K-T, Xiao H, Rich EA. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J Biol Chem. 1999;274(41):28837–40.
Kurnaeva MA, Sheval EV, Musinova YR, Vassetzky YS. Tat basic domain: a “Swiss army knife” of HIV-1 Tat? Rev Med Virol. 2019;29(2): e2031.
Mediouni S, Marcondes MC, Miller C, McLaughlin JP, Valente ST. The cross-talk of HIV-1 Tat and methamphetamine in HIV-associated neurocognitive disorders. Front Microbiol. 2015;6:1164.
Grégoire C, Péloponèse JM Jr, Esquieu D, Opi S, Campbell G, Solomiac M, Lebrun E, Lebreton J, Loret EP. Homonuclear 1H-NMR assignment and structural characterization of human immunodeficiency virus type 1 Tat Mal protein. Biopolymers. 2001;62(6):324–35.
Watkins JD, Campbell GR, Halimi H, Loret EP. Homonuclear 1H NMR and circular dichroism study of the HIV-1 Tat Eli variant. Retrovirology. 2008;5(1):1–11.
Frankel AD, Biancalana S, Hudson D. Activity of synthetic peptides from the Tat protein of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1989;86(19):7397–401.
Churcher MJ, Lamont C, Hamy F, Dingwall C, Green SM, Lowe AD, Butler PJG, Gait MJ, Karn J. High affinity binding of TAR RNA by the human immunodeficiency virus type-1 tat protein requires base-pairs in the RNA stem and amino acid residues flanking the basic region. J Mol Biol. 1993;230(1):90–110.
Hauber J, Malim MH, Cullen BR. Mutational analysis of the conserved basic domain of human immunodeficiency virus tat protein. J Virol. 1989;63(3):1181–7.
Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276(8):5836–40.
Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem. 2001;276(5):3254–61.
Ruiz AP, Ajasin DO, Ramasamy S, DesMarais V, Eugenin EA, Prasad VR. A naturally occurring polymorphism in the HIV-1 Tat basic domain inhibits uptake by bystander cells and leads to reduced neuroinflammation. Sci Rep. 2019;9(1):3308.
Loret E. HIV extracellular Tat: myth or reality? Curr HIV Res. 2015;13(2):90–7.
King JE, Eugenin EA, Buckner CM, Berman JW. HIV tat and neurotoxicity. Microbes Infect. 2006;8(5):1347–57.
Mahlknecht U, Dichamp I, Varin A, Van Lint C, Herbein G. NF-kappaB-dependent control of HIV-1 transcription by the second coding exon of Tat in T cells. J Leukoc Biol. 2008;83(3):718–27.
Neuveut C, Scoggins RM, Camerini D, Markham RB, Jeang KT. Requirement for the second coding exon of Tat in the optimal replication of macrophage-tropic HIV-1. J Biomed Sci. 2003;10(6 Pt 1):651–60.
Sodroski J, Patarca R, Rosen C, Wong-Staal F, Haseltine W. Location of the trans-activating region on the genome of human T-cell lymphotropic virus type III. Science. 1985;229(4708):74–7.
Brake DA, Debouck C, Biesecker G. Identification of an Arg-Gly-Asp (RGD) cell adhesion site in human immunodeficiency virus type 1 transactivation protein, tat. J Cell Biol. 1990;111(3):1275–81.
Barillari G, Gendelman R, Gallo RC, Ensoli B. The Tat protein of human immunodeficiency virus type 1, a growth factor for AIDS Kaposi sarcoma and cytokine-activated vascular cells, induces adhesion of the same cell types by using integrin receptors recognizing the RGD amino acid sequence. Proc Natl Acad Sci USA. 1993;90(17):7941–5.
Kamori D, Ueno T. HIV-1 Tat and viral latency: what we can learn from naturally occurring sequence variations. Front Microbiol. 2017;8:80.
Spector C, Mele AR, Wigdahl B, Nonnemacher MR. Genetic variation and function of the HIV-1 Tat protein. Med Microbiol Immunol. 2019;208(2):131–69.
Williams ME, Zulu SS, Stein DJ, Joska JA, Naudé PJW. Signatures of HIV-1 subtype B and C Tat proteins and their effects in the neuropathogenesis of HIV-associated neurocognitive impairments. Neurobiol Dis. 2020;136: 104701.
Campbell GR, Pasquier E, Watkins J, Bourgarel-Rey V, Peyrot V, Esquieu D, Barbier P, de Mareuil J, Braguer D, Kaleebu P. The glutamine-rich region of the HIV-1 Tat protein is involved in T-cell apoptosis. J Biol Chem. 2004;279(46):48197–204.
Lata S, Ronsard L, Sood V, Dar SA, Ramachandran VG, Das S, Banerjea AC. Effect on HIV-1 gene expression, Tat-Vpr interaction and cell apoptosis by natural variants of HIV-1 Tat exon 1 and Vpr from Northern India. PLoS ONE. 2013;8(12): e82128.
de Almeida SM, Rotta I, Vidal LRR, Dos Santos JS, Nath A, Johnson K, Letendre S, Ellis RJ. HIV-1C and HIV-1B Tat protein polymorphism in Southern Brazil. J Neurovirol. 2021;27(1):126–36.
Kurosu T, Mukai T, Komoto S, Ibrahim MS, Li YG, Kobayashi T, Tsuji S, Ikuta K. Human immunodeficiency virus type 1 subtype C exhibits higher transactivation activity of Tat than subtypes B and E. Microbiol Immunol. 2002;46(11):787–99.
Williams ME, Ruhanya V, Paul RH, Ipser JC, Stein DJ, Joska JA, Naudé PJW. An investigation of the HIV Tat C31S and R57S mutation on peripheral immune marker levels in South African participants: A pilot study. J Med Virol. 2022;94(7):2936–8.
Aksenov MY, Aksenova MV, Mactutus CF, Booze RM. Attenuated neurotoxicity of the transactivation-defective HIV-1 Tat protein in hippocampal cell cultures. Exp Neurol. 2009;219(2):586–90.
Donahue DA, Kuhl BD, Sloan RD, Wainberg MA. The viral protein Tat can inhibit the establishment of HIV-1 latency. J Virol. 2012;86(6):3253–63.
Paul R, Dutta D, Paul R, Dash J. Target-directed azide-alkyne cycloaddition for assembling HIV-1 TAR RNA binding ligands. Angew Chem Int Ed Engl. 2020;59(30):12407–11.
Marino J, Maubert ME, Mele AR, Spector C, Wigdahl B, Nonnemacher MR. Functional impact of HIV-1 Tat on cells of the CNS and its role in HAND. Cell Mol Life Sci. 2020;77(24):5079–99.
Karn J. Tackling tat. J Mol Biol. 1999;293(2):235–54.
Rice AP. The HIV-1 Tat protein: mechanism of action and target for HIV-1 cure strategies. Curr Pharm Des. 2017;23(28):4098–102.
Ning S, Zeng C, Zeng C, Zhao Y. The TAR binding dynamics and its implication in Tat degradation mechanism. Biophys J. 2021;120(23):5158–68.
Michels AA, Nguyen VT, Fraldi A, Labas V, Edwards M, Bonnet F, Lania L, Bensaude O. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol Cell Biol. 2003;23(14):4859–69.
Yik JH, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell. 2003;12(4):971–82.
Romani B, Engelbrecht S, Glashoff RH. Functions of Tat: the versatile protein of human immunodeficiency virus type 1. J Gen Virol. 2010;91(1):1–12.
Hamy F, Asseline U, Grasby J, Iwai S, Pritchard C, Slim G, Butler PJ, Karn J, Gait MJ. Hydrogen-bonding contacts in the major groove are required for human immunodeficiency virus type-1 tat protein recognition of TAR RNA. J Mol Biol. 1993;230(1):111–23.
Ronsard L, Rai T, Rai D, Ramachandran VG, Banerjea AC. In silico analyses of subtype specific HIV-1 Tat-TAR RNA interaction reveals the structural determinants for viral activity. Front Microbiol. 2017;8:1467.
Pantano S, Tyagi M, Giacca M, Carloni P. Amino acid modification in the HIV-1 Tat basic domain: insights from molecular dynamics and in vivo functional studies. J Mol Biol. 2002;318(5):1331–9.
Calnan BJ, Biancalana S, Hudson D, Frankel AD. Analysis of arginine-rich peptides from the HIV Tat protein reveals unusual features of RNA-protein recognition. Genes Dev. 1991;5(2):201–10.
Metzger AU, Bayer P, Willbold D, Hoffmann S, Frank RW, Goody RS, Rösch P. The interaction of HIV-1 Tat (32–72) with its target RNA: a fluorescence and nuclear magnetic resonance study. Biochem Biophys Res Commun. 1997;241(1):31–6.
Tao J, Frankel AD. Electrostatic interactions modulate the RNA-binding and transactivation specificities of the human immunodeficiency virus and simian immunodeficiency virus Tat proteins. Proc Natl Acad Sci. 1993;90(4):1571–5.
Edwards TE, Okonogi TM, Sigurdsson ST. Investigation of RNA-protein and RNA-metal ion interactions by electron paramagnetic resonance spectroscopy. The HIV TAR-Tat motif. Chem Biol. 2002;9(6):699–706.
Emmerich CH, Harris CM. Minimum information and quality standards for conducting, reporting, and organizing in vitro research. Handb Exp Pharmacol. 2019;257:177–96.
Likert R. A technique for the measurement of attitudes. Arch Psychol. 1932;22(140):55–55.
McHugh ML. Interrater reliability: the kappa statistic. Biochem Med (Zagreb). 2012;22(3):276–82.
Cao H, Tamilarasu N, Rana TM. Orientation and affinity of HIV-1 Tat fragments in Tat—TAR complex determined by fluorescence resonance energy transfer. Bioconjug Chem. 2006;17(2):352–8.
Tassew N, Thompson M. Kinetic characterization of TAR RNA–Tat peptide and neomycin interactions by acoustic wave biosensor. Biophys Chem. 2003;106(3):241–52.
Weeks KM, Ampe C, Schultz SC, Steitz TA, Crothers DM. Fragments of the HIV-1 Tat protein specifically biand TAR RNA. Science. 1990;249(4974):1281–5.
Li W, Li G, Steiner J, Nath A. Role of Tat protein in HIV neuropathogenesis. Neurotox Res. 2009;16(3):205–20.
Cordingley MG, LaFemina RL, Callahan PL, Condra JH, Sardana VV, Graham DJ, Nguyen TM, LeGrow K, Gotlib L, Schlabach AJ. Sequence-specific interaction of Tat protein and Tat peptides with the transactivation-responsive sequence element of human immunodeficiency virus type 1 in vitro. Proc Natl Acad Sci. 1990;87(22):8985–9.
Chaloin O, Peter J-C, Briand J-P, Masquida B, Desgranges C, Muller S, Hoebeke J. The N-terminus of HIV-1 Tat protein is essential for Tat-TAR RNA interaction. Cell Mol Life Sci. 2005;62:355–61.
Matsumoto C, Hamasaki K, Mihara H, Ueno A. A high-throughput screening utilizing intramolecular fluorescence resonance energy transfer for the discovery of the molecules that bind HIV-1 TAR RNA specifically. Bioorg Med Chem Lett. 2000;10(16):1857–61.
Long KS, Crothers DM. Interaction of human immunodeficiency virus type 1 Tat-derived peptides with TAR RNA. Biochemistry. 1995;34(27):8885–95.
Gelman MA, Richter S, Cao H, Umezawa N, Gellman SH, Rana TM. Selective binding of TAR RNA by a Tat-derived β-peptide. Org Lett. 2003;5(20):3563–5.
Weiss GA, Watanabe CK, Zhong A, Goddard A, Sidhu SS. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc Natl Acad Sci. 2000;97(16):8950–4.
Borkar AN, Bardaro MF, Camilloni C, Aprile FA, Varani G, Vendruscolo M. Structure of a low-population binding intermediate in protein-RNA recognition. Proc Natl Acad Sci. 2016;113(26):7171–6.
Link RW, Mele AR, Antell GC, Pirrone V, Zhong W, Kercher K, Passic S, Szep Z, Malone K, Jacobson JM. Investigating the distribution of HIV-1 Tat lengths present in the Drexel medicine CARES cohort. Virus Res. 2019;272: 197727.
Jeyapaul J, Reddy MR, Khan SA. Activity of synthetic tat peptides in human immunodeficiency virus type 1 long terminal repeat-promoted transcription in a cell-free system. Proc Natl Acad Sci. 1990;87(18):7030–4.
Weeks B, Lieberman D, Johnson B, Roque E, Green M, Loewenstein P, Oldfield E, Kleinman H. Neurotoxicity of the human immunodeficiency virus type 1 Tat transactivator to PC12 cells requires the Tat amino acid 49–58 basic domain. J Neurosci Res. 1995;42(1):34–40.
Moroder L, Musiol HJ, Götz M, Renner C. Synthesis of single- and multiple-stranded cystine-rich peptides. Biopolymers. 2005;80(2–3):85–97.
Yeo JY, Goh GR, Su CT, Gan SK. The Determination of HIV-1 RT mutation rate, its possible allosteric effects, and its implications on drug resistance. Viruses. 2020;12(3):297.
Williams ME, Cloete R. Molecular modeling of subtype-specific Tat protein signatures to predict Tat-TAR interactions that may be involved in HIV-associated neurocognitive disorders. Front Microbiol. 2022;13:866611–866611.
Acknowledgements
Not applicable.
Funding
Open access funding provided by North-West University. MW was funded by DSI-NRF Research Development Grants for new Generation of Academics Programme (nGAP) Scholars and NRF Thuthuka grant (TTK22031652). PTG was funded by the Poliomyelitis Research Foundation (22/91).
Author information
Authors and Affiliations
Contributions
MEW and PTG conceptualized the study and generated the first draft. MEW and PTG conducted all database scans and articles for inclusion and generated all tables and figures. RvDS provided conceptual input and reviewed all drafts of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Database search terms.
Additional file 2: Table S1. A
Quality of assessment of studies conducted by PTG. B Quality of assessment of studies conducted by MEW.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gotora, P.T., van der Sluis, R. & Williams, M.E. HIV-1 Tat amino acid residues that influence Tat-TAR binding affinity: a scoping review. BMC Infect Dis 23, 164 (2023). https://doi.org/10.1186/s12879-023-08123-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-023-08123-0