
Abstract
Background
Yams (Dioscorea spp.) are economically important food for millions of people in the humid and sub-humid tropics. Dioscorea dumetorum (Kunth) is the most nutritious among the eight-yam species, commonly grown and consumed in West and Central Africa. Despite these qualities, the storage ability of D. dumetorum is restricted by severe postharvest hardening of the tubers that can be addressed through concerted breeding efforts. The first step of any breeding program is bound to the study of genetic diversity. In this study, we used the Genotyping-By-Sequencing of Single Nucleotide Polymorphism (GBS-SNP) to investigate the genetic diversity and population structure of 44 accessions of D. dumetorum in Cameroon. Ploidy was inferred using flow cytometry and gbs2ploidy.
Results
We obtained on average 6371 loci having at least information for 75% accessions. Based on 6457 unlinked SNPs, our results demonstrate that D. dumetorum is structured into four populations. We clearly identified, a western/north-western, a western, and south-western populations, suggesting that altitude and farmers-consumers preference are the decisive factors for differential adaptation and separation of these populations. Bayesian and neighbor-joining clustering detected the highest genetic variability in D. dumetorum accessions from the south-western region. This variation is likely due to larger breeding efforts in the region as shown by gene flow between D. dumetorum accessions from the south-western region inferred by maximum likelihood. Ploidy analysis revealed diploid and triploid levels in D. dumetorum accessions with mostly diploid accessions (77%). Male and female accessions were mostly triploid (75%) and diploid (69%), respectively. The 1C genome size values of D. dumetorum accessions were on average 0.333 ± 0.009 pg and 0.519 ± 0.004 pg for diploids and triploids, respectively.
Conclusions
Germplasm characterization, population structure and ploidy are an essential basic information in a breeding program as well as for conservation of intraspecific diversity. Thus, results obtained in this study provide valuable information for the improvement and conservation of D. dumetorum. Moreover, GBS appears as an efficient powerful tool to detect intraspecific variation.
Similar content being viewed by others

Background
Yams (Dioscorea spp.) constitute a staple food for over 300 million people in the humid and sub-humid tropics. About 600 species are described and are widely distributed throughout the tropics [1]. Dioscorea dumetorum has the highest nutrient value among eight other yam species commonly grown and consumed in West and Central Africa [2]. The species originated in tropical Africa and occurs in both wild and cultivated forms. Its cultivation is restricted to West and Central Africa [3], and widespread in western Cameroon. Tubers of D. dumetorum are protein-rich (9.6%) with fairly balanced essential amino acids and its starch is easily digestible [4,5,6]. Agronomically, D. dumetorum is high-yielding, with yield of 40 tons/hectare recorded in agricultural stations [7]. Dioscorea dumetorum is also recognized for its pharmaceutical properties. A novel bioactive compound dioscoretine has been identified in D. dumetorum [8], which can be used advantageously as a hypoglycemic agent in anti-diabetic medications [9].
Despite these qualities, the storage ability of D. dumetorum is restricted by severe postharvest hardening of the tubers, which begins within 24 h after harvest and renders them unsuitable for human consumption [2]. According to Treche and Delpeuch [10], the usual storage conditions in West Africa (under airy warehouse, shelter from sunlight) induce 100% losses after 4 months of storage. It is manifested by the loss of culinary quality due to a combination of factors resulting from normal but inadvertently deleterious reactions leading to textural changes [11]. Therefore, D. dumetorum is consumed exclusively during its limited harvest period and only freshly harvested tubers are cooked and sold to consumers. To add more value to D. dumetorum as an important source of food and energy, hardened tubers are transformed into instant flour [12]. However, flour obtained directly from hardened tubers has poor organoleptic qualities, such as coarseness in the mouth [4]. Thus, other techniques have been used, such as salt soaking treatment [13] and fermentation [14], but the hardening phenomenon has not been surmounted. Consequently, molecular breeding of D. dumetorum appears as the appropriate method to overcome this phenomenon.
The study of genetic diversity is an important, early step in plant breeding. Highlighting this variability is part of the characterization of germplasm under investigation. In our recent study on the phenotypic diversity of D. dumetorum we found relatively high diversity of morphological characters suggesting high underlying genetic diversity [15]. Indeed, the expression of morphological characters are subject to agro-climatic variations and thus provides limited genetic information. Therefore, molecular markers that are not subject to environmental variations are necessary for estimation of genetic diversity. The development of molecular markers over the last 30 years has enabled the study of diversity and evolution as well as germplasm characterization [16]. Among these markers, Single Nucleotide Polymorphisms (SNPs) have emerged as the most widely used genotyping markers due to their abundance in the genome allowing not only germplasm characterization but also quantification of relative proportions of ancestry derived from various founder genotypes of currently grown cultivars [16]. Moreover, the development of traditional markers like SSRs, RFLPs and AFLPs was a costly, iterative process that involved either time-consuming cloning and enzyme testing or primer design steps that could not easily be parallelized [17].
Genotyping-By-Sequencing (GBS) has emerged as a new approach to mitigate these constraints. The method has been demonstrated to be suitable for population studies, germplasm characterization, genetic improvement, trait mapping in a variety of diverse organisms and thereby, SNP discovery and genotyping of multiple individuals are performed cost-effectively and efficiently [18]. GBS is performed by an initial digest of sample DNA with restriction enzymes reducing genome complexity followed by a round of PCR to generate a high-throughput sequencing library [19]. Reducing genome complexity with restriction enzymes is quick, extremely specific and highly reproducible [19]. Unlike other similar approaches using restriction enzymes, GBS is technically simple [20]. Besides, bioinformatic pipelines are publicly available [21] and GBS can be easily applied to non-model species with limited genomic information [20]. This method has been successfully used on Cassava (Manihot esculenta Crantz) [22], guinea yam [23] and water yam [24], which demonstrated the power of GBS-SNP genotyping as a suitable technology for high-throughput genotyping in yam.
Genetics of yams is least understood and remains largely neglected among the major staple food crops due to several biological constraints and research neglect [25]. Some progress has been made in germplasm characterization and the development of molecular markers for genome analysis. Various dominant molecular markers (AFLP, RAPD) have been used on yam with little success (e.g., [9]). Additionally, genomic microsatellite markers have been developed for yam species [24,25,26,27,28,29,30,31,32]. However, no markers have been developed for D. dumetorum and its genetics is the least known in spite of its qualities among the cultivated yam. Until now, no information is available using SNP genotyping to assess population structure, genetic diversity and the relationship among D. dumetorum cultivars.
A possible additional factor that influences population structure and genetic diversity is polyploidy. Polyploidy has several advantages for plant breeding such as the increment in plant organs (“gigas”effect), buffering of deleterious mutations, increased heterozygosity, and heterosis (hybrid vigor) [33]. In yam, ploidy increase is correlated with growth vigor, higher and more stable tuber yield and increased tolerance to abiotic and biotic stress [33, 34]. Recent studies using flow cytometry revealed diploid and triploid levels in D. dumetorum with predominance of the diploid cytotype [35, 36]. Therefore, the objective of this study is to understand the genetic diversity and the population structure of D. dumetorum using the genotyping-by-sequencing (GBS) in relation to ploidy information.
Methods
Plants materials
Overall, 44 accessions of D. dumetorum were used in this study (Table 1). All these accessions were collected from different localities in the major yam growing regions (western, south-western, and north-western) of Cameroon, with an additional three accessions of D. dumetorum from Nigeria complementing the dataset (Fig. 1). Western and north-western regions belong to agro-ecological zone (AEZ) 3 and the south-western region to AEZ 4 of Cameroon [38]. Most of these accessions were previously used for morphological characterization [15] and hardness assessment [39]. Here, we selected some characters related to tubers (Fig. 2).The yam tubers of these accessions were planted in April 2015 at the “Ferme Ecole de Bokué” in the western region of Cameroon (latitude 05°20.040’ N and longitude 010°22.572 E). Silica-dried young leaves were transported to Oldenburg (Germany) for molecular analyses. Genomic DNA was extracted using an innuPREP Plant DNA kit (Analytik Jena, Jena, Germany).

Sampling map of D. dumetorum accessions in Cameroon. Boundaries of agro-ecological zones (AEZ) were defined according to [38]

D. dumetorum tuber forms. 1a) Accession with few roots on the tuber and yellow flesh colour. 2a) accession with few roots on the tubers and white flesh colour b) accession with many roots on the tuber
Preparation of libraries for next-generation sequencing
A total of 200 ng of genomic DNA for each sample were digested with 1 Unit MslI (New England Biolabs, NEB) in 1x NEB4 buffer in 30 μl volume for 1 h at 37 °C. The restriction enzyme was heat inactivated by incubation at 80 °C for 20 min. Afterwards, 15 μl of digested DNA were transferred to a new 96well PCR plate, mixed and stored on ice first with 3 μl of one of the 192 L2 ligation adaptors (Ovation Rapid DR Multiplex System, Nugen Technologies, Leek, The Netherlands) and then with 12 μl master mix (combination of 4.6 μl D1 water/ 6 μl L1 ligation buffer mix/ 1.5 μl L3 ligation enzyme mix). Ligation reactions were incubated at 25 °C for 15 min followed by inactivation of the enzyme at 65 °C for 10 min. Then, 20 μl of the kits ‘final repair’ master mix were added to each tube and the reaction was incubated at 72 °C for 3 min. For library purification, reactions were diluted with 50 μl TE 10/50 (10 mM Tris/HCl, 50 mM EDTA, pH:8.0) and mixed with 80 μl magnetic beads, incubated for 10 min at room temperature and placed for 5 min on a magnet to collect the beads. The supernatant was discarded and the beads were washed two times with 200 μl 80% Ethanol. Beads were air-dried for 10 min and libraries were eluted in 20 μl Tris Buffer (5 mM Tris/HCl, pH 9). Each of the 45 libraries (including one technical repeat) were amplified with 10 μl of the purified restriction product in 20 μl PCR reactions using 4 μl MyTaq (Bioline) 5x buffer, 0.2 μl polymerase and 1 μl (10 pmol/μl) of standard Illumina TrueSeq amplification primers. Cycle number was limited to ten cycles. Then, 5 μl from each of the 48 amplified libraries were pooled. PCR primers and small amplicons were removed by magnetic bead purification using 0.6 volume of beads. The PCR polymerase was removed by an additional purification on Qiagen MinElute Columns. The pooled library was eluted in a final volume of 20 μl Tris buffer (5 mM Tris/HCl, pH 9). The final library pool was sent to LGC genomics (Berlin, Germany) and sequenced on an Illumina NextSeq with 1.5 million 150 bp paired-end reads for each sample. Additional steps at LGC for the sequencing preparation were normalization, reamplification and size selection. Normalization was conducted using Trimmer Kit (Evrogen). For this 1 μg pooled GBS library in 12 μl was mixed with 4 μl 4x hybridization buffer, denatured for 3 min at 98 °C and incubated for 5 h at 68 °C to allow re-association of DNA fragments. 20 μl of 2 x DSN master buffer was added and the samples were incubated for 10 min at 68 °C. One unit of DSN enzyme (1 U/μl) was added and the reaction was incubated for another 30 min. The reaction was terminated by the addition of 20 μl DSN Stop Solution, purified on a Qiagen MinElute Column and eluted in 10 μl Tris Buffer (5 mM Tris/HCl pH 9).The normalized library pools were re-amplified in 100 μl PCR reactions using MyTaq (Bioline). Primer i5-Adaptors were used to include i5-indices into the libraries, allowing parallel sequencing of multiple libraries on the Illumina NextSeq 500 sequencer. Cycle number was limited to 14 cycles. The nGBS libraries were size selected using Blue Pippin, followed by a second size selection on a LMP-Agarose gel, removing fragments smaller than 300 bp and those larger than 400 bp. Libraries were sequenced on an Illumina NextSeq 500 using Illumina V2 Chemistry.
GBS data analysis
GBS data were analyzed using the custom software pipeline iPyrad (Versions: 0.7.19 and 0.7.28) developed by Eaton and Ree [21] for population genetic and phylogenetic studies. It includes seven steps to demultiplex and quality filtering, cluster loci with consensus alignments and SNP calling with SNP filtering to the final SNP matrix, which can be transferred into various output formats. We have conducted demultiplexing and QC separately to retrieve fastq sequences as input for iPyrad. The restriction sites and barcodes were trimmed for each sequence, bases with a quality score less than PHRED 20 were changed into N and sequences having more than 5% of N were discarded. Step 3 of iPyrad used in our de-novo SNP analysis VSEARCH [40] for dereplication and merging of paired reads and for clustering reads per sample into putative loci with 85% sequence similarity. Alignments of consensus sequences of the putative loci were built with MUSCLE [41]. After estimation of sequencing errors (Π) and heterozygosity (ɛ), consensus alleles were estimated with these estimated parameters and the number of alleles was recorded. Resulting consensus alleles were again clustered with VSEARCH and aligned with MUSCLE. Base SNPs were called when loci were observed in at least 75% of the samples, had not more than 20 SNPs and eight indels and heterozygous sites in 50% of the samples, but all samples were treated as diploid, thus allowing two haplotypes per polymorphic site.
Phylogenetic inference
An unrooted tree was generated using the neighbor-net method in SplitsTree (Version 4.14.6) [42] based on the concatenated GBS data. To control whether or not the introduction of triploid accessions affected our phylogenetic analysis, we constructed dendrograms with and without triploid accessions.
Historical relationship between accessions (TreeMix)
Historical relationships between D. dumetorum accessions including possible gene flow events was assessed through the maximum likelihood method implemented in TreeMix (version 1.13) [43]. TreeMix reconstructs the possible migrations between populations based on allele frequency of genomic data. It uses a method that allows for both population splits and gene flow. We defined the population parameter as 0, because we worked at the individual level. Of 25,541 SNPs loci investigated, 157 SNPs were filtered to get a gap free matrix and used to determine the relationships between the accessions. The tree was built with the confidence of 1000 bootstrap replicates and visualized with toytree (version 0.1.4) and toyplot (version 0.16.0).
Population structure analysis
Analysis of population structure was performed using the software STRUCTURE [44] and MavericK [45]. Structure uses a Bayesian model-based clustering method with a heuristic approach for estimation whereas MavericK uses a computation technique called Thermodynamic Integration (TI). However, the mixture modeling framework is identical in both programs [45]. The analysis was carried out in STRUCTURE using the admixture model across 10 replicates (K of 2 to 5) of sampled unlinked SNPs (one randomly chosen SNP per ipyrad-cluster). A burn-in period of 10,000 iterations and 100,000 Markov Chain Monte Carlo (MCMC) replicates were run. The true number of clusters (K) was detected using Evanno’s method [46] implemented in STRUCTURE HARVESTER [47]. The MCMC implementation of MavericK differs slightly, although the core model assumed is identical to that used in Structure [45]. Thus, the admixture model across five replicates (K of 2 to 5) was run with a burn-in period of 2000 iterations and 10,000 MCMC. The best value of K was detected in 25 TI rungs each for a range of K (2 to 5) with default settings.
Ploidy/genome size estimation
For each accession, about 1 cm2 of young leaf was co-chopped with a standard using a razor blade in a Petri dish containing 1.1 mL ice-cold Otto I buffer (0.1 M citric acid monohydrate and 5% Triton X-100). We used Solanum lycopersicum L. ‘Stupicke’ (1C = 0.98 pg; [48] as the internal standard. The chopped material and buffer were then filtered through a Cell-Tric 30-μm filter into a plastic tube, and 50 μL RNase were added. After incubation in a water bath for 30 min at 37 °C, 450 μL of the solution were transferred to another tube, to which 2 mL Otto II (propidium iodide + Na2HPO4) were added. This solution was placed at 4 °C for 1 h. The samples were analyzed using a CyFlow flow cytometer (Partec GmbH, Münster, Germany). For each accession, three replicates comprising 5000 counts were measured. We measured the genome size of 17 out 44 D. dumetorum accessions due to the loss of certain accessions, in which the sex has been identified. Ploidy level of the remaining accessions (27) was assessed using the R package gbs2ploidy [49]. This method infers cytotypes based on the allelic ratios of heterozygous SNPs identified during variant calling within each individual. Data was prepared by acquiring a *.vcf output file for all specimens from iPyrad using VCFConverter2.py (https://github.com/dandewaters/VCF-File-Converter) as in [50]. Cytotypes were estimated in two ways: 1) without reference to accessions of known ploidy and 2) with reference of 17 accessions for which ploidy is known) from flow cytometry as set of triploids and diploids to the 27 remaining accessions.
Results
GBS data analysis summary
We generated an average of 2.2 million raw reads per D. dumetorum accessions by Illumina sequencing (Table 2). After filtering we obtained on average 1.3 × 104 reads clustered at 85%, with an average depth per accession of 53. The maximum likelihood average estimate of heterozygosity (ɛ = 1.1 × 10− 2) was greater than the sequence error rate (Π = 6 × 10− 3). Consensus sequences were called for each cluster, yielding on average 32,532 reads per accession. We recorded on average 6371 loci recovered in at least 75% of accessions. Accession D09S had a markedly higher proportion of missing data.
Phylogenetic inference
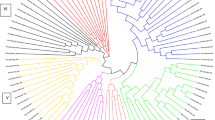
The unrooted neighbor-net clustered the 44 accessions of D. dumetorum into four groups: a western/north-western group, a western group, a southwestern group and a mixed group (Fig. 3). However, two accessions (E10S and H06N) were not clustered in these groups. Triploid accessions did not affect the topology of the network (Additional file 1: Figure S1).

Phylogenetic relationships within D. dumetorum based on multilocus concatenated SNP sequences alignment from GBS data of 44 accessions
The western/north-western group had 16 accessions, 88% were from the western and north-western regions (50% were from West and 50% from North-west). Remaining accessions (12%) were from the southwestern region (H11S) and Nigeria (A09I). In this group, accessions are characterized by yellow flesh color with few roots on the tuber and were from high altitude regions except A09I. Here, all the accessions hardened after harvest except A09I from Nigeria.
The western group consisted of eight accessions; almost all were from the western region and one from the north-western region (G07 N). This group was constituted by accessions with yellow flesh color and many roots on the tuber. They originate all from high altitude regions and hardened after harvest. The western group was closely related to the western/north-western group and differed in the number of roots on the tubers.
The south-western group had 12 accessions originating from the south-western region except C08I from Nigeria. Unlike the western/north-western group and western group, all accessions were from low altitude regions and had white flesh color. However, all accessions hardened after harvest. The fourth group was a mixed group consisting of six accessions, among which four were from the Southwest, one from the West (F08 W) and one from Nigeria (E08I). As compared to the others, the group is variable with respect to tuber characters. Here again, all accessions hardened after harvest.
Population structure
We determined the population structure of D. dumetorum using both a Bayesian approach and Thermodynamic Integration (TI) as implemented in STRUCTURE and MavericK, respectively. The STRUCTURE and MavericK results revealed that D. dumetorum accessions can be clustered into populations. The delta K (∆K) of Evanno’s method and TI estimator of the evidence for K showed strong peaks at K = 4 and K = 2 respectively (Additional file 2: Figure S2). The K value (K = 4) is the most likely number of populations (Fig. 4), because the existence of four groups was also supported by the neighbor-net method (Fig. 3). In total, 33 accessions (75%) were assigned to one of the first three populations with at least 60% of their inferred ancestry derived from one of the three populations. No accession was assigned to the fourth population with at least 60 of the inferred ancestry. The populations P1, P2 and P3 contained 16, 8, and 9 accessions respectively. The remaining accessions (11) were the result of admixture between the populations.

STRUCTURE plot of 44 accessions of D. dumetorum with K = 4 clusters based on 6457 unlinked SNPs. Each accession is represented by a single row, which is partitioned into colored segments in proportion to the estimated membership in the three subpopulations
In population P1, accessions were from the western and north-west region except accessions A09I (Nigeria) and H11S (south-western region). Here, three accessions were assigned 100% to P1, twelve as admixture between P1 and P4 and one accession A09I as admixture of P1xP2xP3xP4. In contrast, all the accessions of population P2 were from the south-western region except H06N (North-west). Four accessions were assigned 100% to P2, two accessions as admixture P2xP4, while two each as admixture P1xP2xP4 and P1xP2. Regarding P3, almost all accessions (8) were from the western region except G07 N from the north-western region. Conversely, no accession was assigned 100% to P3. Five were assigned as admixture P1xP3, three classified as P1xP2xP3 and one as P1xP2xP3xP4. Moreover, the population structure did not change with the increased values of K = 5 (Additional file 3: Figure S3). Comparing results from the STRUCTURE analysis with the neighbor-net, we found generally similar results. Thus, P1 corresponds to the west/north-western population, P2 to the south-western population, and P3 to the western population. No accessions belonging to P4 were identified.
Ploidy/genome size estimation
We found that 13 (76%) accessions of D. dumetorum were diploid (2x) and four (24%) were triploid (3x) (Table 3). The 1C genome size values for D. dumetorum measured here were on average 0.333 ± 0.009 pg and 0.519 ± 0.004 pg for diploids and triploids, respectively. The standard coefficient of variation (CV) of each measurement was < 5% for all runs (Additional file 4: Table S1). Comparing the data with sex, we found that diploid accessions, were 69% female and 31% were male. For triploid accessions, 75% were male and 25% female. With respect to geographic origin, all triploid accessions come from the southwestern region.
Using the R package gbs2ploidy on accessions with known ploidy (17), we assessed the sensitivity of gbs2ploidy on our GBS data. The probability of concurrence between flow cytometry and gbs2ploidy was 35%, with 8 of 17 accessions assigned to the opposite cytotype and three (A09I, B09W, E08I) being inconclusive. The probability of correct diploid and triploid assignments was 38 and 25%, respectively. Training gbs2ploidy with reference accessions from flow cytometry on the remaining accessions (27), we found that 21 (78%) accessions were diploids and 6 (22%) triploids with the mean assignment probability of 74 and 73%, respectively. Regarding diploid accessions, seven, five and nine accessions originated from western, north-western and south-western regions, respectively. For triploids, three were from north-western, two from western and one from south-western regions. In summary, 34 accessions of D. dumetorum (77%) were diploid (2x) and 10 (23%) were triploid (3x). Triploid accessions originated mainly (70%) from the south-western region.
Historical relationship between accessions
We used TreeMix in order to determine splits and gene flow between D. dumetorum accessions. We constructed the tree allowing between no migration and ten migration events. We found eight gene flow events between D. dumetorum accessions (Fig. 5). Despite the likelihood for the tree with nine migration events being highest (but almost similar to eight migrations), we chose the one with eight events because the ninth migration was redundant (Additional file 5). The migration events involved eleven accessions from the south-western region and two (G10 N and H06N) from the north-western region. We did not find a migration event involving A08, which does not harden after harvest, as well accessions originating from the western region and Nigeria. C12S (2x, few root and white flesh) was possibly the result of gene flow between D07S (2x, female, few root and white flesh) and D09S (3x, male, few root and white flesh) or their ancestors; C07S (3x, male, few roots and white flesh) and E07S (2x, male, many root and yellow flesh) were possibly the result of a introgression between H06N (2x, few roots and yellow flesh) and H07S (2x, male, many roots and yellow flesh). Furthermore, allowing migrations altered the topology of the tree compared to the tree with no migrations events (Additional file 6: Figure S4).

Maximum likelihood tree of the inferred gene flow within D. dumetorum species on 157 SNPs. The colored lines represent the possible gene flow events
Discussion
Genotyping-by-sequencing is an innovative, robust and cost-effective approach allowing multiplexing individuals in one library to generate thousands to millions of SNPs across a wide range of species [51]. In our study, we identified on average 30,698 reads per accession. After filtering to avoid the effect of missing data, 5054 loci were kept for the analyses. In total, 26,325 SNPs were investigated. These numbers are similar to a previous study using the same pipeline in another non-model species [21].
The unrooted neighbor-net tree (Fig. 3) clustered D. dumetorum accessions into four groups: A western/north-western group, a western group, a south-western group and a mixed group. The West and North-west belong to agro-ecological zone III (Western Highlands) and the Southwest belongs to agro-ecological zone IV. This result disagrees with previous results using morphological characters [15], in which there was no clear separation of D. dumetorum accessions according to agro-ecological zone. However, morphological markers are subject to environmental conditions and thus provide limited genetic information. Moreover, Sonibare et al. [9] using AFLP on D. dumetorum accessions from three countries didn’t find a clear separation according to the area of collection. However, SNP markers are the most abundant in a genome and suitable for analysis on a wide range of genomic scales [52, 53]. In combination with high-throughput sequencing, thousands to millions of SNP generated using GBS [54] allow assessing more efficiently the genetic diversity compared to AFLP. This was already suggested by Saski et al. [24], who stated that GBS is a powerful tool for high-throughput genotyping in yam.
Our assignment test results based on STRUCTURE also separate D. dumetorum accessions into four populations in which three were clearly identified, the western/north-western population, the western population and the south-western population. On the contrary, MavericK revealed that D. dumetorum was structured into two populations in accordance with known agro-ecological zones (Additional file 2, Figure S2). However, the number of loci investigated was large (more than hundreds of loci). In this situation, the heuristic approximation implemented in STRUCTURE appears to be better [45]. Furthermore, tuber flesh color of all accessions in the western and northwestern region was yellow whereas the majority of accessions from south-western have white tuber flesh. Our results suggest that altitude and farmers-consumers preference played a role as a barrier between D. dumetorum populations. Indeed, AEZ 3 corresponds to western highlands covering the western and northwestern region. It is characterized by high altitude (1000–2740 m), low temperature (annual mean 19 °C) and annual rainfall of 1500 to 2000 mm. In contrast, AEZ 4 comprises mainly humid forest covering the south-western and littoral regions. It is characterized by low altitude (< 700 m except a few mountains), with an annual rainfall of 2500 to 4000 mm and a mean temperature of 26 °C [38]. All three regions of Cameroon belong to the yam belt, where the species occurs in both wild and cultivated forms. Nevertheless, its center of origin remains unknown so far excluding an explanation for the origin of the separation of populations in Cameroon. Tuber quality is an important criterion of adoption of yam varieties by farmers and consumers [55]. Thus, the difference regarding tuber flesh color in the western/north-western and south-western regions could be explained by different preferences of consumers in these regions, which also depends on yam food form. In the western and north-western regions, yam tubers are almost exclusively consumed as boiled tubers contrary to the South-west where tubers are consumed either boiled or pounded. Consumers in Cameroon probably prefer yellow tubers in the boiled and white tubers in the pounded form. Indeed, Egesi et al. [56] demonstrated that flesh color determines a general preference for boiled or pounded yam in D. alata. Assuming white flesh as the ancestral character state based on its predominant occurrence in other yams species, we assumed that the yellow flesh color has evolved several times (probably four times) because it is present in our four groups inferred, although a single origin with subsequent intraspecific hybridization or losses cannot be excluded. Yams with many roots have likely evolved once, in the western region probably due to environmental conditions of the highland with occasional scarcity of water. The root system has an important physiological function in nutrient and water absorption. It is well known, that several root system traits are considered to be important in maintaining plant productivity under drought stress [57]. The occurrence of mutations related to yellow flesh color and many roots on the tuber in the south-western region (mixed group) was probably caused by artificial crossing of genetically diverse accessions in the region.
The importance of gene flow within and between our four main groups in D. dumetorum can be seen in the high proportion of admixture. This observation could be explained by the efforts, which have been made in the past in Cameroon, especially in the South-west to improve D. dumetorum [7]. Indeed, genetic diversity can be increased by breeding activities [58]. Especially noteworthy is the fourth groups with all individuals assigned to it being admixed, suggesting the absence of genetically unambiguous accessions belonging to this group from Cameroon (Fig. 3). It is possibly that genetically unambiguous individuals of this group was not sampled within Cameroon or went extinct, but our preferred hypothesis is that such plants originated from Nigeria. This finding further corroborates a close relationship between D. dumetorum accessions from Nigeria and Cameroon. The South-west and North-west regions of Cameroon share a common border with Nigeria. Exchanges of D. dumetorum accessions between farmers on both side of the border are well-known, providing gene flow and interbreeding. Indeed, Sonibare et al. [9] reported that introduction of the D. dumetorum germplasm to Central African countries has been affected by the activities of farmers from Nigeria.
TreeMix results obtained in our study also indicate that there was more gene flow between accessions from the south-western region than in the western/north-western region. These findings support the admixture result of STRUCTURE discussed above and allow refinement of our understanding of genotypes crossed in the past. However, regarding the sample with non-postharvest hardening, we did not detect any gene flow. This suggests that the sample was not used, yet, in any breeding in Cameroon and that non-postharvest hardening appears yet to be restricted in D. dumetorum to Nigeria. Thus, a broader study on the genetic diversity involving samples across the distribution range of the species is needed to track the origin of this character and the ancestry of this sample.
Ploidy is another factor possibly relevant for population structure and breeding causing hybrid vigor (heterosis) and buffering of deleterious mutations. Our analysis revealed that 77% of D. dumetorum accessions were diploid and 23% were triploid. This result is broadly consistent with previous findings, in which 83% were diploid and 17% triploid [36] and 60% diploid and 40% triploid [37]. However, the probability of concurrence between flow cytometry and gbs2ploidy was low (35%). In fact, a limitation of the gbs2ploidy method is low coverage, especially if possible ploidy levels for the species are unknown [49]. The authors reported that this problem could be resolved by including validated reference samples with known cytotypes in the analysis as done in our study.
The association between sex and ploidy showed a predominance of triploids for male accessions and diploids for female accessions. These findings partially contradict those of Adaramola et al. [37] in which a predominance of diploid for male accessions has been reported. However, Adaramola et al. [37] outlined that a more systematic sampling method that ensures an equal number of D. dumetorum accession may change their results, which was the case in our study. The 1C genome size values of D. dumetorum accessions ranged on average from 0.33 to 0.52 pg for diploids and triploids, respectively. This supports the results of Obidiegwu et al. [36], who found that the 1C genome of five diploid and one triploid D. dumetorum clones ranged from 0.35 to 0.53 pg, respectively. Thus, D. dumetorum appears to have a very small size genome (1C-value ≤1.4 pg) following the categories of [59]. TreeMix results suggested admixture of some accessions between different ploidy levels. Triploid accessions may either be the result of a possible admixture between triploid (3x) or diploid (2x) male with diploid (2x) females, although sex of accessions H06N and C12S has not been determined. Similar results were reported in D. alata [60]. This suggests that the occurrence of triploid accessions in D. dumetorum is most likely due to the involvement of unreduced (2n) gametes in the pollen rather than the egg cell. This was confirmed by artificial crossing of triploid (3x) male and diploid (2x) female we performed in the field (Siadjeu unpublished data, Additional file 7: Figure S5). Finally, the predominant occurrence of triploid accessions in the southwestern region coincides with the more intensive breeding program in the region and may be explained by it since it is known that hybridization between genetically diverse accessions of a species may increase the number of unreduced gametes [61].
Conclusions
In this study, we reported population structure, genetic diversity and ploidy/genome size of D. dumetorum in Cameroon using GBS. We demonstrated that D. dumetorum is structured into populations. There is a high genetic variability of D. dumetorum accessions in Cameroon. We revealed intraspecific hybridization and provided useful information regarding ploidy/genome size of D. dumetorum. All this information is relevant for conservation and a breeding program of D. dumetorum. However, we did not infer a firm relationship of the sample with postharvest hardening, the character most important for future breeding efforts, suggesting a broad study with respect to this character in West and Central Africa will be needed to elucidate its origin. Finally, GBS appears as an efficient powerful tool for phylogeographic studies in yams.
Abbreviations
- AEZ:
-
Agro-ecological zone
- AFLP:
-
Amplified Fragment Length Polymorphism
- CV:
-
Coefficient of Variation
- EDTA:
-
Ethylenediaminetetra-acetic acid
- GBS:
-
Genotyping-By-Sequencing
- MCMC:
-
Markov Chain Monte Carlo
- P:
-
Population
- PCR:
-
Polymerase Chain Reaction
- RAPD:
-
Random Amplified Polymorphic DNA
- RFLP:
-
Restriction Fragment Length Polymorphism
- SNP:
-
Single Nucleotide Polymorphism
- SSR:
-
Single Sequence Repeats
- TI:
-
Thermodynamic Integration
References
Viruel J, Forest F, Paun O, Chase MW, Devey D, Sousa Couto R, et al. A nuclear Xdh analysis of yams (Dioscorea, Dioscoreaceae) congruent with plastid trees reveals a new Neotropical lineage. Bot J Linn Soc. 2018; in press.
Sefa-Dedeh S, Afoakwa EO. Biochemical and textural changes in trifoliate yam Dioscorea dumetorum tubers after harvest. Food Chem. 2002;79:27–40.
Degras L. The yam: a tropical root crop. Yam a trop. Root crop. London: Macmilan Press Ltd.; 1993.
Medoua GN, Mbome IL, Agbor-Egbe T, Mbofung CMF. Physicochemical changes occurring during postharvest hardening of trifoliate yam (Dioscorea dumetorum) tubers. Food Chem. 2005;90:597–601.
Mbome Lape I, Treche S. Nutritional quality of yam (Dioscorea dumetorum and D rotundata) flours for growing rats. J Sci Food Agric. 1994;66:447–55.
Afoakwa EO, Sefa-Dedeh S. Chemical composition and quality changes occurring in Dioscorea dumetorum pax tubers after harvest. Food Chem. 2001;75:85–91.
Treche S. Potentialités nutritionnelles des ignames (Dioscorea spp.) cultivées au Cameroun. Editions de l’ORSTOM CE et T, editor. Paris; 1989.
Iwu MM, Okunji C, Akah P, Corley D, Tempesta S. Hypoglycaemic Activity of Dioscoretine from Tubers of Dioscorea dumetorum in Normal and Alloxan Diabetic Rabbits. Planta Med. 1990;56:264–7.
Sonibare MA, Asiedu R, Albach DC. Genetic diversity of Dioscorea dumetorum (Kunth) Pax using amplified fragment length polymorphisms (AFLP) and cpDNA. Biochem Syst Ecol. 2010;38:320–34.
Treche S, Delpeuch F. Evidence for the development of membrane thickening in the parenchyma of tubers of Dioscorea dumetorum during storage. C R Acad Sc Paris t. 1979;288:67–70.
Medoua GN. Potentiels nutritionnel et technologique des tubercules durcis de l ‘ igname Dioscorea dumetorum ( Kunth ). Cameroon: Doctoral thesis, Ngaoundere University; 2005.
Treche S, Mbome Lape I, Agbor-Egbe T. Variations de la valeur nutritionnelle au cours de la preparation des produits seches a partir d’ignames cultivees. Rev Sci Tech (Sci Santé). 1984:7–22.
Medoua GN, Mbome IL, Egbe TA, Mbofung CMF. Salts soaking treatment for improving the textural and functional properties of trifoliate yam (Dioscorea dumetorum) hardened tubers. J Food Sci. 2007;72:E464–E469.
Medoua GN, Mbome IL, Agbor-Egbe T, Mbofung CMF. Influence of fermentation on some quality characteristics of trifoliate yam (Dioscorea dumetorum) hardened tubers. Food Chem. 2008;107:1180–6.
Siadjeu C, MahbouSomoToukam G, Bell JM, Nkwate S. Genetic diversity of sweet yam Dioscorea dumetorum (Kunth) Pax revealed by morphological traits in two agro-ecological zones of Cameroon. African J Biotechnol. 2015;14:781–93.
Deschamps S, Llaca V, May GD. Genotyping-by-sequencing in plants. Biology (Basel). 2012;1:460–83.
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12:499–510.
De Donato M, Peters SO, Mitchell SE, Hussain T, Imumorin IG. Genotyping-by-sequencing (GBS): a novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS One. 2013;8:1–8.
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6:1–10.
Wickland DP, Battu G, Hudson KA, Diers BW, Hudson ME. A comparison of genotyping-by-sequencing analysis methods on low-coverage crop datasets shows advantages of a new workflow, GB-eaSy. BMC Bioinformatics. 2017;18:586.
Aeton DAR, Ree RH. Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae). PLoS One. 2013;62:689–706.
Rabbi IY, Kulakow PA, Manu-Aduening JA, Dankyi AA, Asibuo JY, Parkes EY, et al. Tracking crop varieties using genotyping-by-sequencing markers: a case study using cassava (Manihot esculenta Crantz). BMC Genet. 2015;16:1–11.
Girma G, Hyma KE, Asiedu R, Mitchell SE, Gedil M, Spillane C. Next generation sequencing based genotyping, cytometry and phenotyping for understanding diversity and evolution of Guinea yams. Theor Appl Genet. 2014;127.
Saski CA, Bhattacharjee R, Scheffler BE, Asiedu R. Genomic resources for water yam (Dioscorea alata L.): analyses of estsequences, de novo sequencing and GBS libraries. PLoS One. 2015;10:1–14.
Mignouna HD, Abang MM, Asiedu R. Harnessing modern biotechnology for tropical tuber crop improvement: yam (Dioscorea spp.) molecular breeding. African J Biotechnol. 2003;2:475–85.
Terauchi R, Konuma A. Microsatellite polymorphism in Dioscorea tokoro, a wild yam species - genome. Genome. 1994;37:794–801.
Mizuki I, Tani N, Ishida K, Tsumura Y. Development and characterization of microsatellite markers in a clonal plant, Dioscorea japonica Thunb. Mol Ecol Notes. 2005;5:721–3.
Tostain S, Scarcelli N, Brottier P, Marchand JL, Pham JL, Noyer JL. Development of DNA microsatellite markers in tropical yam (Dioscorea sp.). Mol Ecol Notes. 2006;6:173–5.
Hochu I, Santoni S, Bousalem M. Isolation, characterization and cross-species amplification of microsatellite DNA loci in the tropical American yam Dioscorea trifida. Mol Ecol Notes. 2006;6:137–40.
Siqueira MVBM, Marconi TG, Bonatelli ML, Zucchi MI, Veasey EA. New microsatellite loci for water yam (Dioscorea alata, Dioscoreaceae) and cross-amplification for other Dioscorea species. Am J Bot. 2011;98:e144–e146.
Silva LRG, Bajay MM, Monteiro M, Mezette TF, Nascimento WF, Zucchi MI, Pinheiro JB, Veasey EA. Isolation and characterization of microsatellites for the yam Dioscorea cayenensis (Dioscoreaceae) and cross-amplification in D. rotundata. Genet Mol Res. 2014;13(2):2766–71.
Tamiru M, Yamanaka S, Mitsuoka C, Babil P, Takagi H, Lopez-Montes A, et al. Development of genomic simple sequence repeat markers for yam. Crop Sci. 2015;55:2191–200.
Sattler MC, Carvalho CR, Clarindo WR. The polyploidy and its key role in plant breeding. Planta. 2016;243:281–96.
Malapa R, Arnau G, Noyer JL, Lebot V. Genetic diversity of the greater yam (Dioscorea alata L.) and relatedness to D. nummularia lam. and D. transversa Br. As revealed with AFLP markers. Genet Resour Crop Evol. 2005;52:919–29.
Lebot V. Tropical root and tuber crops. In: Verheye WH, editor. oils, plant growth crop prod. Encyclopei. Oxford: Eloss Publishers; 2010. p. 9.
Obidiegwu JE, Rodriguez E, Loureiro J, Muoneke CO, Asiedu R. Estimation of the nuclear DNA content in some representative of genus Dioscorea. Sci Res Easay. 2009;4:448–52.
Adaramola TF, Sonibare MA, Sartie A, Lopez-Montes A, Franco J, Albach DC. Integration of ploidy level, secondary metabolite profile and morphological traits analyses to define a breeding strategy for trifoliate yam (Dioscorea dumetorum (Kunth) Pax). Plant Genet Resour. 2014;14:1–10.
IRAD. Second report on the state of plant genetic resources for food and agriculture in Cameroon. Yaoundé: Institute of Agricultural Research and Development (IRAD); 2008. Available from: http://www.fao.org/docrep/013/i1500e/Cameroun.pdf
Siadjeu C, Panyoo EA, Mahbou Somo Toukam G, Bell M, Nono B, Medoua GN. Influence of cultivar on the postharvest hardening of trifoliate yam (Dioscorea dumetorum) tubers. Adv Agric. 2016;2016:1–18.
Rognes T, Flouri T, Nichols B, Quince C, Mahé FVSEARCH. A versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
Edgar RCMUSCLE. A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:1–19.
Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–67.
Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8:1–17.
Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.
Verity R, Nichols RA. Estimating the number of subpopulations (K) in structured populations. Genetics. 2016;203:1827–35.
Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.
Earl DA, vonHoldt BM. Structure harvester: a website and program for visualizing Structure output and implementing the Evanno method. Conserv Genet Resour. 2012;4:359–61.
Dolezel J, Greilhuber J, Lucretti S, Meister A, Lysak MA, Nardi L, Obermayer R. Plant genome size estimation by flow cytometry: inter-laboratory comparison. Ann Bot. 1998;82:17–26.
Gompert Z, Mock KE. Detection of individual ploidy levels with genotyping-by-sequencing (GBS) analysis. Mol Ecol Resour. 2017;17:1156–67.
Burns M, Hedin M, Tsurusaki N. Population genomics and geographical parthenogenesis in Japanese harvestmen (Opiliones, Sclerosomatidae, Leiobunum). Ecol Evol. 2018;8:36–52.
Torkamaneh D, Laroche J, Belzile F. Genome-wide SNP calling from genotyping by sequencing (GBS) data: a comparison of seven pipelines and two sequencing technologies. PLoS One. 2016;11:1–14.
Rafalski A. Applications of single nucleotide polymorphisms in crop genetics. Curr Opin Plant Biol. 2002;5:94–100.
Zhu YL, Song QJ, Hyten DL, Van Tassell CP, Matukumalli LK, Grimm DR, et al. Single-nucleotide polymorphisms in soybean. Genetics. 2003;163:1123–34.
DaCosta JM, Sorenson MD. DdRAD-seq phylogenetics based on nucleotide, indel, and presence-absence polymorphisms: analyses of two avian genera with contrasting histories. Mol Phylogenet Evol. 2016;94:122–35.
Asiedu R, Wanyera NM NS and NQ. Yams. In: Fuccillo , Dominic, Sears, Linda, Stapleton P, editor. Biodivers trust Conserv use plant genet Resour CGIAR centres. Cambridge: Cambridge University Press; 1997. p. 371.
Egesi CN, Asiedu R, Egunjobi JK, Bokanga M. Genetic diversity of organoleptic properties in water yam (Dioscorea alata L). J Sci Food Agric. 2003;83:858–65.
Janiak A, Kwas̈niewski M, Szarejko I. Gene expression regulation in roots under drought. J Exp Bot. 2016;67:1003–14.
Arnau G, Bhattacharjee R, Mn S, Malapa R, Lebot V, Abraham K, et al. Understanding the genetic diversity and population structure of yam (Dioscorea alata L .) using microsatellite. Mark Theory. 2017;12:1–17.
Leitch IJ, Chase MW, Bennett MD. Phylogenetic analysis of DNA C-values provides evidence for a small ancestral genome size in flowering plants. Ann Bot. 1998;82:85–94.
Nemorin A, David J, Maledon E, Nudol E, Dalon J, Arnau G. Microsatellite and flow cytometry analysis to help understand the origin of Dioscorea alata polyploids. Ann Bot. 2013;112:811–9.
Ramsey J, Schemske DW. Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu Rev Ecol Syst. 1998;29:467–501.
Acknowledgements
We thank Silvia Kempen, for her support in the analysis of genome size. We also thank Dr. Robert Verity for his support to deal with bad rungs of TI after analysis on MavericK and two anonymous reviewers who helped to improve this paper.
Funding
This study was financially supported by the German Academic Exchange Service (DAAD), and Appropriate Development for Africa foundation (ADAF) for the yam accessions collection and the field work in Cameroon. Moreover, the funders had no role in the design of the study and collection, analysis and interpretation of data, decision to publish, or the preparation of the manuscript.
Availability of data and materials
The GBS data was deposited in European Nucleotide Archive (ENA) (Access No. PRJEB27526; https://www.ebi.ac.uk/ena/data/search?query=PRJEB27526). All data generated during the current study are included in this published article and its supplementary information as Additional files 1, 2, 3, 4, 5, 6, and 7.
Author information
Authors and Affiliations
Contributions
CS contributed to design of the experiments, collection of yam accessions, data analysis and interpretation and manuscript writing. EMQ: DNA extraction and sequencing, DCA: data interpretation and manuscript writing. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Phylogenetic relationships within D. dumetorum based on multilocus concatenated SNP sequences alignment from GBS data of 34 diploid accessions. (PDF 259 kb)
Additional file 2:
Figure S2. Estimates of the model evidence for K = 2:5 using TI estimator a) log-evidence and b) the evidence and Structure estimator Delta K ∆K c) (PDF 95 kb)
Additional file 3:
Figure S3. STRUCTURE plot of 44 accessions of D. dumetorum with K = 2, 3, 5 clusters based on 6457 unlinked SNPs. (PDF 133 kb)
Additional file 4:
Table S1. Coefficient of variation of ploidy measurements using flow cytometric and ploidy level per accessions estimated by gbs2ploidy. * Ploidy level assessed by gbs2ploidy (PDF 47 kb)
Additional file 5:
Edges, weight of migrations (8 and 9) and likelihood for migrations 0:9. (TREEOUT 1 kb)
Additional file 6:
Figure S4. Maximum likelihood tree of the inferred gene flow within D. dumetorum species with no gene flow events. (PDF 125 kb)
Additional file 7:
Figure S5. Flowing and fructification of D. dumetorum. a) male flower, b) female flower. Bar scale = 3 cm. c) fruits, d) seeds. Bar scale = 2 cm (PDF 288 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Siadjeu, C., Mayland-Quellhorst, E. & Albach, D.C. Genetic diversity and population structure of trifoliate yam (Dioscorea dumetorum Kunth) in Cameroon revealed by genotyping-by-sequencing (GBS). BMC Plant Biol 18, 359 (2018). https://doi.org/10.1186/s12870-018-1593-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-018-1593-x