
Abstract
Background
Exposure to cold weather is associated with infaust cardiovascular responses, including myocardial infarction and arrhythmias. However, the exact mechanisms of these adverse changes in the myocardium under cold stress are unknown. This study was designed to investigate the mechanisms of cardiac injury induced by cold stress in mice.
Methods
The mice were randomly divided into three groups, normal control (no handling), 1-week cold stress and 2-week cold stress. We observed physiological changes of the mice and morphological changes of myocardium tissues, and we measured the changes of 3′-nitrotyrosine and 4-hydroxynonenal, the expression levels of superoxide dismutase-1, superoxide dismutase-2, Bax, Bad, Bcl-2, Nuclear factor erythroid-derived 2-like 2 (Nrf2) and Kelch like-ECH-associated protein 1 (Keap1) in myocardium by western blot. Besides, we detected mRNA of superoxide dismutase-1, superoxide dismutase-2, Bax, Bad, Bcl-2, Nrf2 and Keap1 by real-time PCR. One-way analysis of variance, followed by LSD-t test, was used to compare each variable for differences among the groups.
Results
Echocardiography analyses demonstrated left ventricle dysfunction in the groups receiving cold stress. Histological analyses witnessed inflammation, vacuolar and eosinophilic degeneration occurred in left ventricle tissues. Western blotting results showed increased 3′-nitrotyrosine and 4-hydroxynonenal and decreased antioxidant enzymes (superoxide dismutase-1 and superoxide dismutase-2) in the myocardium. Expression of Nrf2 and Keap1 followed a downward trend under cold exposure, as indicated by western blotting and real-time PCR. Expression of anti-apoptotic protein Bcl-2 also showed the same trend. In contrast, expression of pro-apoptotic proteins Bax and Bad followed an upward trend under cold exposure. The results of real-time PCR were consistent with those of western blotting.
Conclusions
These findings were very significant, showing that cold exposure induced cardiac injury by inhibiting the Nrf2-Keap1 signaling pathway.
Similar content being viewed by others


Background
Exposure to cold weather is broadly considered to be a global challenge to human health, affecting Europe, the United States and many other countries [1,2,3,4,5]. The highest seasonal death rates occur in winter months and the relationship between an extreme cold climate and increased cardiovascular morbidity and mortality is apparent [2, 6,7,8]. A retrospective analysis in Canada suggested that a 3.0% increase in non-accidental deaths occurred with each 5 °C increase in daily temperature [8]. A cold climate has an especially adverse effect on cardiovascular morbidity and death among people under age 65 [7, 8]. Many clinical and epidemiological studies confirmed an association of cold exposure with infaust cardiovascular responses, for example, increased blood pressure and viscosity and compromised myocardial hemodynamics and performance, all potentially leading to myocardial infarction and arrhythmias [2, 3, 9,10,11,12]. Nevertheless, the exact mechanisms of these adverse changes in the myocardium under cold stress remain unknown.
During environmental stresses, oxidative stress is implicated in cellular activities in inflammatory responses, apoptosis and damage to multiple systems, including in cardiovascular disease [13,14,15,16]. Nrf2, a transcription factor, plays a protective role by regulating expression of antioxidant proteins that act against oxidative damage [17]. Nrf2 is co-localized in the cytoplasm with Keap1 and Cullin 3 (Cul3). These promote its rapid degradation by ubiquitination under physiological conditions. However, when the Keap1-Cul3 ubiquitination system is disrupted by oxidative stress, Nrf2 translocates into the nucleus from the cytoplasm to bind to a DNA promoter, the antioxidant response element (ARE), in the upstream region of several antioxidant genes, thus initiating their transcription [18, 19]. However, specific mechanisms involving oxidative stress under cold exposure require further exploration.
Methods
Mice and cold exposure model
The study was approved by the Animal Care and Use Committee of the General Hospital of Shenyang Military Command. Thirty 8–10 w old Kunming mice were obtained from Liaoning Provincial Laboratory Animal Public Service Center (China, Benxi city). An equal number of male and female mice were maintained at 22 ± 2 °C for the control group and exposed to low ambient temperature in an artificial cold climate chamber (− 20 °C) for the experimental groups. The experimental protocol involved exposure at − 20 °C for 4 h per day for 1 or 2 w, for the two respective experimental groups. Body weights were the same among groups. All animals were raised in the Animal Facility, supplied with food and water ad libitum under a 12-h light—dark cycle, before assessments of myocardial morphology and function. For these assessments, mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg).
Echocardiographic assessment
Mice were anesthetized and evaluated for cardiac geometry and function by animal echocardiography (Vevo 770, a 12 MHz transducer). During diastole and systole, the anterior and posterior wall dimensions of the left ventricle (LV) were determined from three consecutive cycles. Fractional shortening was defined as the ratio of the duration from LV end-diastolic to end-systolic diameters. Positive and negative LV dP/dtmax were measured through the right common carotid artery into the ascending aorta by pressure catheter (Scisense Inc., Canada).
Histopathological assessment
Cardiac tissues were excised, fixed in 10% formaldehyde buffer at room temperature and embedded in paraffin by Leica Microsystem tissue processor (ASP 300S, Germany). After sectioning at 3 μm, they were stained with hematoxylin and eosin (HE) and examined under a light microscope. Masson’s trichrome staining was also performed to detect fibrosis by modified masson’s trichrome stain Kit (Sigma, USA).
Western blot analysis
Myocardial tissue was solubilized with a lysis buffer contained protease and phosphotase inhibitors to prepare protein extracts. For western blotting, equal amount of proteins were resolved on sodium dodecyl sulfate polyacrylamide gel electrophoresis and bands then transferred to polyvinylidene difluoride membranes. The membranes were blocked with blocking buffer (5% nonfat milk in tris-buffered saline (TBS-T)) for 1 h, then incubated overnight at 4 °C with the appropriate primary antibodies (Abcam, UK). After washed by TBS-T, blot was incubated with the secondary antibodies (Abcam, UK) for 2 h at room temperature. After immunoblotting, stained bands were detected by enhanced chemiluminescence using the ECL detection kit (Bio-Rad, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abcam, UK) was used as the loading control.
Real-time quantitative PCR analysis
Total RNA was extracted and reverse-transcribed by TRIzol (Takara Biotechnology, Tokyo, Japan) and complementary DNA reverse transcription kit (Invitrogen, USA), respectively. The real-time PCR reaction was performed using the real-time PCR thermocycler (Bio-Rad, USA). The comparative threshold cycle method was used to calculate the mRNA expression of the target genes normalized to GAPDH. The primer sequences were as follows: SOD-1 5′-GAACCATCCACTTCGAGCAG-3′ (forward), 5′-GATGGACGTGGAACCCATGC-3′ (reverse); SOD-2 5′-CGGCCTACGTGAACAATCTC-3′ (forward), 5′-TTAGGGCTCAGGTTTGTCCAG-3′ (reverse); Bax 5′-CTCCGGCGAATTGGAGATGAA-3′ (forward), 5′-CAGTTGAAGTTGCCATCAGC-3′ (reverse); Bad 5′-GAGCAGGAAGACGCTAGTGC-3′ (forward), 5′-GGTACGAACTGTGGCGACTC-3′ (reverse); Bcl-2 5′-TGAGTACCTGAACCGGCATC-3′ (forward), 5′-AAGCCCAGACTCATTCAACCA-3′ (reverse); Nrf2 5′-CCCAGCAGGACATGGATTTGA-3′ (forward), 5′-AGCTCATAGTCCTTCTGTCGC-3′ (reverse); Keap1 5′-CCAGATTGACAGCGTGGTTC-3′ (forward), 5′-GGTTGAAGAACTCCTCCTGCT-3′ (reverse); GAPDH 5′-GGTCCCAGCTTAGGTTCATCA-3′ (forward), 5′-CCGTTCACACCGACCTTCA-3′ (reverse).
Data and statistical analysis
All values are expressed as means ± standard deviation (SD). Statistical significance was defined as a two-sided P value < 0.05. One-way analysis of variance (ANOVA), followed by LSD-t test, was used to compare each variable for differences among the groups, using SPSS 20.0 software (IBM, USA).
Results
Cold exposure induced cardiac injury
Under cold exposure, body weight was increased with exposure time, so the 1 w and 2 w cold exposure groups were heavier than the control groups (P < 0.05, Fig. 1a). Ratio of left ventricular weight to body weight in the two experimental groups were also a little higher than in the control group (Fig. 1b), but not significantly, indicating that cold stress maybe exacerbated myocardial hypertrophy slightly. Cold stress also led to increased ratios of lung weight to body weight, suggesting that LV dysfunction occurred after cold exposure for 2 weeks (Fig. 1c).

Physiological changes of the mice under cold exposure. a Body weight. b Ratio of LV weight to body weight. c: Ratio of lung weight to body weight. d LV ejection fraction. e LV fraction shortening. f LVPWS of body weight. g LVPWD of body weight. h LV dP/dtmax. (i) LV dP/dtmin. Data are mean ± SD. * p < 0.05, compared to control group
Echocardiography showed that cold exposure decreases in LV ejection fraction and LV fractional shortening (Fig. 1d and e). And the left ventricular posterior wall thickness at end systole (LVPWS) and diastole (LVPWD) of the body weight showed the opposite trend, but not significantly (Fig. 1f and g). LV dP/dtmax was decreased under cold exposure, further demonstrating LV dysfunction in the experimental groups (Fig. 1h). However, LV dP/dtmin was just a little increased (Fig. 1i).
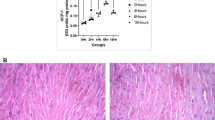
Histological staining of LV tissues showed that inflammation and vacuolar degeneration occurred in cardiac myocyte in the 1-week cold exposed mice. And eosinophilic degeneration was witnessed in the 2-week cold exposed mice (Fig. 2a). There were more blue stained area in the 2-week cold exposed mice, which demonstrated that myocardial fibrosis was observed by Masson’s trichrome staining (Fig. 2b).

Histopathological changes of myocardium under cold exposure. a HE staining of myocardium in different groups. b Masson stained sections of left ventricles in different groups
Cold exposure induced cardiac oxidative stress
To examine the potential mechanism of action of cold exposure-induced myocardial contractile dysfunction, oxidation markers were examined in the myocardium. The protein and lipid oxidation markers 3′-nitrotyrosine (3’-NT) and 4-hydroxynonenal (4-HNE) were measured to assess effects of cold stress on myocardial oxidative stress. Western blotting for 3′-NT and 4-HNE showed that both increased under cold exposure (Fig. 3a and b), indicating oxidative stress in the myocardium.

Oxidative stress markers in myocardium under cold exposure. a Western blot of 3’-NT and 4-HNE. b Relative density of 3’-NT and 4-HNE. Data are mean ± SD. * p < 0.05, compared to control group, ** p < 0.01, compared to control group
Cold exposure affected myocardial antioxidant enzymes
To assess effects on antioxidant enzymes during oxidative stress in the myocardium under cold exposure, protein and mRNA levels of these enzymes were measured in tissue from the LV myocardium. Western blotting results demonstrated that superoxide dismutase (SOD)-1 and SOD-2 levels were increased in 1-week cold exposed and decreased in 2-week cold exposed mice (Fig. 4a). Expression of SOD-1 and SOD-2 mRNA in the LV myocardium, detected by real-time RT-PCR, also indicated the same changes as observed with the proteins (Fig. 4b).

Antioxidant enzymes changes in myocardium under cold exposure. a Western blot of SOD-1 and SOD-2. b Real time PCR of SOD-1 and SOD-2. Data are mean ± SD. * p < 0.05, compared to control group, ** p < 0.01, compared to control group
Cold exposure increased apoptosis in the myocardium
Apoptosis can be triggered as a result of oxidation. So we examined expression of pro-apoptotic proteins and anti-apoptotic protein in myocardium under cold exposure. Expression of Bax and Bad increased in 1-week and 2-week cold exposed mice, and Bcl-2 decreased in two experimental groups (Fig. 5a). And expression of mRNA in Bax and Bad was significantly elevated in the cold exposed groups, which opposite to expression of mRNA in Bcl-2 (Fig. 5b).

Pro-apoptotic proteins in myocardium under cold exposure. a Western blot of Bax, Bad and Bcl-2. b Real time PCR of Bax, Bad and Bcl-2. Data are mean ± SD. * p < 0.05, compared to control group, ** p < 0.01, compared to control group
Cold exposure induced oxidative stress and apoptotic injury by inhibiting the Nrf2-Keap1 signaling pathway
In our study, western blotting showed that levels of the cellular sensor protein for oxidative stress, Nrf2, were increased in the 1-week, then decreased in the 2-week, exposure group. Levels of Keap1, a promoter of Nrf2 degradation by autophagy, were decreased under cold exposure (Fig. 6a). The same trends were observed measuring mRNA levels of the two genes (Fig. 6b).

Pro-apoptotic proteins in myocardium under cold exposure. a Western blot of Nrf2 and Keap-1. b Real time PCR of Nrf2 and Keap-1. Data are mean ± SD. * p < 0.05, compared to control group, ** p < 0.01, compared to control group
Discussion
The major findings of our study were that cold exposure induced: (i) an oxidative stress response, indicated by subsequent effects on myocardial antioxidant enzymes; (ii) apoptosis in the myocardium; and (iii) inhibition of the Nrf2-Keap1 signaling pathway by decreasing Nrf2 expression. These findings implied that cold stress induced oxidative stress, apoptosis and LV dysfunction by regulating the Nrf2-Keap1 signaling pathway.
In recent years, several reports were focus on chronic low temperature exposure induced myocardial dysfunction, and witnessed cardiac hypertrophy and fibrosis by intracellular Ca2+ derangement, oxidative stress, mitochondrial damage and apoptosis [20,21,22]. But extreme cold weather induced cardiac injury was seldom reported. In this study, we tried to explore the changes in myocardium under extreme cold weather and found followings. Increased ratios of lung weight to body weight, decreased LV ejection fraction and LV fractional shortening to body weight and decreased LV dP/dtmax were witnessed in the 2-week cold exposure group of this study (Fig.1). All these physiological changes demonstrated LV dysfunction induced by cold exposure. Histological analyses witnessed inflammation, vacuolar degeneration, eosinophilic degeneration and fibrosis occurred in the myocardium (Fig.2). As a result, they further demonstrated that 2-week cold exposure could lead to cardiac injury.
Injury induced by myocardial oxidative stress is determined by the balance between oxydate and their elimination by various antioxidants in the tissue. In recent studies, antioxidant defense enzymes exhibited a compensatory increase following long-term cold exposure, probably indicating an important role of oxidative stress in cold-related health problems [21, 23]. Oxidative metabolism in the mitochondria is the major source of myocardial ATP. Similarly, the primary site for reactive oxygen species (ROS) generation in cardiomyocytes is the mitochondria [24, 25]. 4-HNE is a α, β-unsaturated hydroxyalkenal, generated by peroxidation of n-6 polyunsaturated fatty acids. High 4-HNE levels, under conditions of high oxidative stress, can induce a variety of biological responses, such as cellular dysfunction and apoptosis [26,27,28,29,30,31]. We observed increased levels of 3’-NT and 4-HNE, two widely used markers for myocardial oxidative stress. Their levels increased with length of cold exposure (Fig 3).
As a member of the bZIP transcription factor family, Nrf2 plays a well-established role against oxidative stress. Nrf2 remains at low levels under normal conditions because of its rapid ubiquitylation and proteasome dependent degradation. During oxidative stress, Nrf2 degradation was inhibited in various organs, such as the heart, brain, kidney, pancreas and liver [32,33,34,35,36,37,38,39]. Keap1 modification by HNE enables release and nuclear translocation of Nrf2, and this process is possibly modulated by mitochondrial ROS production. Oxidative stress led to oxidation of critical cysteine residues in Keap1, disrupting the Keap1 ubiquitination system [40]. It was confirmed that Nrf2/Keap1 signaling pathway could be activated to regulate protective effect against cardiovascular disease [41]. In our study, Nrf2 levels in the nucleus increased in the 1-week and decreased in the 2-week cold exposed group. Keap1 levels followed a downward trend with longer cold exposure, suggested that prolonged cold stress would impede transcriptional activation by Nrf2. Higher Bax expression with longer cold stress exposure was further evidence for apoptosis, likely triggered by the oxidative stress (Fig. 5). These results were different from those in other reports [38,39,40] and we propose two possible reasons for the discrepancy. One is that Nrf2 levels were exhausted under long-term stress. Two-week’s continuous stimulation was given in our study, which opposite to short-term stress was given in several Nrf2-related studies [41,42,43]. Moreover, it was suggested that persistent accumulation of Nrf2 in the nucleus has toxic effects, such as inducing apoptosis and aggravating oxidative stress [44, 45]. So such effects might have caused negative feedback, leading to decreased Nrf2 in the 2-week cold exposed group.
Various mitochondrial antioxidant enzymes, including manganese SOD-2, are important defenses against mitochondrial ROS. In many studies, these mitochondrial antioxidant enzymes protected the myocardium against oxidative injury and ventricular dysfunction under oxidative stress [46,47,48,49,50,51]. Such protection was also a result of Nrf2 activation [52,53,54]. Our findings showed that levels of SOD enzymes were increased in 1-week and decreased in 2-week cold exposed groups, likely because of Nrf2 levels (Figs. 4 and 6).
Our results implied that the cold exposure first triggered oxidative stress, leading to active 4-HNE-dependent modifications in the myocardium. At the same time, the Nrf2 signaling pathway was activated and oxidative stress disrupted the critical cysteine residues in Keap1, enabling Nrf2 to translocate into the nucleus from the cytoplasm. Progressively, Nrf2 exerted its anti-oxidative effects, activating transcription of antioxidant enzymes, including SOD-1 and SOD-2. However, with longer cold exposure, Nrf2 levels were suppressed, leading to decreased levels of antioxidant enzymes, enabling aggravation of oxidative stress and apoptosis.
Conclusion
In summary, we demonstrated that cold exposure induced cardiac injury by inhibited the Nrf2-Keap1 signaling pathway. Our data indicated that apoptosis, 4-HNE production, decreased Nrf2 levels and the resulting cardiac injury may be essential to the adverse effects of cold exposure. Given the role of antioxidants in protection against cardiac injury, our data supported the therapeutic potential of antioxidants for managing cold stress-associated myocardial complications.
Abbreviations
- 3’-NT:
-
3′-nitrotyrosine
- 4-HNE:
-
4-hydroxynonenal
- Cul3:
-
Cullin 3
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- HE:
-
Hematoxylin and eosin
- Keap1:
-
Kelch like-ECH-associated protein 1
- LV:
-
Left ventricle
- LVPWD:
-
Left ventricular posterior wall thickness at end diastole
- LVPWS:
-
Left ventricular posterior wall thickness at end systole
- Nrf2:
-
Nuclear factor erythroid-derived 2-like 2
- SOD:
-
Superoxide dismutase
- TBS-T:
-
Tris-buffered saline
References
Mercer JB. Cold--an underrated risk factor for health. Environ Res. 2003;92(1):8–13.
Cheng X, Su H. Effects of climatic temperature stress on cardiovascular diseases. Eur J Intern Med. 2010;21(3):164–7.
Yu W, Mengersen K, Wang X, Ye X, Guo Y, Pan X, et al. Daily average temperature and mortality among the elderly: a meta-analysis and systematic review of epidemiological evidence. Int J Biometeorol. 2012;56(4):569–81.
Analitis A, Katsouyanni K, Biggeri A, Baccini M, Forsberg B, Bisanti L, et al. Effects of cold weather on mortality: results from 15 european cities within the phewe project. Am J Epidemiol. 2008;168(12):1397–408.
von Klot S, Zanobetti A, Schwartz J. Influenza epidemics, seasonality, and the effects of cold weather on cardiac mortality. Environ Health. 2012;11:74.
Okamoto-Mizuno K, Tsuzuki K, Mizuno K, Ohshiro Y. Effects of low ambient temperature on heart rate variability during sleep in humans. Eur J Appl Physiol. 2009;105(2):191–7.
Song X, Wang S, Hu Y, Yue M, Zhang T, Liu Y, et al. Impact of ambient temperature on morbidity and mortality: an overview of reviews. Sci Total Environ. 2017;586:241–54.
Chen H, Wang J, Li Q, Yagouti A, Lavigne E, Foty R, et al. Assessment of the effect of cold and hot temperatures on mortality in ontario, canada: a population-based study. CMAJ Open. 2016;4(1):E48–58.
Howard BV, Comuzzie A, Devereux RB, Ebbesson SO, Fabsitz RR, Howard WJ, et al. Cardiovascular disease prevalence and its relation to risk factors in alaska eskimos. Nutr Metab Cardiovasc Dis. 2010;20(5):350–8.
Medina-Ramon M, Zanobetti A, Cavanagh DP, Schwartz J. Extreme temperatures and mortality: assessing effect modification by personal characteristics and specific cause of death in a multi-city case-only analysis. Environ Health Perspect. 2006;114(9):1331–6.
Wang X, Li G, Liu L, Westerdahl D, Jin X, Pan X. Effects of extreme temperatures on cause-specific cardiovascular mortality in china. Int J Environ Res Public Health. 2015;12(12):16136–56.
Diaz J, Carmona R, Miron IJ, Ortiz C, Linares C. Comparison of the effects of extreme temperatures on daily mortality in madrid (spain), by age group: the need for a cold wave prevention plan. Environ Res. 2015;143(Pt A):186–91.
Devasagayam TP, Tilak JC, Boloor KK, Sane KS, Ghaskadbi SS, Lele RD. Free radicals and antioxidants in human health: current status and future prospects. J Assoc Physicians India. 2004;52:794–804.
Lobo V, Patil A, Phatak A, Chandra N. Free radicals, antioxidants and functional foods: impact on human health. Pharmacogn Rev. 2010;4(8):118–26.
Kim HJ, Kim CH, Ryu JH, Kim MJ, Park CY, Lee JM, et al. Reactive oxygen species induce antiviral innate immune response through ifn-lambda regulation in human nasal epithelial cells. Am J Respir Cell Mol Biol. 2013;49(5):855–65.
Deffert C, Cachat J, Krause KH. Phagocyte nadph oxidase, chronic granulomatous disease and mycobacterial infections. Cell Microbiol. 2014;16(8):1168–78.
Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, et al. DEFINE study investigators. Placebo-controlled phase 3 study of oral bg-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–107.
Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, et al. Physiological significance of reactive cysteine residues of keap1 in determining nrf2 activity. Mol Cell Biol. 2008;28(8):2758–70.
Sekhar KR, Rachakonda G, Freeman ML. Cysteine-based regulation of the cul3 adaptor protein keap1. Toxicol Appl Pharmacol. 2010;244(1):21–6.
Yin K, Zhao L, Feng D, Ma W, Liu Y, Wang Y, et al. Resveratrol attenuated low ambient temperature-induced myocardial hypertrophy via inhibiting Cardiomyocyte apoptosis. Cell Physiol Biochem. 2015;35(6):2451–62.
Zhang Y, Hu N, Hua Y, Richmond KL, Dong F, Ren J. Cardiac overexpression of metallothionein rescues cold exposure-induced myocardial contractile dysfunction through attenuation of cardiac fibrosis despite cardiomyocyte mechanical anomalies. Free Radic Biol Med. 2012;53(2):194–207.
Zhang Y, Li L, Hua Y, Nunn JM, Dong F, Yanagisawa M, et al. Cardiac-specific knockout of ET(a) receptor mitigates low ambient temperature-induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Biol. 2012;4(2):97–107.
Hong JH, Kim KJ, Suzuki K, Lee IS. Effect of cold acclimation on antioxidant status in cold acclimated skaters. J Physiol Anthropol. 2008;27(5):255–62.
Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–95.
Babizhayev MA, Yegorov YE. Reactive oxygen species and the aging eye: specific role of metabolically active mitochondria in maintaining lens function and in the initiation of the oxidation-induced maturity onset cataract--a novel platform of mitochondria-targeted antioxidants with broad therapeutic potential for redox regulation and detoxification of oxidants in eye diseases. Am J Ther. 2016;23(1):e98–117.
Chapple SJ, Cheng X, Mann GE. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biol. 2013;1:319–31.
Csala M, Kardon T, Legeza B, Lizák B, Mandl J, Margittai É, et al. On the role of 4-hydroxynonenal in health and disease. Biochim Biophys Acta. 2015;1852(5):826–38.
Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med Res Rev. 2008;28(4):569–631.
Asselin C, Ducharme A, Ntimbane T, Ruiz M, Fortier A, Guertin MC, et al. Circulating levels of linoleic acid and hdl-cholesterol are major determinants of 4-hydroxynonenal protein adducts in patients with heart failure. Redox Biol. 2014;2:148–55.
Leonarduzzi G, Chiarpotto E, Biasi F, Poli G. 4-hydroxynonenal and cholesterol oxidation products in atherosclerosis. Mol Nutr Food Res. 2005;49(11):1044–9.
Ross MK, Borazjani A, Mangum LC, Wang R, Crow JA. Effects of toxicologically relevant xenobiotics and the lipid-derived electrophile 4-hydroxynonenal on macrophage cholesterol efflux: silencing carboxylesterase 1 has paradoxical effects on cholesterol uptake and efflux. Chem Res Toxicol. 2014;27(10):1743–56.
Ke B, Shen XD, Zhang Y, Ji H, Gao F, Yue S, et al. Keap1-nrf2 complex in ischemia-induced hepatocellular damage of mouse liver transplants. J Hepatol. 2013;59(6):1200–7.
Deng C, Sun Z, Tong G, Yi W, Ma L, Zhao B, et al. Alpha-lipoic acid reduces infarct size and preserves cardiac function in rat myocardial ischemia/reperfusion injury through activation of pi3k/akt/nrf2 pathway. PLoS One. 2013;8(3):e58371.
Chai J, Luo L, Hou F, Fan X, Yu J, Ma W, et al. Agmatine reduces lipopolysaccharide-mediated oxidant response via activating pi3k/akt pathway and up-regulating nrf2 and ho-1 expression in macrophages. PLoS One. 2016;11(9):e0163634.
Wu J, Li Q, Wang X, Yu S, Li L, Wu X, et al. Neuroprotection by curcumin in ischemic brain injury involves the akt/nrf2 pathway. PLoS One. 2013;8(3):e59843.
He M, Pan H, Chang RC, So KF, Brecha NC, Pu M. Activation of the nrf2/ho-1 antioxidant pathway contributes to the protective effects of lycium barbarum polysaccharides in the rodent retina after ischemia-reperfusion-induced damage. PLoS One. 2014;9(1):e84800.
Masuda Y, Vaziri ND, Li S, Le A, Hajighasemi-Ossareh M, Robles L, et al. The effect of nrf2 pathway activation on human pancreatic islet cells. PLoS One. 2015;10(6):e0131012.
Shelton LM, Park BK, Copple IM. Role of nrf2 in protection against acute kidney injury. Kidney Int. 2013;84(6):1090–5.
Porritt MJ, Andersson HC, Hou L, Nilsson Å, Pekna M, Pekny M, et al. Photothrombosis-induced infarction of the mouse cerebral cortex is not affected by the nrf2-activator sulforaphane. PLoS One. 2012;7(7):e41090.
Barancik M, Gresova L, Bartekova M, Dovinová I. Nrf2 as a key player of redox regulation in cardiovascular diseases. Physiol Res. 2016;65(Suppl 1):S1–S10.
Cui G, Luk SC, Li RA, Chan KK, Lei SW, Wang L, et al. Cytoprotection of baicalein against oxidative stress-induced cardiomyocytes injury through the Nrf2/Keap1 pathway. J Cardiovasc Pharmacol. 2015;65(1):39–46.
Rao J, Qian X, Li G, Pan X, Zhang C, Zhang F, et al. ATF3-mediated NRF2/HO-1 signaling regulates TLR4 innate immune responses in mouse liver ischemia/reperfusion injury. Am J Transplant. 2015;15(1):76–87.
Li H, Song F, Duan LR, Sheng JJ, Xie YH, Yang Q, et al. Paeonol and danshensu combination attenuates apoptosis in myocardial infarcted rats by inhibiting oxidative stress: roles of Nrf2/HO-1 and PI3K/Akt pathway. Sci Rep. 2016;6:23693.
Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the keap1-nrf2-are pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116.
Zhang C, Li XN, Xiang LR, Qin L, Lin J, Li JL. Atrazine triggers hepatic oxidative stress and apoptosis in quails (coturnix c. Coturnix) via blocking nrf2-mediated defense response. Ecotoxicol Environ Saf. 2017;137:49–56.
Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2006;113(14):1779–86.
Nojiri H, Shimizu T, Funakoshi M, Yamaguchi O, Zhou H, Kawakami S, et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. J Biol Chem. 2006;281(44):33789–801.
Nickel AG, von Hardenberg A, Hohl M, Löffler JR, Kohlhaas M, Becker J, et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22(3):472–84.
Korge P, Calmettes G, Weiss JN. Increased reactive oxygen species production during reductive stress: the roles of mitochondrial glutathione and thioredoxin reductases. Biochim Biophys Acta. 2015;1847(6–7):514–25.
Yin B, Barrionuevo G, Batinic-Haberle I, Sandberg M, Weber SG. Differences in reperfusion-induced mitochondrial oxidative stress and cell death between hippocampal ca1 and ca3 subfields is due to the mitochondrial thioredoxin system. Antioxid Redox Signal. 2017; [Epub ahead of print]
Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation. 2015;131(12):1082–97.
Branco V, Godinho-Santos A, Goncalves J, Lu J, Holmgren A, Carvalho C. Mitochondrial thioredoxin reductase inhibition, selenium status, and nrf-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Radic Biol Med. 2014;73:95–105.
Danielli NM, Trevisan R, Mello DF, Fischer K, Deconto VS, da Silva AD, et al. Upregulating nrf2-dependent antioxidant defenses in pacific oysters crassostrea gigas: investigating the nrf2/keap1 pathway in bivalves. Comp Biochem Physiol C Toxicol Pharmacol. 2017;195:16–26.
Ahn CB, Je JY, Kim YS, Park SJ, Kim BI. Induction of nrf2-mediated phase ii detoxifying/antioxidant enzymes in vitro by chitosan-caffeic acid against hydrogen peroxide-induced hepatotoxicity through jnk/erk pathway. Mol Cell Biochem. 2017;424(1–2):79–86.
Acknowledgments
Not applicable.
Funding
The study was supported by the PLA foundation of Key issues of logistical support department of the Central Military Commission (BWS16J010). The funding sources had no involvement in the conduct of the research or preparation of the article.
Availability of data and materials
The datasets generated and analysed during the current study are not publicly available due the principle of confidentiality of funding but are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: MH, YL, PC. Conducted the experiments: PC, NL, YZ, CT, LS, XL, XS, YL, ZT. Analysed the data: PC. Contributed reagents/materials/analysis tools: MH, YL. Wrote the paper: PC. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Animal Care and Use Committee of the General Hospital of Shenyang Military Command.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cong, P., Liu, Y., Liu, N. et al. Cold exposure induced oxidative stress and apoptosis in the myocardium by inhibiting the Nrf2-Keap1 signaling pathway. BMC Cardiovasc Disord 18, 36 (2018). https://doi.org/10.1186/s12872-018-0748-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-018-0748-x