
Abstract
Background
The efficacy of early treatment with convalescent plasma in patients with COVID-19 is debated. Nothing is known about the potential effect of other plasma components other than anti-SARS-CoV-2 antibodies.
Methods
To determine whether convalescent or standard plasma would improve outcomes for adults in early phase of Covid19 respiratory impairment we designed this randomized, three-arms, clinical trial (PLACO COVID) blinded on interventional arms that was conducted from June 2020 to August 2021. It was a multicentric trial at 19 Italian hospitals. We enrolled 180 hospitalized adult patients with COVID-19 pneumonia within 5 days from the onset of respiratory distress. Patients were randomly assigned in a 1:1:1 ratio to standard of care (n = 60) or standard of care + three units of standard plasma (n = 60) or standard of care + three units of high-titre convalescent plasma (n = 60) administered on days 1, 3, 5 after randomization. Primary outcome was 30-days mortality. Secondary outcomes were: incidence of mechanical ventilation or death at day 30, 6-month mortality, proportion of days with mechanical ventilation on total length of hospital stay, IgG anti-SARS-CoV-2 seroconversion, viral clearance from plasma and respiratory tract samples, and variations in Sequential Organ Failure Assessment score. The trial was analysed according to the intention-to-treat principle.
Results
180 patients (133/180 [73.9%] males, mean age 66.6 years [IQR 57–73]) were enrolled a median of 8 days from onset of symptoms. At enrollment, 88.9% of patients showed moderate/severe respiratory failure. 30-days mortality was 20% in Control arm, 23% in Convalescent (risk ratio [RR] 1.13; 95% confidence interval [CI], 0.61–2.13, P = 0.694) and 25% in Standard plasma (RR 1.23; 95%CI, 0.63–2.37, P = 0.544). Time to viral clearance from respiratory tract was 21 days for Convalescent, 28 for Standard plasma and 23 in Control arm but differences were not statistically significant. No differences for other secondary endpoints were seen in the three arms. Serious adverse events were reported in 1.7%, 3.3% and 5% of patients in Control, Standard and Convalescent plasma arms respectively.
Conclusions
Neither high-titer Convalescent nor Standard plasma improve outcomes of COVID-19 patients with acute respiratory failure.
Trial Registration Clinicaltrials.gov Identifier: NCT04428021. First posted: 11/06/2020
Similar content being viewed by others

Background
Given the lack of evidence for effective treatment of COVID-19 during the first wave of pandemic, empirical and historical interventions have re-emerged as options for the control of the disease. That is the case of convalescent plasma, which has been considered an emergency intervention in several pandemics [1,2,3,4,5]. Initially available observational or control matched studies on COVID-19 patients were encouraging, suggesting that COVID-19 Convalescent Plasma (CCP) could reduce mortality, improve clinical outcomes, and confirming its safety [6,7,8,9,10,11,12,13]. The majority of those studies suggested that treatment in early phases of infection and high titer antibodies could represent the keys for its efficacy. However, at the time the current trial was designed, it had not been investigated whether the potential efficacy of CCP could be attributable only to its specific antibody content or if other substances in plasma, as anti-inflammatory cytokines and natural or acquired antibodies, could exert positive immunomodulation effects. Thus, due to tolerability and potential benefits at that time, we designed a 3-arms randomized trial to explore the effectiveness of high titer CCP or Standard Plasma (SP) in early phases of infection as therapeutic options to add to Standard of Care (SC) to control short and long-term progression of the disease.
Preliminary results on 14-days mortality of control and COVID-19 Convalescent Plasma arms have been included in an international metanalysis on published, unpublished and ongoing randomized trials all over the world [14].
Methods
This study was a randomized, three-arms, blinded on interventional arms, multicentric trial conducted at 19 hospitals (listed in the Study Protocol in Additional file 1) in Piedmont and Valle d’Aosta Regions (North-Western Italy).
An independent Data and Safety Monitoring Committee was settled to verify study protocol, trial conduction and perform an interim analysis to assess safety and efficacy at 40% of enrolment. The authors take full responsibility for the design, conduct, and analysis of the trial in adherence to the study protocol and guarantee the accuracy and completeness of the data.
Hospitalized adults (age > 18 yrs) with a reverse-transcriptase–polymerase-chain-reaction (RT-PCR) confirmed SARS-CoV-2 infection on a nasopharyngeal swab or bronchoalveolar lavage, and a radiologically confirmed pneumonia with a respiratory impairment onset within five days were eligible for enrollment.
Exclusion criteria were: pregnancy, previous severe reactions to plasma infusion, and unavailability of AB0 compatible CCP.
After assessing eligibility and availability of AB0 compatible CCP, treating physicians informed hospitalized patients about the trial protocol and asked to sign a written informed consent. Those who accepted, after entering the baseline data on EPICLIN (https://new.epiclin.it/it/placo/), a website-based platform, were automatically stratified by severity of respiratory impairment in three groups:
-
mild: partial pressure of oxygen (PaO2) ≥ 60 mmHg in ambient air (aa) with non-invasive supplemental oxygen
-
moderate: PaO2 < 60 mmHg in aa in non-invasive ventilation (NIV) or in Continuous Positive Airway Pressure (CPAP)
-
severe: suspected or confirmed acute respiratory distress syndrome (ARDS) in CPAP or mechanical ventilation (MV) ± Extra Corporeal Membrane Oxygenation (ECMO). ARDS (according to Berlin definition) was suspected when a rapid reduction of PaO2/FIO2 towards 300 mmHg was observed.
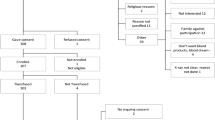
and then randomized in a 1:1:1 ratio according to a computerized generated sequence (for details on randomization see the Study Protocol available in the online version—see Supplementary Information). Study flow is presented in Fig. 1.

Study flow with patients enrolled, randomized and analyzed for primary endpoint. SC: Standard of Care therapy (control arm). SP: Standard Plasma (experimental arm). CCP: Covid-19 Convalescent Plasma (experimental arm). *The patient withdrew the consent the day after randomization before starting treatment. **Four patients died after second SP infusion. ***2 patients died after first and 2 after second CCP infusion, 1 patient withdrew the consent after second infusion because of a moderate allergic reaction
The trial used a blinded interventional arm design. The web-based random procedure was unpredictable by all those involved in the study, who received only the assignment either to the standard arm or to the experimental arms with plasma. Only the transfusion Centers knew the type of plasma (SP or CCP) assigned to patients in the experimental arms. They were responsible for blinding the three plasma bags (SP or CCP) and accompanying certificates, with black tags reporting “TRIAL PLASMA”.
Participants were randomized to receive either Standard of care or SC + three units of Standard Plasma (SP) collected in the pre-COVID-19 era (before September 2019), or SC + three units of high-titre CCP.
The SC was not strictly defined, but the trial protocol recommended to follow national or international updated guidelines for COVID-19. CCP was collected in May and June 2020 from donors recovered from first-wave COVID-19 infection, when the predominant variant in our area was the 20A S614G lineage (https://clades.nextstrain.org/). The plasma units were administered on days 1, 3, and 5 after randomization. Plasma infusion was discontinued whether a severe life-threatening reaction to transfusion happened or in case of withdrawal of written consent for any reason.
97% of the units used in the trial had IgG anti-SARS-CoV-2 > 40 Arbitrary Units (AU)/ml by a quantitative Chemiluminescence-Immunoassay (CLIA) (LIAISON® SARS-CoV-2 S1/S2 IgG) showing to be concordant with Plaque Reduction Neutralization Test (PRNT): 40AU/ml = PRNT titer > 1:80. The three doses (100–300 ml each) of CCP were chosen possibly from different donors to reach a total median administered amount of 70.000 AU of neutralizing antibodies.
A detailed description of COVID-19 convalescent donors and of CCP process methods are presented in Additional file 1.
The primary outcome was 30-days mortality rate.
Secondary outcomes were: incidence of mechanical ventilation (MV) or death at day 30, 6-month mortality rate, proportion of days with MV (originally defined as days in ICU) on total length of hospital stay, proportion of patients showing seroconversion to IgG anti-SARS-CoV-2, viral clearance by RT-PCR on plasma and respiratory tract samples, and variations in Sequential Organ Failure Assessment (SOFA) score from randomization, with assessments on day 2, 4, 6, 10, 14, 21, 28 or until discharge or death. Laboratory methods of SARS-CoV-2 RNA extraction and quantitation, and SOFA score are described in Additional file 1.
Complete data were not available for the other endpoint defined in the protocol (proportion of patients with drug treatment modification).
Safety outcome was the proportion of patients developing any Adverse Events (AEs) assessed daily from randomization to day 30 or discharge or death. Definition of AEs is reported in Additional file 1, as well as complete participant timeline for blood tests and clinical parameters to be reported on daily Case Report Forms.
The study design involves comparing each of the two experimental arms with the control without correction for multiplicity [15]. We calculated a sample size of 180 patients (58 per arm, rounded to 60) to assess a reduction from 25 to 10% of 30-days mortality (primary endpoint), with an alpha error (1-tail) of 0.10 and a statistical power of 80%.
The trial was analysed according to the intention-to-treat principle.
The comparisons of the proportion of deaths and of MV or death at 30 days and proportion of deaths at 6 months were stratified (by the severity of respiratory failure) and estimated in terms of RR with a Mantel–Haenszel Chi-square test. A secondary analysis, adjusted for the stratification criterion and few unbalanced critical prognostic factors (age, sex, BMI, CCI, blood group), was conducted with a Poisson regression model to estimate adjusted RRs. Subgroup analyses for the primary endpoint were performed, including in the same model the interaction terms between treatment arms and the following variables: (a) planned: stratification level, age group (< 65; 65–74; 75 + years), sex, and (b) exploratory: blood group (A vs others), days from symptom onset to randomization (0–5, 6–10, ≥ 11) and the viraemic and serologic test results (positive, negative) at baseline. Cumulative incidence of virus clearance from plasma and respiratory tract samples and seroconversion to IgG anti-SARS-CoV-2 were compared in terms of sub-distribution Hazard Ratios (sHR) with Fine and Gray models, considering death or discharge as competing events.
We compared the percentage of MV days using an ordinal logistic model and variations of serum IgG anti-SARS-CoV-2 levels and SOFA scores during hospitalization using generalized linear mixed models for repeated measures. Statistical analyses were performed with SAS v. 9.4 and STATA v.15.
Results
From June 2020 to February 2021, 180 patients (73.9% males) were enrolled in the trial, the majority between October 2020 and January 2021, during the second pandemic wave; follow-up ended in December 2021. Demographic, clinical characteristics and treatment at baseline of the enrolled patients are listed in Table 1. The median age was 66.6 years (IQR 57.0–73.0). Most patients (88.9%) showed moderate to severe respiratory failure at enrollment, with a mean SOFA score of 2.99 (SD 1.66).
The three arms were well balanced for COVID-19 related variables, with some unbalances for age, sex, BMI and blood groups.
The mean amount of IgG anti-SARS-CoV-2 administered to a single patient with three doses of CCP was 93,431 AU, comparable to three 350 ml units with a PRNT > 1:160. 56/60 patients (93.3%) in the SP arm and 54/60 (90%) in the CCP arm completed plasma infusion. Eight patients (four in each experimental arm) died within day 5 (2 deaths after the first infusion in CCP arm and 6 deaths after the second infusion: 4 in the SP arm and 2 in CCP arm). Furthermore, in the CCP arm, two patients withdrew consent for infusion, one, the day after enrollment, before the first infusion and one after the second infusion, because of a moderate allergic reaction.
Overall, with 41 deaths out of 180, the overall 30-days mortality rate was 22.8% (95%CI: 17.3–29.4), with increasing risks according to the severity of respiratory failure at enrollment (mild: 10%, intermediate: 13.7% and severe: 40%).
In comparison with patients treated with SC, who experienced a 30-days mortality of 20%, no reductions were seen for patients treated with CCP (23.3%; RR 1.13; 95%CI, 0.61–2.13, P = 0.694) or with SP (25.0%; RR 1.23; 95%CI, 0.63–2.37, P = 0.544) (Fig. 2A and Table 2). These results were confirmed with a multivariable model including age, sex, BMI, CCI, blood group, and the stratification variable (severity of respiratory failure) (Additional file 1: Table 1s) as well as by subgroup analyses (Fig. 3).

Cumulative incidence of death (A) and mechanical ventilation or death (B) by treatment arm. *Risk ratio of 30-day mortality, stratified by severity of respiratory impairment. **Risk ratio of 30-day mechanical ventilation or death, stratified by severity of respiratory impairment

Forest plot with subgroup comparisons of Standard plasma vs Control (A) and COVID-19 Convalescent plasma vs Control (B). *Not estimated because no events were observed in Standard Plasma arm
Incidence of the composite endpoint of MV or death within 30 days was not improved in the experimental arms compared to SC (Fig. 2B and Table 2).
At 6 months, with 46 deaths out of 180, the overall mortality rate was 25.6% (95%CI 19.7–32.4). In comparison with patients treated with SC, no clear differences were seen for patients treated with SP (RR 0.98; 95%CI, 0.55–1.76, P = 0.951) or with CCP (RR 0.85; 95%CI, 0.48–1.53, P = 0.600).
Sixty-eight patients (38%) had undetectable IgG anti SARS-CoV-2 at enrollment, 51 patients (28%) had an antibody titer lower than that of CCP units (< 40 AU/ml), while 61 (34%) had an antibody titer > 40 AU/ml. Time to seroconversion was slightly shorter for CCP (sHR 1.54) and SP (sHR 1.38), but the differences between medians were minor (1 day) and statistically weak (Additional file 1: Figure 1sA). 127 patients (70.6%) showed the presence of SARS-CoV-2-RNA by RT-PCR in plasma samples at baseline and other 17 during follow-up. Viremia became negative in a median time of 5 or 6 days (IQR 4–6), without differences between arms (Additional file 1: Figure 1sB).
Median time to viral clearance in the respiratory tract was reached in a slightly shorter time in the CCP arm (21 days) than in SC (23 days), but the difference was not statistically sound (Additional file 1: Figure 1sC).
The median length of hospital stay for the entire population was 15 days, slightly shorter in patients who received CCP or SP than SC (14 and 15.5 vs 17.5, respectively).
The proportion of days of MV on the total length of hospital stay was 10.2%, without meaningful differences between the three arms. A mean reduction from baseline of − 0.70 (95% CI, 1.57–0.15, P = 0.107) of the SOFA score during hospitalization was recorded in patients in the CCP arm compared to the SC arm. In contrast, no apparent differences in IgG seroconversions between arms were recorded (Additional file 1: Figure 2sA and B).
A descriptive analysis of the frequency and percentage of patients with altered laboratory values at baseline and during hospitalization (within 30 days since randomization) and 30-day mortality, by treatment arm, is reported in Additional file 1: Table 2s. As expected, for all the variables analysed, especially for D-Dimer and CRP, a strong positive association between altered values and increased mortality was evident.
Details of AEs are described in the Additional file 1: Table 3s. We observed 4 AEs to plasma infusion, 2 in each arm. Severe AEs were: 3 pulmonary thromboembolism, 1 massive cerebral hemorrhage during ECMO, 1 myocardial infarction, 1 iatrogenic pneumothorax (reported in 1.7%, 3.3% and 5% of patients in Control, SP and CCP arms respectively) and 41 deaths (for respiratory failure in all cases).
Discussion
At variance with most previous non randomized studies, but in accordance with nearly all randomized controlled trials, we failed to demonstrate any significant improvement of relevant clinical outcomes adding COVID-19 convalescent plasma to the standard of care even though we tried to anticipate as much as possible the treatment and to use high doses of antibodies [10, 16,17,18,19,20,21,22,23,24,25,26,27,28]. A trend to a shorter length of hospital stays and a reduction in MV incidence and duration through the hospital stay was seen for CCP treatment, but differences were small and statistically weak. Moreover, no improvement was seen in outcomes by adding SP to SC, thus proving that anti-inflammatory cytokines and natural or acquired antibodies contained in standard plasma did not help in this phase of COVID-19 disease.
Our inclusion criteria permitted enrollment within 5 days since onset of respiratory impairment that was the shortest possible interval considering standards for inpatients in our hospitals (patients were advised to come to the hospital only if respiratory impairment was present). This led to a median interval of 8 days (range 5–12) since the onset of symptoms. Clinical results of our trial are in accordance with those of most randomized controlled studies published to date [14, 17,18,19, 24, 26, 27, 29, 30], that showed no efficacy of CCP for patients with comparable time since onset of symptoms. Different results, with a reduced risk of evolution of the disease, have only been shown in a single randomized trial [25] that used CCP within 3 days since onset of symptoms, before pneumonia and its complications became clinically evident at variance with other two more recent papers that failed to confirm the efficacy of early use [22, 31]. Our subgroup analyses only suggest a decreasing effect of CCP with increasing time from symptom onset, but the evidence is weak.
In contrast to a randomized trial that was interrupted because 79% of patients at enrollment were showing comparable antibodies titers than CCP [18], in our study only 34% of patients had antibodies titers higher than the lower CCP antibody titers (40 AU/ml) so most patients being in a very early phase of infection before an immune response was appreciable. Nevertheless, even in this early phase, when antibodies titers are not yet raised, passive immunotherapy didn’t seem to play a crucial role in shortening disease history, preventing complication or ameliorating clinical outcomes.
It has also been suggested that the titer of neutralizing antibodies plays a crucial role in the effectiveness of CCP treatment [28]. Our trial neutralizing antibodies total dose, even though no precise comparison amongst trials can be made to date, can be considered one of the highest administered in a randomized trial so far. We enlarged the total volume of CCP administered to patients compared to other studies (3 units). All our plasma units were tested with an ELISA assay that correlates with PRNT, and we estimated a mean infusion of three 350 ml units with a PRNT > 1:160. All participants received plasma from at least two donors (15 patients from 3 donors) trying to increase antibodies heterogenicity. Furthermore, what differs from other treatments is the attempt to standardize the total amount of antibodies administered per patient. A mean total dose of 93,000 AU of antibodies was administered to patients in CCP arm. Nevertheless, no advantage in outcomes was seen with this high dose strategy compared to SC in this phase of the disease. In light of suggestions from a recent paper [29] showing that patients treated with high levels of anti-Spike protein CCP showed worse outcomes, the ELISA test we used, detecting anti-Spike-protein antibodies, despite the excellent correlation with PRNT, could have selected CCP with unfavorable antibody profile for COVID-19 patients’ treatment.
In our study, most enrolled patients (88.9%) were affected by moderate to severe respiratory failure. This selection reflects the greater propensity of physicians to propose study participation to most severe than to mild cases that could be considered a limit of our study. The high prevalence of patients (80%) with detectable SARS-CoV-2 viremia, one of the highest described in the literature, confirms the severity of the disease in our cohort of patients. Our results confirm previous studies showing worse outcomes and increased mortality in plasma RNA + patients, irrespective of treatment [32,33,34,35,36]. Furthermore, CCP did not increase the clearance of SARS-CoV-2 viremia from plasma, indicating that passive immunization does not play a key role for infection in this phase of the disease.
A slightly faster clearance of virus from respiratory tract samples was seen in CCP patients in accordance with other data [6, 7, 16, 18]. Still, this difference was not statistically sound and is of questionable clinical relevance.
On the other side, no meaningful difference was seen in our trial in number or types of AEs between three arms of treatment confirming the safety of SP and CCP in this subset of patients [37, 38]. The three cases of thromboembolism observed in our trial, one in each treatment arm, were not related to plasma infusion. With strict daily monitoring of possible AEs, our trial confirms that this amount of plasma (mean = 230 ml, equivalent to 3 ml per Kg) is neutral in terms of coagulation processes in vivo, probably providing a balanced amount of procoagulants and anticoagulant factors.
The inclusion of a study arm with SP, the careful selection of CCP units to administer a comparable dose of antibodies to all treated patients, the masking of the plasma bags, the evaluation of SARS-CoV-2-RNA on patients plasma and in the respiratory tract over time, the strict monitoring of clinical data and the 6 months follow-up represent the originality and the strengths of our study. Due to the substantial expected benefits, the relatively small sample size is the major limitation of our study.
Conclusions
Our study supports the findings from almost all randomized controlled trials that CCP does not offer meaningful therapeutical advantages over standard care in fighting against COVID-19 disease and its complications after the onset of respiratory failure and confirms that there is no reason to continue to use CCP in this subset of patients. Furthermore, it underlines that SP and its potential immune-modulatory effect has no impact on this clinical condition.
Availability of data and materials
The datasets used and/or analysed during the current study are not publicly available due to database complexity for structure and amount of collected data, that request a precise and clear definition of dataset requested and precise objectives before sharing. Data are in any case available from the corresponding author on reasonable request.
Abbreviations
- CCP:
-
COVID-19 convalescent plasma
- SP:
-
Standard plasma
- SC:
-
Standard of care
- RT-PCR:
-
Reverse-transcriptase–polymerase-chain-reaction
- PaO2:
-
Partial pressure of oxygen
- AA:
-
Ambient air
- NIV:
-
Non-invasive ventilation
- CPAP:
-
Continuous Positive Airway Pressure
- ARDS:
-
Acute respiratory distress syndrome
- MV:
-
Mechanical ventilation
- ECMO:
-
Extra Corporeal Membrane Oxygenation
- AU:
-
Arbitrary units
- CLIA:
-
Chemiluminescence-Immunoassay
- PRNT:
-
Plaque Reduction Neutralization Test
- SOFA score:
-
Sequential Organ Failure Assessment score
- AEs:
-
Adverse events
- sHR:
-
Sub-distribution Hazard Ratios
References
Maiztegui JI, Fernandez NJ, de Damilano AJ. Efficacy of immune plasma in treatment of Argentine haemorrhagic fever and association between treatment and a late neurological syndrome. Lancet Lond Engl. 1979;2:1216–7.
Ko J-H, Seok H, Cho SY, Eun Ha Y, Baek JY, Kim SH, et al. Challenges of convalescent plasma infusion therapy in Middle East respiratory coronavirus infection: a single centre experience. Antivir Ther. 2018;23:617–22.
Cheng Y, Wong R, Soo YOY, Wong WS, Lee CK, Ng MHL, et al. Use of convalescent plasma therapy in SARS patients in Hong Kong. Eur J Clin Microbiol Infect Dis. 2005;24:44–6.
Hung IF, To KK, Lee C-K, Lee K-L, Chan K, Yan W-W, et al. Convalescent plasma treatment reduced mortality in patients with severe pandemic influenza A (H1N1) 2009 virus infection. Clin Infect Dis. 2011;52:447–56.
Zhou B, Zhong N, Guan Y. Treatment with convalescent plasma for influenza A (H5N1) infection. N Engl J Med. 2007;357:1450–1.
Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J, et al. Treatment of 5 critically ill patients with covid-19 with convalescent plasma. JAMA. 2020;323:1582–9.
Duan K, Liu B, Li C, Zhang H, Yu T, Qu J, et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc Natl Acad Sci. 2020;117:9490–6.
Joyner MJ, Senefeld JW, Klassen SA, Mills JR, Johnson PW, Theel ES, et al. Effect of convalescent plasma on mortality among hospitalized patients with COVID-19: initial three-month experience. Infect Dis (except HIV/AIDS); 2020.
Abolghasemi H, Eshghi P, Cheraghali AM, Imani Fooladi AA, Bolouki Moghaddam F, Imanizadeh S, et al. Clinical efficacy of convalescent plasma for treatment of COVID-19 infections: results of a multicenter clinical study. Transfus Apher Sci. 2020;59.
Hegerova L, Gooley TA, Sweerus KA, Maree C, Bailey N, Bailey M, et al. Use of convalescent plasma in hospitalized patients with COVID-19: case series. Blood. 2020;136:759–62.
Perotti C, Baldanti F, Bruno R, Del Fante C, Seminari E, Casari S, et al. Mortality reduction in 46 severe COVID-19 patients treated with hyperimmune plasma. A proof of concept single arm multicenter interventional trial. Infect Dis (except HIV/AIDS); 2020.
Liu STH, Lin H-M, Baine I, Wajnberg A, Gumprecht JP, Rahman F, et al. Convalescent plasma treatment of severe COVID-19: a matched control study. Infect Dis (except HIV/AIDS); 2020.
Salazar E, Christensen PA, Graviss EA, Nguyen DT, Castillo B, Chen J, et al. Treatment of coronavirus disease 2019 patients with convalescent plasma reveals a signal of significantly decreased mortality. Am J Pathol. 2020;190:2290–303.
Axfors C, Janiaud P, Schmitt AM, van’t Hooft J, Smith ER, Haber NA, et al. Association between convalescent plasma treatment and mortality in COVID-19: a collaborative systematic review and meta-analysis of randomized clinical trials. BMC Infect Dis. 2021;21:1170.
Odutayo A, Gryaznov D, Copsey B, Monk P, Speich B, Roberts C, et al. Design, analysis and reporting of multi-arm trials and strategies to address multiple testing. Int J Epidemiol. 2020;49:968–78.
Li L, Zhang W, Hu Y, Tong X, Zheng S, Yang J, et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: a randomized clinical trial. JAMA. 2020;324:460.
Gharbharan A, Jordans CCE, Geurtsvankessel C, den Hollander JG, Karim F, Mollema FPN, et al. Convalescent Plasma for COVID-19. A randomized clinical trial. Infect Dis (except HIV/AIDS); 2020.
Agarwal A, Mukherjee A, Kumar G, Chatterjee P, Bhatnagar T, Malhotra P. Convalescent plasma in the management of moderate covid-19 in adults in India: open label phase II multicentre randomised controlled trial (PLACID Trial). BMJ. 2020;m3939.
Simonovich VA, Burgos Pratx LD, Scibona P, Beruto MV, Vallone MG, Vázquez C, et al. A randomized trial of convalescent plasma in COVID-19 severe pneumonia. N Engl J Med. 2021;384:619–29.
Bajpai M, Kumar S, Maheshwari A, Chhabra K, kale P, Gupta A, et al. Efficacy of convalescent plasma therapy compared to fresh frozen plasma in severely ill COVID-19 patients: a pilot randomized controlled trial. Infect Dis (except HIV/AIDS); 2020.
AlQahtani M, Abdulrahman A, Almadani A, Alali SY, Al Zamrooni AM, Hejab AH, et al. Randomized controlled trial of convalescent plasma therapy against standard therapy in patients with severe COVID-19 disease. Infect Dis (except HIV/AIDS); 2020.
Balcells ME, Rojas L, Le Corre N, Martínez-Valdebenito C, Ceballos ME, Ferrés M, et al. Early versus deferred anti-SARS-CoV-2 convalescent plasma in patients admitted for COVID-19: a randomized phase II clinical trial. PLoS Med. 2021;18.
Ray Y, Paul SR, Bandopadhyay P, D’Rozario R, Sarif J, Lahiri A, et al. Clinical and immunological benefits of convalescent plasma therapy in severe COVID-19: insights from a single center open label randomised control trial. Infect Dis (except HIV/AIDS); 2020.
Avendaño-Solà C, Ramos-Martínez A, Muñez-Rubio E, Ruiz-Antorán B, de Molina RM, Torres F, et al. Convalescent plasma for COVID-19: a multicenter, randomized clinical trial. Infect Dis (except HIV/AIDS); 2020.
Libster R, Pérez Marc G, Wappner D, Coviello S, Bianchi A, Braem V, et al. Early high-titer plasma therapy to prevent severe COVID-19 in older adults. N Engl J Med. 2021;384:610–8.
O’Donnell MR, Grinsztejn B, Cummings MJ, Justman J, Lamb MR, Eckhardt CM, et al. A randomized, double-blind, controlled trial of convalescent plasma in adults with severe COVID-19. Infect Dis (except HIV/AIDS); 2021.
RECOVERY Collaborative Group. Convalescent plasma in patients admitted to hospital with COVID-19 (RECOVERY): a randomised controlled, open-label, platform trial. Lancet Lond Engl. 2021;397:2049–59.
Joyner MJ, Carter RE, Senefeld JW, Klassen SA, Mills JR, Johnson PW, et al. Convalescent plasma antibody levels and the risk of death from COVID-19. N Engl J Med. 2021;384:1015–27.
Bégin P, Callum J, Jamula E, Cook R, Heddle NM, Tinmouth A, et al. Convalescent plasma for hospitalized patients with COVID-19: an open-label, randomized controlled trial. Nat Med. 2021. https://doi.org/10.1038/s41591-021-01488-2.
Menichetti F, Popoli P, Puopolo M, Spila Alegiani S, Tiseo G, Bartoloni A, et al. Effect of high-titer convalescent plasma on progression to severe respiratory failure or death in hospitalized patients with COVID-19 pneumonia: a randomized clinical trial. JAMA Netw Open. 2021;4.
Korley FK, Durkalski-Mauldin V, Yeatts SD, Schulman K, Davenport RD, Dumont LJ, et al. Early convalescent plasma for high-risk outpatients with COVID-19. N Engl J Med. 2021;385:1951.
Chen X, Zhao B, Qu Y, Chen Y, Xiong J, Feng Y, et al. Detectable serum severe acute respiratory syndrome coronavirus 2 viral load (RNAemia) is closely correlated with drastically elevated interleukin 6 level in critically ill patients with coronavirus disease 2019. Clin Infect Dis. 2020;71:1937–42.
Chen W, Lan Y, Yuan X, Deng X, Li Y, Cai X, et al. Detectable 2019-nCoV viral RNA in blood is a strong indicator for the further clinical severity. Emerg Microbes Infect. 2020;9:469–73.
Hogan CA, Stevens BA, Sahoo MK, Huang C, Garamani N, Gombar S, et al. High frequency of SARS-CoV-2 RNAemia and association with severe disease. Clin Infect Dis. 2021;72:e291–5.
Hagman K, Hedenstierna M, Gille-Johnson P, Hammas B, Grabbe M, Dillner J, et al. Severe acute respiratory syndrome coronavirus 2 RNA in serum as predictor of severe outcome in coronavirus disease 2019: a retrospective cohort study. Clin Infect Dis. 2020;73:e2995.
Eberhardt KA, Meyer-Schwickerath C, Heger E, Knops E, Lehmann C, Rybniker J, et al. RNAemia corresponds to disease severity and antibody response in hospitalized COVID-19 patients. Viruses. 2020;12:1045.
Joyner MJ, Bruno KA, Klassen SA, Kunze KL, Johnson PW, Lesser ER, et al. Safety update: COVID-19 convalescent plasma in 20,000 hospitalized patients. Mayo Clin Proc. 2020;95:1888–97.
Pathak EB. Convalescent plasma is ineffective for covid-19. BMJ. 2020;m4072.
Acknowledgements
The authors acknowledge all COVID-19 recovered plasma donors who donated their plasma; all patients that participated in the study; the staff of transfusion centers involved in recruitment, collection, qualification, and distribution of CCP; the staff of microbiology and virology Laboratory for testing all donors and patient’s follow-up samples; Medical doctors, nurses, and all staff of COVID-19 wards that collaborated for patients recruiting and monitoring. A special acknowledgment goes to the Independent Data and Safety Monitoring Committee: Rodolfo Saracci, Valter Torri, Luca Mascaretti, and Antonio Pesenti.
PLACO COVID STUDY GROUP authors: Franco Castagno MD 1, Adriano Valfrè MD 1, Gabriella Rizzioli MSc 1, Teresa D’Amato MSc 1, Cristina Crocillà MSc 1, Silvana Naselli MSc 1, Valentino Granero MSc 1, Grazia Cornagliotto MSc 1, Graziella Lucania MSc 1, Cristiana Scaglia MSc 1, Francesca Ferro MSc 1, Carmela Solimine MSc 1, Monica Ricotti MSc 1, Cristina Gilestro BLT 1, Remigio Roncato NP 1, Angela Palladino NP 1, Daniela Ongaro NP 1, Giulia Anna Poggio MD 18, Chiara Chiappero MD 17, Simone Mornese Pinna 19, Silvia Scabini MD 19, Federico Vischia MD 32, Maria Grazia Gregoretti MD 33, Enrico Lupia MD 16, Luca Brazzi Prof, MD, PhD 34, Carlo Albera Prof, MD, PhD 17, Luca Scaglione MD 33, Valter Gallo MD 35, Claudio Norbiato MD 21, Roberto Albiani MD 10, Bruno Lucio Sini MD 10, Andrea Fassiola MD 36, Alessandro Locatelli MD 37, Giovanni Di Perri MD 38, Mauro Navarra MD 39, Isabella Gardini MD 9, Aurora Ciardiello MD 9, Rita La Grotta MD 9, Anna De Rosa BLT 9, Paola Pasquino MD 20, Gilberto Fiore MD 40, Orietta Franza MD 41, Paola Artoni MD 42, Stefano Meinardi MD 43, Liliana Calosso MD 44, Paola Molino MD 45, Maria Grazia Veglio MD 46, Tiziana Beltramo MD 11, Odetta Camerini MD 11, Karol Giancaspero MD 11, Franca Napoli MD 11, Alberto Perboni MD 47, Emanuela Messa MD 48, Fabrizio Buffolo MD 48, Fiammetta Pagnozzi MD 48, Stefania Bertone MD 22, Lorenzo Lutri MD 22, Umberto Gravante MD 22, Petros Sacchetti MD 22, Alessandra Pavan MD 22, Enzo Castenetto MD 22, Marco Novelli MD 49, Marco Tucciarone MD 14, Patrizia Ocello MD 14, Giulia Guido MD 14, Chiara Frascaroli MD 14, Daniela Maria Luisa Vivenza PhD 12, Francesca Patti MD 27, Laura Lorenzelli MD 50, Guido Balduzzi MD 51, Deborah Ratti MD 52, Laura Mazzucco MSc 13, Valeria Balbo MSc 13, Francesca Pollis MD 13, Sabrina Leoncino MD 13, Chiara Lupo MD 13, Daniele Romano MD 13, Silvia Ziccardi MD 13, Melania Marmifero MD 13, Guido Chichino MD 53, Mario Salio MD 54, Giuseppe Aiosa MD 55, Riccardo Boverio MD 56, Ilaria Avonto MD 7, Sara Ghiotto MD 7, Riccardo Balbo MD 7, Vincenza Nico MD 94, Chiara Aguzzi MD 60, Maria Chiara Pellegrino MD 7, Maristella Prucca Msc 7, Lucia Assunta Longa MSc 7, Laura Perotti MSc 7, Federica Piovano MSc 7, Luca Ambrogio MD 57, Marco Formica MD 58, Elisa Monge MD 26, Flavia Arena MD 26, Nicoletta Barzaghi MD 59, Silvia Tavera MD 60, Mariaelisa Canepari MD 60, Guido Strani MD 60, Fulvio Pomero MD 61, Maria Grazia Cianci MD 62, Mariella Gianarda MD 9, Leonardo Ruscitto MD 63, Daniel De Martino MD 8, Sandro Macchi MD 8, Michele Montagnana MD 15, Vladimiro Grandinetti MD 15, Silvia Magnani MD 64, Elisabetta Radin MD 65, Valentina Pellu MD 65, Monica Meucci MD 66, Erika Noè MD 66, Paola Torti MD 66, Luca Montagnani MD 66, Giulio Doveri MD 67, Gabriella Giustetto BLT 1, Costantino Avdis DBA 1, Marco Prina BCS 1, Franco Eliantonio ITPM 68, Francesco Lemut MD 69, Giuseppe Semino MD 70, Palmina Spidalieri A 1, Domenico Vallino MD 71, Roberto Prota MD 72, Gabriella Buono MD 73, Vincenzo Segala MD 73, Maria Grazia Milia MSc PhD 5, Franco Aprà MD 74, Sergio Livigni MD 25, Emilpaolo Manno MD 36, Giuseppe Caula MD 75, Emanuela Vitali MD 75, Nicola Liuzzi MD 76, Mauro Pastorelli MD 77, Pietro Caironi MD 78, Federica Gamna MD 79, Bruno Scapino MD 80, Lorenzo Gurioli MD 81, Emanuele Magro MD 82, Giuseppe Roberti MD 83, Gian Mario Santamaria MD 84, Antonella Daffonchio MD 85, Paola Varese MD 86, Gianfranco Ghiazza MD 87, Margherita Girino MD 88, Carolina Pelazza MSc 89, Fabrizio Racca MD 90, Mirco Grillo MD 59, Valerio Del Bono MD 25, Giorgio Gianotto MD 91, Enzo Aluffi MD 92, Enrico Ravera MD 93.
PLACO COVID STUDY GROUP authors’ affiliations are as follows:
32 Internal Medicine 3U, University Hospital City of Science and Health Turin, Italy, 33 Internal Medicine 5, University Hospital City of Science and Health Turin, Italy, 34 University Intensive Care Unit, University Hospital City of Science and Health Turin, Italy, 35 Pulmunology Division, Ordine Mauriziano di Torino Hospital, Turin, Italy, 36 Intensive Care Unit, Maria Vittoria Hospital, Turin, Italy, 37 Intensive Care and Emergency Department, S Croce and Carle Cuneo Hospital District, Cuneo, Italy, 38 Infectious Diseases Unit, Amedeo di Savoia Hospital, Turin, Italy, 39 Intensive Care Unit, Martini Hospital, Turin, Italy, 40 Intensive care Unit, Santa Croce Hospital of Moncalieri, Moncalieri, Italy, 41 Internal Medicine Department, San Lorenzo Hospital, Carmagnola, Italy, 42 Internal Medicine, Maggiore Hospital, Chieri, Italy, 43 Intensive Care Unit, Maggiore Hospital, Chieri, Italy, 44 Transfusion Medicine, Edoardo Agnelli Hospital, Pinerolo, Italy, 45 Internal and Emergency Medicine, Infermi Rivoli Hospital, Rivoli, Italy, 46 COVID Emergency Medicine, Infermi Rivoli Hospital, Rivoli, Italy, 47 Pulmunology Division, San Luigi Gonzaga University Hospital, Orbassano, Italy, 48 Internal Medicine, Chivasso Hospital, Chivasso, Italy, 49 Internal Medicine, Lanzo Hospital, Lanzo, Italy, 50 Intensive Care Unit, Cardinal Massaia Hospital of Asti, Asti, Italy, 51 Immunohematology and Transfusion Medicine, ASL Alessandria, Alessandria, Italy, 52 Internal Medicine, SS Antonio and Margherita Hospital, Tortona, Italy, 53 Infectious Diseases Unit, Saints Anthony and Biagio and Cesare Arrigo Alessandria National Hospital, Alessandria, Italy, 54 Pulmunology Unit Saints Anthony and Biagio and Cesare Arrigo Alessandria National Hospital, Alessandria, Italy, 55 Internal Medicine, Saints Anthony and Biagio and Cesare Arrigo Alessandria National Hospital, Alessandria, Italy, 56 Emergency Department Saints Anthony and Biagio and Cesare Arrigo Alessandria National Hospital, Alessandria, Italy, 57 Medical Department, S Croce and Carle Cuneo Hospital District, Cuneo, Italy, 58 General Medical Department and Rehabilitation, S Croce and Carle Cuneo Hospital District, Cuneo, Italy, 59 Intensive Care and Emergency Department, S Croce and Carle Cuneo Hospital District, Cuneo, Italy, 60 Immunohematology and Transfusion Medicine, ASL CN1 Savigliano Savigliano, Italy, 61 Internal Medicine, Michele e Pietro Ferrero Hospital, Verduno, Italy, 62 Immunohematology and Transfusion Medicine, ASL Vercelli, Vercelli, Italy, 63 Immunohematology and Transfusion Medicine, ASL VCO, Omegna, Italy, 64 Infectious Diseases Unit, Umberto Parini Hospital, Aosta, Italy, 65 Nephrology unit, Umberto Parini Hospital, Aosta, Italy, 66 Intensive Care Unit, Umberto Parini Hospital, Aosta, Italy, 67 Internal Medicine, Umberto Parini Hospital, Aosta, Italy, 68 Information technology and clinical engineering Unit, University Hospital City of Science and Health Turin, Italy, 69 Intensive Care Unit, ASL Alessandria, Alessandria, Italy, 70 Transfusion Medicine, SS Antonio and Margherita Hospital, Tortona, Italy, 71 Medical Emergency Division, Ordine Mauriziano di Torino Hospital, Turin, Italy, 72 Semi-intensive Pulmunology Division, Ordine Mauriziano di Torino Hospital, Turin, Italy, 73 Intensive Care Unit, Ordine Mauriziano di Torino Hospital, Turin, Italy, 74 Medical Department, San Giovanni Bosco Hospital, Turin, Italy, 75 Medical emergency Division, Martini Hospital, Turin, Italy, 76 Internal Medicine, Edoardo Agnelli Hospital, Pinerolo, Italy, 77 Intensive Care Unit, Edoardo Agnelli Hospital, Pinerolo, Italy, 78 Intensive Care Unit, San Luigi Gonzaga University Hospital, Orbassano, Italy, 79 Rehabilitative Unit, San Luigi Gonzaga University Hospital, Orbassano, Italy, 80 Intensive Care Unit, Ivrea Hospital, Ivrea, Italy, 81 COVID Medicine Unit 2, Ivrea Hospital, Ivrea, Italy, 82 Internal Medicine, Ciriè Hospital, Ciriè, Italy, 83 Intensive Care Unit Ciriè Hospital, Ciriè, Italy, 84 Internal Medicine, SS Antonio and Margherita Hospital, Tortona, Italy, 85 Internal Medicine, San Giacomo Hospital, Novi Ligure, Italy, 86 Internal Medicine, Ovada Hospital, Ovada, Italy, 87 Internal Medicine, Acqui Terme Hospital, Acqui Terme, Italy, 88 Internal Medicine, Santo Spirito Hospital, Casale M.to, Italy, 89 Research and Innovation Unit, Saints Anthony and Biagio and Cesare Arrigo Alessandria National Hospital, Alessandria, Italy, 90 Intensive Care Unit Saints Anthony and Biagio and Cesare Arrigo Alessandria National Hospital, Alessandria, Italy, 91 Transfusion Service, Michele e Pietro Ferrero Hospital, Verduno, Italy, 92 Medical Emergency Division, Michele e Pietro Ferrero Hospital, Verduno, Italy, 93 Intensive Care Unit, Michele e Pietro Ferrero Hospital, Verduno Italy, 94 Immunohematology and Transfusion Medicine, ASL CN1 Mondovì, Mondovì, Italy.
Funding
Funding for the study was provided by: Piemonte Regional Health System that was funding for IgG anti-SARS-CoV-2 testing, SARS-CoV-2-RNA testing by RT-PCR on plasma, on nasopharyngeal swab, or bronchoalveolar lavage. University Hospital City of Science and Health Turin that was funding for Plasma inactivation procedures (Mirasol, TerumoTM) and extra screening requested by the Italian Blood Authority (Centro Nazionale Sangue): Hepatitis E virus and Hepatitis A virus RNA (HAV-RNA, HEV-RNA), and ParvoB19-DNA by RT-PCR on donor’s collected plasma.
Author information
Authors and Affiliations
Consortia
Contributions
PMM is the Chief Investigator; she conceived the study, led the proposal and protocol development and wrote the manuscript. GCi, FGDR, CG, SDA, FS, ACas, ML, GCa, OG, AMB, AT, LB, LS, Cav, MPri, FE, contributed to study design and to development of the proposal. GCi was the lead trial methodologist. FS, ACas were trial metodologists and they analyzed results. FD, LL, designed logistic organization for CCP preparation and distribution. TF, HH, CP, MP, PO, ITS, RG, RF, PB, AN, LMa, FPM, SR, AB, AR, MMi, GD, FCas, AV, RR, AP, DO, RA, BLS, IG, ACi, RLG, ADR, LC, TB, OC, KG, FN, MT, PO, GBa, LMaz, VB, MPr, LAL, MGC, MGia, LR, DDM, SMa, MMo, GSe, PSp, GGia organized donors’ selection and recruitment and plasma and CCP collection in their hospitals. TF, HH, CP, GD, GGu, CF, DMLV, FPol, SLe, ChL, DRo, SZ, MeMa, IA, SG, RBa, VN, CA, MCP, ST, MCa, GSt, VlG, organized patients’ data collection for their centers. BM, GSc, CP were responsible for coordinating Hospital organization for the trial. RC, VG, FD, LL, FP, MA, CC, MGM, organized and performed all donors and patients serological and RT-PCR tests. GC, GL, CSc, FF, CSo, MR, CG, organized plasma and CCP fractionation, treatment and stockage. GR, TDA, CC, SN, VaG, LP, FPio, GGiu, organized CCP and plasma blinding and assignment. AGDM, AM, SBa, FC, MLR, DB, FVit, MML, ACh, ACa, MCo, CAL, VL, IB, VB, GAP, ChCh, SMP, SS, FVis, MGG, EL, LB, CAl, LS, VG, CN, AF, AL, GDP, MNa, PP, GF, OF, PA, SMe, PM, MGV, APe, EMe, FB, FP, SBe, LL, UG, PSa, APa, EC, MNo, FPat, LLo, DRa, GCh, MS, GA, RBo, LA, MF, EMo, FAr, NBa, FPo, SM, ER, VP, MMe, EN, PT, LMo, GD, FL, DV, RP, GBu, VS, FAp, SLi, EMan, GCa, EV, NL, MPas, PC, FG, BS, LG, EMag, GR, GMS, AD, PV, GGh, MGir, FR, MGri, VDB, EA, ER were responsible for organization of recruitment and follow-up of patients, preparation and transmission of daily CRFs at their Hospitals. NBi was responsible for collection and analysis of adverse events. GCi, CG, SDA, ACas, ML were major contributors in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The trial protocol was approved by the Ethical Committee of the coordinating center: Comitato Etico Interaziendale A.O.U. Città della Salute e della Scienza di Torino—A.O. Ordine Mauriziano di Torino—A.S.L. Città di Torino and then by all ethical Committee at each participating center and was conducted following the principles stated in the declaration of Helsinki and the Good Clinical Practice Guidelines. A written informed consent was obtained from all subjects (donors and patients) and/or their legal guardian(s) before enrollment. The informed consent is available in the Study Protocol available in the online version—see Supplementary Information).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary appendix.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Manzini, P.M., Ciccone, G., De Rosa, F.G. et al. Convalescent or standard plasma versus standard of care in the treatment of COVID-19 patients with respiratory impairment: short and long-term effects. A three-arm randomized controlled clinical trial. BMC Infect Dis 22, 879 (2022). https://doi.org/10.1186/s12879-022-07716-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-022-07716-5