
Abstract
Background
Preterm labor syndrome is associated with high perinatal morbidity and mortality, and intra-amniotic infection is a cause of preterm labor. The standard identification of causative microorganisms is based on the use of biochemical phenotypes, together with broth dilution-based antibiotic susceptibility from organisms grown in culture. However, such methods could not provide an accurate epidemiological aspect and a genetic basis of antimicrobial resistance leading to an inappropriate antibiotic administration. Hybrid genome assembly is a combination of short- and long-read sequencing, which provides better genomic resolution and completeness for genotypic identification and characterization. Herein, we performed a hybrid whole genome assembly sequencing of a pathogen associated with acute histologic chorioamnionitis in women presenting with PPROM.
Results
We identified Enterococcus faecium, namely E. faecium strain RAOG174, with several antibiotic resistance genes, including vancomycin and aminoglycoside. Virulence-associated genes and potential bacteriophage were also identified in this genome.
Conclusion
We report herein the first study demonstrating the use of hybrid genome assembly and genomic analysis to identify E. faecium ST17 as a pathogen associated with acute histologic chorioamnionitis. The analysis provided several antibiotic resistance-associated genes/mutations and mobile genetic elements. The occurrence of E. faecium ST17 raised the awareness of the colonization of clinically relevant E. faecium and the carrying of antibiotic resistance. This finding has brought the advantages of genomic approach in the identification of the bacterial species and antibiotic resistance gene for E. faecium for appropriate antibiotic use to improve maternal and neonatal care.
Similar content being viewed by others

Background
Preterm labor is the leading cause of perinatal morbidity and mortality worldwide [1,2,3,4,5,6,7,8]. Two-thirds of preterm deliveries occur after the spontaneous onset of preterm labor, with either intact or ruptured membranes [7, 9,10,11]. Intraamniotic infection is causally linked to spontaneous preterm delivery/PPROM [12,13,14,15,16]. One of every three preterm infants is born to a mother with intraamniotic infection that is largely subclinical [12,13,14,15,16]. Microorganisms isolated from the amniotic fluid are similar to those found in the lower genital tract; therefore, an ascending pathway is considered the most frequent route of infection [17]. The most common microorganisms identified in the amniotic cavity in women presenting with preterm labor/PPROM include Ureaplasma urealyticum, Mycoplasma hominis, Bacteroides spp., Gardnerella vaginalis, Neisseria gonorrhoeae, Chlamydia trachomatis, Trichomonas vaginalis, and group B hemolytic streptococci [18]. In 30% of cases with intraamniotic infection, bacteria are identified in fetal circulation [19], resulting in FIRS [20, 21]. Such fetuses have multi-organ involvement and are at risk for long-term complications, such as cerebral palsy and chronic lung disease, underscoring that complications of infants born preterm are not only due to immaturity but also to the inflammatory process responsible for preterm labor [14, 21, 22]. Therefore, accurate identification of a causative pathogen is essential for the eradication of microbial invasion of the amniotic cavity with antibiotics [23,24,25,26,27,28,29,30,31,32].
In clinical medicine, identification of the presence of bacteria and bacterial species is based on cultivation and the use of biochemical phenotypes, together with broth dilution-based antibiotic susceptibility. However, such methods are time-consuming, and they could not provide the epidemiological aspect and genetic basis of antimicrobial resistance [33, 34]. Knowledge of the presence of specific bacterial species and AMR genes can guide decision-making to deliver or to treat intraamniotic infection with a particular antibiotic. In addition, the understanding of the specific microorganism and the AMR gene profile could be helpful to the neonatologist to tailor antimicrobial agents appropriate for each newborn [35, 36].
Our group recently reported the use of the 16S nanopore sequencing method, a long-read sequencing, for rapid identification of intraamniotic infection in patients with PPROM [37]. This method allows identification of bacteria at the species level within 5–9 h from DNA extraction, demonstrating that this sequencing technique is effective for clinical use in a timely manner. We have extended the study by performing whole genome sequencing since whole genome sequencing and comparative genomic analysis allow insightful information, i.e., microbiological diagnosis and infectious outbreak investigations [38,39,40]. Several studies utilize whole genome sequencing to identify causative pathogens from clinical specimens [39, 41]. Hybrid genome assembly, which is a combination of short- and long-read sequencing, provides better genomic resolution and completeness for genotypic identification and characterization [42]. A recent study demonstrated that hybrid assembly using Illumina and Nanopore sequencing elucidated genomic insight of multidrug-resistant bacteria and provided epidemiological data of the pathogen [43]. Herein, we performed hybrid whole genome assembly sequencing of a pathogen associated with acute histologic chorioamnionitis in women presenting with PPROM.
Results
During the clinical microbiological laboratory investigation, the sample of the chorioamniotic membranes was cultured under an aerobic condition. When the culture was negative on day-3 after cultivation, the placental tissue was then cultured under an anaerobic condition. The bacteria were finally recovered and identified as E. faecium by conventional phenotypic methods on the 10th day after anaerobic cultivation (13th day after specimen collection). However, antibiotic susceptibility test was not performed because the susceptibility test of anaerobic culture was done by request only according to our hospital protocol. The colonies were also collected for genomic DNA extraction. This isolate was listed as the E. faecium RAOG174 strain.
Whole genome sequencing was performed on colonies recovered from the anaerobic culture of chorioamniotic membranes. ONT sequencing and assembly delivered a total number of 3,150,009 bp, comprised of 11 contigs, largest contigs of 2,852,659 bp, N50 of 2,852,659 bp, and GC content of 37.8%. Genome annotation of E. faecium RAOG174, using Dfast software [44], resulted in 3,038 CDSs, 18 rRNAs, and 67 tRNAs. Species identification, using a whole genome sequence, agreed with E. faecium with ANI of 98.98%, calculated by fasANI. Additional whole genome analysis was performed to illustrate the molecular identification (Supplementary Figures. 1 and 2). The isolate was assigned to ST17, based on MLST scheme. Global genetic epidemiological analysis of E. faecium using bacWGSTdb 2.0 provided MLST-based typing similar to a previous analysis (ST17),however, the most closely related isolate was not available.
Genome features
Resistance
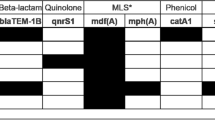
Potential antibiotic resistance genes were predicted with CARD and ResFinder 4.1. Genes included several resistance mechanisms to beta-lactam, quinolone, aminoglycoside, macrolide, tetracycline, and vancomycin (Table 1). Mutations in pbp5, parC and gyrA genes were considered intrinsic resistance. The others were considered acquired resistance (Table 1 and Supplementary Table 1).
Detection of mobile genetic elements and phages
Using Mobile Element Finder, a total number of 61 IS elements were predicted (shown as the black arc in Supplementary Figure 2). The most abundant type of IS was identified as ISEfa11, followed by ISEfa5 and ISEnfa3. PHASTER identified 7 phage regions on the chromosome: 2 intact regions (PHAGE_Lister_2389 and PHAGE_Lister_LP_101) (red arcs in Supplementary Figure 1), 4 incomplete regions (PHAGE_Paenib_Xenia, PHAGE_Lactoc_bIL311, 2 regions of PHAGE_Bacill_vB_BtS_BMBtp14), and 1 questionable region (PHAGE_Entero_EFAP_1). CRISPRCasFinder identified 1 region that was similar to the CRISPR-Cas region, but it was categorized into “level 1,” which is unlikely.
Virulence
For the virulence-associated genes, the VFanalyzer and Virulence Finder identified a total of 22 virulence genes (Table 2). Of them, 12 genes were previously described in E. faecium DO. The other 10 genes were similar to previously defined virulence genes in other genera, including Streptococcus, Staphylococcus, Listeria, and Vibrio.
Discussion
This study demonstrates the first use of hybrid genome assembly to identify the potential virulence, the AMR gene profile, and the sequence type of bacterial pathogen associated with acute histologic chorioamnionitis in women presenting with PPROM. Identification of a causative pathogen is the key for obstetricians to administer proper antibiotics to the mother in order to eradicate intraamniotic infection or to prevent postpartum infectious complications such as endometritis and pelvic abscess [45]. Currently, cultivation-based species identification generally takes 48 h (including biochemical assay and antibiotic susceptibility test). Whole genome sequencing using nanopore method could reveal the result by, at least, 2 h with more informative data, including sequence type, antibiotic resistance-associated genes and virulence genes. In addition, an understanding of the accurate bacterial species and its genomic information is essential for neonatologists to initiate appropriate antibiotic agents to the neonate. This work demonstrates that the potential causative pathogen carried several genes associated with antibiotic resistance and that proper antibiotic selection was crucial in an individual case.
In this study, the patient presented with PPROM and suspected intraamniotic infection. The definite diagnosis of intraamniotic infection was difficult due to anhydramnios; therefore, the microbiologic work-up from amniotic fluid was unsuccessful. However, there was evidence of maternal systemic inflammation as shown by the elevated WBC and CRP as well as acute histologic chorioamnionitis and acute funisitis. The latter two represent the placental histologic landmarks of intraamniotic infection and FIRS [46]. Microbiological identification eventually recovered E. faecium from the anaerobic culture of chorioamniotic membranes.
Enterococcus species are gram-positive bacteria that are abundant in the gastrointestinal tract of a wide range of animals [47]. In humans, Enterococci are one of the earliest colonizers in the gut. Currently, more than 50 species of Enterococcus have been identified. However, a few species are clinically important, including E. faecium [48]. In general, E. faecium is a commensal and does not harm a healthy host, but it becomes pathogenic when the host is immunocompromised [49]. This bacterium is associated with urinary tract and surgical site infections and bacteremia, especially in a hospital-acquired setting [50]. In pregnancy, Enterococcal infection is uncommon; however, it has been identified in the amniotic fluid of women diagnosed with clinical chorioamnionitis [51]. Ncib et al. demonstrated that the presence of vaginal-derived Enterococcus spp. is associated with recurrent pregnancy loss [52], PPROM [53], and bacterial vaginosis in pregnant women [54]. Seliga-Siwecka and Kornacka reported that the presence of E. faecalis in amniotic fluid significantly increases the risk for acute placental inflammation, necrotizing enterocolitis, and bronchopulmonary dysplasia in neonates [55, 56].
To investigate genomic insight of the bacteria, hybrid assembly, using ONT and Illumina sequencing, was then performed to obtain the complete genome, which was matched to E. faecium with a high similarity index (ANI = 98.98%). Genome assembly resulted in a genome size of 2.8 Gbp and the GC content of 37.8%, similar to the previously reported genome characteristic of E. faecium (2.5–2.9 Gbp for genome size and 37.8–38.5% for GC content) [57]. Whole genome sequencing has been used as a diagnostic tool for clinical microbiology to understand genomic insight of the pathogen and has become a “gold standard” technique [58, 59]. In microbial genomics, hybrid genome assembly utilizes short- and long-reads to construct the complete or nearly complete genome with better resolution and less error [60,61,62]. Hybrid assembly uses long reads as scaffolds for short read-based contigs to be rearranged into a correct direction [63]. Khezri et al. demonstrated that hybrid genome assembly has better performance in constructing the complete genome, more accurately resulting in the identification of genes associated with virulence and drug resistance [42]. By incorporating the sequence information from both generations of sequencing, hybrid assembly can provide the sequence with the following improvements: reducing error rates, minimizing gaps, and unveiling sequences that are not covered by short-read sequencing alone [64].
It has been demonstrated that hybrid assembly could provide genomic importance for epidemiological study, particularly antibiotic resistance genes [65, 66]. The in silico sequence typing, using pubMLST, revealed that the E. faecium strain RAOG174 belongs to ST17. E. faecium ST17 is clinically relevant [67, 68] and is defined as an ancestor of CC17 [69]. The CC17 belongs to clade A, which is associated with human infection (as opposed to clade B, which is the commensal clade) [70, 71], however, it is later identified in non-clinical specimens, including wastewater and domestic animals [69]. The characteristics of CC17 are ampicillin and quinolone resistance, and most isolates have a putative pathogenicity island harboring the esp gene, which was also present in the isolate RAOG174 (Table 2) [72]. Some isolates additionally carry the vanA gene conferring vancomycin resistance [71]. The vanA typically situates in vanHAX gene clusters where vanH, vanA, and vanX encode for D-alanyl-D-lactate ligase, α-keto acid reductase, and Zn2 + -dependent d-Ala-d-Ala dipeptidase, respectively [73]. The isolate RAOG174 is likely ampicillin- and vancomycin-resistant due to the presence of pbp mutation and vanHAX, respectively (Table 1), although vanA-positive vancomycin-susceptible E. faecium was also previously identified [74, 75].
Our work revealed that IS and phages were predicted in the E. faecium RAOG174 genome. E. faecium is highly evolved and adaptable, as previously illustrated by its open pan-genome [76]. With this genomic characteristic, E. faecium can receive and donate genetic elements, including antibiotic resistance genes, with other cells or environments. In this study, major antibiotic groups, including beta-lactam, glycopeptide, aminoglycoside, fluoroquinolone, and macrolide, were predicted. AMR gene analysis identified point mutations and genes that are associated with antibiotic resistance, and these genes/mutations indicated intrinsic resistance and acquired resistance. E. faecium has been known to exhibit several antibiotic resistance phenotypes.
“Empirical antibiotics with ampicillin and erythromycin was administered for prolong latency period in women with preterm PROM. Postpartum period was uneventful. Neonate was diagnosed with suspected sepsis based on mother’s history (preterm PROM) and was administered with routine antibiotics (ampicillin and gentamicin) for 7 days.” The standard management of clinical chorioamnionitis is the administration of antibiotics and augmentation of labor [45, 77,78,79]. The American College of Obstetricians and Gynecologists recommended the use of ampicillin and gentamicin (ampicillin 2 g IV every 6 h combined with gentamicin 5 mg/kg every 24 h) whenever an intraamniotic infection is suspected or confirmed [80]. Such antibiotics would not have been effective against the E. faecium reported herein. For the neonates, antibiotic prescription is recommended only in symptomatic neonates and in neonates with risk of early-onset sepsis due to an avoidance of inappropriate antibiotic exposure [81, 82] with the standard regimen of ampicillin and gentamicin [83]. Interestingly, a case of early-onset sepsis caused by vancomycin-resistant E. faecium in a newborn, who was born from a mother without any sign of clinical chorioamnionitis, has been reported. The newborn was initially diagnosed with meconium aspiration syndrome and neonatal sepsis and then was treated with the standard antibiotic regimen for early-onset neonatal sepsis. Then antibiotic-resistant E. faecium was identified from the blood culture of the baby. Subsequently, the antibiotic was changed to linezolid according to its antibiotic susceptibility. The neonate was discharged in normal condition after receiving linezolid for 2 weeks [84].
The vanA-positive E. faecium is mostly associated with hospital-acquired infection. The carriage rates in a given community vary from 0%- to -13% depending on the geographic region [85,86,87,88,89,90], and community-acquired infections are rare. In our study, we could not identify the source of E. faecium. Our hospital has a standard protocol for regular surveillance of VRE. We confirmed that VRE was not detected in the clinical microbiology lab and the ward (where the patient stayed) during the duration of sample collection and patient’s hospitalization.
In general, VRE should not be identified in the labour room, according to the hospital’s infection control surveillance. According to the history that documented the patient had been previously admitted at her provider hospital prior to transferring to our hospital, the patient could have contracted VRE colonization from the previous hospital. As E. faecium is one of the colonizers in human gastrointestinal tract, the bacteria could migrate from maternal gut to maternal blood stream and then enter the chorioamniotic membranes transplacentally [91,92,93].
Alternatively, samples with low bacterial biomass, such as the amniotic fluid and placenta, are commonly vulnerable to bacterial contamination from the environment [94]. VRE is one of the most common laboratory contaminants, and the prevalence of VRE contamination ranges from 10%- to -60% [95, 96]. The most common contaminated sites are the work surface and the hands of the healthcare worker [97]. However, in our case, we believed that this is a true pathogen, as the patient had clear evidence of systemic maternal inflammatory response and her placenta showed acute chorioamnionitis as well as acute funisitis. In addition, she had a history of multiple hospital admissions prior to the last visit. Therefore, despite the possibility of lab contamination, the VRE strain RAOG174 could be the true pathogen associated with acute chorioamnionitis.
Our results suggest that the precise identification of a pathogen and its antibiotic susceptibility is essential for administration of appropriate antibiotics since the routine antibiotic regimen for chorioamnionitis is not effective against this microorganism. Whole genome sequencing, together with an established bioinformatic analysis pipeline, could provide more robust bacterial identification of causative pathogens. Importantly, one study illustrated that the whole genome sequencing approach took approximately 36 to 42 h, compared to conventional (cultivation-based) identification in 48 to 72 h [39]. It is important to note that the nanopore sequencing achieved nearly 30 × coverage of the VRE genomes within a sequencing run of only 40 min. With this depth of coverage, we were able to obtain similar contigs as observed in the hybrid assembly. Furthermore, the advantage of the long-read assembly lies in its rapidity. Our results show that the long-read assembly method can deliver results swiftly, suggesting its potential for future applications in clinical testing. These advantages may lead to appropriate antibiotic use that may enhance maternal and neonatal care by reducing identification time and by providing more precise microorganism determination.
Conclusions
Chorioamnionitis is a global health care problem, and the accurate identification of the causative pathogen is beneficial for the pregnant woman and her neonate. This study is the first to demonstrate the use of hybrid genome assembly and genomic analysis to identify E. faecium ST17 as a pathogen associated with acute histologic chorioamnionitis. The analysis provided several antibiotic resistance-associated genes/mutations and mobile genetic elements. Although the source of infection could not be identified, the occurrence of the vancomycin-resistant gene carrying E. faecium ST17 raised awareness of the colonization and the infection of highly resistant bacteria in a pregnancy-related setting in which resistance identification is critical. This finding spotlights the advantages of the hybrid genome assembly approach in bacterial species and in antibiotic-resistance gene identification for appropriate antibiotic use to improve maternal and neonatal care.
Methods
Patient history and clinical information
A 39-year-old para 0 at 30+5 weeks of gestation, presented to the Labor and Delivery Unit at our hospital due to leakage of fluid and the onset of abdominal cramps every 5 min. At 28+5 weeks of gestation, she had an episode of vaginal bleeding and was diagnosed with placenta previa. The patient was admitted for 3 days at her provider hospital and vaginal bleeding was lessened. At 30 weeks of gestation, she was re-admitted at her provider hospital due to a second episode of vaginal bleeding. Five days later, she experienced rupture of membranes and was transferred to our hospital. Upon arrival, her vital signs were normal without a fever or tachycardia. Transabdominal ultrasound demonstrated a single viable fetus with an estimated fetal weight of 1,507 g (percentile 50–90) and anhydramnios. The patient’s laboratory examination showed anemia (Hb 8.7 g/dL) and an elevated white blood cell (WBC) count as well as C-reactive protein (CRP) [WBC 29,990 cells/microliters (neutrophils 91%) and CRP 138.35 mg/L]. The diagnosis was PPROM, placenta low-lying with preterm labor. Expectant management was undertaken with the administration of steroids to promote fetal lung maturity, antibiotic agents (ampicillin and erythromycin) to prolong the latency period, and magnesium sulfate for tocolysis. Five days after admission, the patient developed regular uterine contractions every 5 min. A Cesarean section was performed due to preterm labor with placenta previa. A female fetus was delivered. The neonate was diagnosed with suspected sepsis (according to mother history) and was administered ampicillin and gentamicin for 7 days. Placental histopathology demonstrated acute histologic chorioamnionitis with acute funisitis. The placental chorioamniotic membranes culture demonstrated Enterococcus faecium (E. faecium). This enrollment and the use of clinical specimens of this patient were approved by the Institutional Research Board of the Faculty of Medicine, Ramathibodi Hospital, Mahidol University (COA.MURA2021/254 and COA.MURA 2022/675).
Placental cultivation
Placental tissue derived from the chorioamniotic membranes was obtained after cesarean delivery with an aseptic technique and kept in a sterile, capped container within the sterile operating field. The placental sample was then brought directly to a biosafety cabinet located at the microbiology unit in the same building by using a standard transportation process. The placental tissue was divided into approximately 1 cm2 in size and inoculated on blood agar, MacConkey agar, and chocolate agar under an aerobic condition and on thioglycolate broth under an anaerobic condition.
Isolation and sequencing
Bacteria were recovered from the placental tissue by cultivating under aerobic and anaerobic conditions, using an aseptic technique throughout the processes. Genomic DNA was extracted from bacterial colonies obtained from the anaerobic culture. The purity of the extracted DNA was observed by a Nanodrop Spectrophotometer (Thermo Fisher Scientific, USA), and the quantity was checked by a Qubit® 4.0 Fluorometer (Invitrogen, USA). Whole genome sequencing was conducted with single molecular sequencing (ONT UK) and Illumina short-read sequencing. Briefly, DNA library preparation was performed by using the Rapid Barcoding Sequencing Kit (SQK-RBK004; ONT). A total of 200 ng of genomic DNA was cleaved with transposase enzyme to produce chemically modified ends and a barcode was added to each DNA sample, finally ligated with an adapter. The library was loaded into the R9.4.1 flow cell (FLO-MIN106 version; ONT) and sequenced with the GridION device (ONT) with 72-h sequencing. For Illumina short-read sequencing, 150-bp paired-end libraries were prepared with a TruSeq DNA PCR-free Kit and sequenced with the Illumina™ Novaseq sequencer (Illumina Inc., San Diego, CA, USA). To obtain high-quality reads, adapter sequences were trimmed by using Skewer v0.2.2. The sequence data from both platforms were combined for hybrid genome assembly. This protocol was approved by Institutional Biosafety Committee of the Faculty of Medicine at Ramathibodi Hospital, Mahidol University (RAMA-IBC 2022–009).
Assembly and annotation
A hybrid genome assembly approach was selected to produce a de novo assembly of the E. faecium genome. This approach involved the use of Illumina short-read and Nanopore long-read sequencing technologies to produce a complete and accurate genome assembly. The hybrid genome assembly procedures were depicted as a series of flowing steps. For short reads QC, we used the Fastp v0.20.1 tool to trim sequencing adapters and low-quality reads, followed by quality assessment with FastQC v0.11.8 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). For long reads, raw signals were processed, demultiplexed, and adapter trimmed, using Guppy v6.2.1 with the super accurate model (–c dna_r9.4.1_450bps_sup.cfg -r –trim_barcodes –barcode_kits SQK-RBK004) and Porechop v0.2.4 (https://github.com/rrwick/Porechop). The quality of the ONT raw reads was assessed by using NanoPlot v1.28.1. The reads were filtered by NanoFilt v2.5.0 [98], based on a mean quality score of 9, and only reads with a length of 1,000 bases were retained for the de novo assembly. Finally, we constructed the genomes with Unicycler v0.4.8 [63], which incorporates hybrid assembly, correction, circularization, and rotation to produce high-quality genome assemblies. Gene annotation was performed in Dfast [44]. MLST assignment was performed by using FastMLST software [99]. Analysis of mobile genetic element, phage, potential antimicrobial resistance genes, and virulence-associated genes was executed by using the Mobile Element Finder, PHASTER, ResFinder version 4.1, CARD and MLST service, respectively [100,101,102,103]. CRISPRCasFinder was used to determine potential CRISPR-Cas region on the chromosome [104]. Virulence-associated genes were obtained by using VFanalyzer [105] and Virulence Finder [106]. As E. faecium is clinically important, especially in hospital-associated infection, genomic epidemiology analysis was performed via web-based platforms in bacWGSTdb 2.0 [107]. The visualization of comparative genomics analysis was completed in BRIG [108]. Phylogenetic tree was constructed by using PhyML with 1000 bootstrap repeats [109] and visualized with Figtree version 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/). The tree was constructed using type strains: Enterococcus thailandicus DSM 21767 (Accession no:SAMN03267187), Enterococcus ratti DSM 15687 (Accession no:SAMN03267184), Enterococcus mundtii DSM 4838 (Accession no:SAMN03267178), Enterococcus xinjiangensis JCM 30200 (Accession no:SAMD00255146), Enterococcus lactis DSM 23655 (Accession no:IMG ID 2928549275), Enterococcus lactis CCM 8412 (Accession no:SAMD00255145), Enterococcus villorum NBRC 100699 (Accession no:SAMD00166261), Enterococcus porcinus ATCC 700913 (Accession no:SAMN02596958), Enterococcus hirae ATCC 9790 (Accession no:SAMN02604142), Enterococcus canis NBRC 100695 (Accession no:SAMD00046312), Enterococcus casseliflavus NBRC 100478 (Accession no:SAMD00045727), Enterococcus durans NBRC 100479 (Accession no:SAMD00045728), Enterococcus faecium NBRC 100486 (Accession no:SAMD00045730), Enterococcus gallinarum NBRC 100675 (Accession no:SAMD00045734).
Availability of data and materials
The genome used in this study was available in the Genbank repository (BioProject PRJNA930917; Reviewer’s link is https://dataview.ncbi.nlm.nih.gov/object/PRJNA930917?reviewer=rn64h3763n6525qpdvobsb0igc).
Abbreviations
- AMR:
-
Antimicrobial resistance
- ANI:
-
Average Nucleotide Identity
- BRIG:
-
BLAST Ring Image Generator
- CARD:
-
Comprehensive Antibiotics Resistance Database
- CC:
-
Clonal complex
- CRISPR:
-
Clustered Regularly Interspaced Short Palindromic Repeats
- CRP:
-
C-reactive protein
- FIRS:
-
Fetal systemic inflammatory response syndrome
- MLST:
-
Multi-locus sequence typing
- ONT:
-
Oxford Nanopore Technologies
- PPROM:
-
Preterm prelabor rupture of membranes
- ST:
-
Sequence Type
- VRE:
-
Vancomycin-resistant E. faecium
- WBC:
-
White Blood Cell
References
Walani SR, Biermann J. March of dimes foundation: leading the way to birth defects prevention. Public Health Rev. 2017;38:1–7.
Blencowe H, Cousens S, Oestergaard MZ, Chou D, Moller AB, Narwal R, et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet. 2012;379:2162–72.
Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu J, et al. Global, regional, and national causes of under-5 mortality in 2000–15: an updated systematic analysis with implications for the Sustainable Development Goals. Lancet. 2016;388:3027–35.
de Costa A, Moller AB, Blencowe H, Johansson EW, Hussain-Alkhateeb L, Ohuma EO, et al. Study protocol for WHO and UNICEF estimates of global, regional, and national preterm birth rates for 2010 to 2019. PLoS One. 2021;16:e0258751.
Chawanpaiboon S, Vogel JP, Moller AB, Lumbiganon P, Petzold M, Hogan D, et al. Global, regional, and national estimates of levels of preterm birth in 2014: a systematic review and modelling analysis. Lancet Glob Health. 2019;7:e37–46.
Cao G, Liu J, Liu M. Global, regional, and national incidence and mortality of neonatal preterm birth, 1990–2019. JAMA Pediatr. 2022;176:787–96.
Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science. 2014;345:760–5.
Muglia LJ, Katz M. The enigma of spontaneous preterm birth. N Engl J Med. 2010;362:529–35.
Romero R, Mazor M, Munoz H, Gomez R, Galasso M, Sherer DM. The preterm labor syndrome. Ann N Y Acad Sci. 1994;734:414–29.
Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371:75–84.
Berkowitz GS, Blackmore-Prince C, Lapinski RH, Savitz DA. Risk factors for preterm birth subtypes. Epidemiology. 1998;9:279–85.
Gomez R, Romero R, Edwin SS, David C. Pathogenesis of preterm labor and preterm premature rupture of membranes associated with intraamniotic infection. Infect Dis Clin North Am. 1997;11:135–76.
Gonçalves LF, Chaiworapongsa T, Romero R. Intrauterine infection and prematurity. Ment Retard Dev Disabil Res Rev. 2002;8:3–13.
Romero R, Gotsch F, Pineles B, Kusanovic JP. Inflammation in pregnancy: its roles in reproductive physiology, obstetrical complications, and fetal injury. Nutr Rev. 2007;65(12 Pt 2):S194–202.
Gomez-Lopez N, Galaz J, Miller D, Farias-Jofre M, Liu Z, Arenas-Hernandez M, et al. The immunobiology of preterm labor and birth: intra-amniotic inflammation or breakdown of maternal-fetal homeostasis. Reproduction. 2022;164:R11–45.
Romero R, Gómez R, Chaiworapongsa T, Conoscenti G, Kim JC, Kim YM. The role of infection in preterm labour and delivery. Paediatr Perinat Epidemiol. 2001;15(Suppl 2):41–56.
Romero R, Gomez-Lopez N, Winters AD, Jung E, Shaman M, Bieda J, et al. Evidence that intra-amniotic infections are often the result of an ascending invasion - a molecular microbiological study. J Perinat Med. 2019;47:915–31.
Romero R, Espinoza J, Kusanovic JP, Gotsch F, Hassan S, Erez O, et al. The preterm parturition syndrome. BJOG. 2006;113 Suppl 3(Suppl 3):17–42.
Carroll SG, Ville Y, Greenough A, Gamsu H, Patel B, Philpott-Howard J, et al. Preterm prelabour amniorrhexis: intrauterine infection and interval between membrane rupture and delivery. Arch Dis Child Fetal Neonatal Ed. 1995;72:F43–6.
Gomez R, Romero R, Ghezzi F, Yoon BH, Mazor M, Berry SM. The fetal inflammatory response syndrome. Am J Obstet Gynecol. 1998;179:194–202.
Jung E, Romero R, Yeo L, Diaz-Primera R, Marin-Concha J, Para R, et al. The fetal inflammatory response syndrome: the origins of a concept, pathophysiology, diagnosis, and obstetrical implications. Semin Fetal Neonatal Med. 2020;25:101146.
Gotsch F, Romero R, Kusanovic JP, Mazaki-Tovi S, Pineles BL, Erez O, et al. The fetal inflammatory response syndrome. Clin Obstet Gynecol. 2007;50:652–83.
Romero R, Scioscia AL, Edberg SC, Hobbins JC. Use of parenteral antibiotic therapy to eradicate bacterial colonization of amniotic fluid in premature rupture of membranes. Obstet Gynecol. 1986;67(3 Suppl):15S–17S.
Romero R, Hagay Z, Nores J, Sepulveda W, Mazor M. Eradication of Ureaplasma urealyticum from the amniotic fluid with transplacental antibiotic treatment. Am J Obstet Gynecol. 1992;166:618–20.
Mazor M, Chaim W, Horowitz S, Leiberman JR, Glezerman M. Successful treatment of preterm labour by eradication of Ureaplasma urealyticum with erythromycin. Arch Gynecol Obstet. 1993;253:215–8.
Mazor M, Chaim W, Meirovitz M, Yohay D, Leiberman JR, Glezerman M. Eradication of viridans streptococci from the amniotic cavity by parenteral antibiotic administration. A case report. J Reprod Med. 1995;40:820–2.
Morency A-M, Rallu F, Laferrière C, Bujold E. Eradication of intra-amniotic Streptococcus mutans in a woman with a short cervix. J Obstet Gynaecol Can. 2006;28:898–902.
Yeo L, Romero R, Chaiworapongsa T, Para R, Johnson J, Kmak D, et al. Resolution of acute cervical insufficiency after antibiotics in a case with amniotic fluid sludge. J Matern Fetal Neonatal Med. 2022;35:5416–26.
Yoon BH, Romero R, Park JY, Oh KJ, Lee J, Conde-Agudelo A, et al. Antibiotic administration can eradicate intra-amniotic infection or intra-amniotic inflammation in a subset of patients with preterm labor and intact membranes. Am J Obstet Gynecol. 2019;221:142.e1–142.e22.
Oh KJ, Romero R, Park JY, Lee J, Conde-Agudelo A, Hong J-S, et al. Evidence that antibiotic administration is effective in the treatment of a subset of patients with intra-amniotic infection/inflammation presenting with cervical insufficiency. Am J Obstet Gynecol. 2019;221:140.e1–140.e18.
Lee J, Romero R, Kim SM, Chaemsaithong P, Park C-W, Park JS, et al. A new anti-microbial combination prolongs the latency period, reduces acute histologic chorioamnionitis as well as funisitis, and improves neonatal outcomes in preterm PROM. J Matern Fetal Neonatal Med. 2016;29:707–20.
Kacerovsky M, Romero R, Stepan M, Stranik J, Maly J, Pliskova L, et al. Antibiotic administration reduces the rate of intraamniotic inflammation in preterm prelabor rupture of the membranes. Am J Obstet Gynecol. 2020;223:114.e1–114.e20.
Fournier P-E, Dubourg G, Raoult D. Clinical detection and characterization of bacterial pathogens in the genomics era. Genome Med. 2014;6:114.
Abayasekara LM, Perera J, Chandrasekharan V, Gnanam VS, Udunuwara NA, Liyanage DS, et al. Detection of bacterial pathogens from clinical specimens using conventional microbial culture and 16S metagenomics: a comparative study. BMC Infect Dis. 2017;17:631.
Oeser C, Pond M, Butcher P, Russell AB, Henneke P, Laing K, et al. PCR for the detection of pathogens in neonatal early onset sepsis. PLoS ONE. 2020;15:e0226817.
Salsabila K, Toha NMA, Rundjan L, Pattanittum P, Sirikarn P, Rohsiswatmo R, et al. Early-onset neonatal sepsis and antibiotic use in Indonesia: a descriptive, cross-sectional study. BMC Public Health. 2022;22:1–12.
Chaemsaithong P, Romero R, Pongchaikul P, Vivithanaporn P, Lertrut W, Jaovisidha A, et al. Rapid diagnosis of intra-amniotic infection using nanopore-based sequencing. J Perinat Med. 2022. https://doi.org/10.1515/jpm-2022-0504.
Didelot X, Bowden R, Wilson DJ, Peto TEA, Crook DW. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet. 2012;13:601.
Hasman H, Saputra D, Sicheritz-Ponten T, Lund O, Svendsen CA, Frimodt-Moller N, et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J Clin Microbiol. 2014;52:139.
Tagini F, Greub G. Bacterial genome sequencing in clinical microbiology: a pathogen-oriented review. Eur J Clin Microbiol Infect Dis. 2017;36:2007–20.
Taxt AM, Avershina E, Frye SA, Naseer U, Ahmad R. Rapid identification of pathogens, antibiotic resistance genes and plasmids in blood cultures by nanopore sequencing. Sci Rep. 2020;10:1–11.
Khezri A, Avershina E, Ahmad R. Hybrid assembly provides improved resolution of plasmids, antimicrobial resistance genes, and virulence factors in escherichia coli and klebsiella pneumoniae clinical isolates. Microorganisms. 2021;9:2560.
Ruan Z, Wu J, Chen H, Draz MS, Xu J, He F. Hybrid genome assembly and annotation of a Pandrug-Resistant Klebsiella pneumoniae strain using Nanopore and Illumina sequencing. Infect Drug Resist. 2020;13:199–206.
Tanizawa Y, Fujisawa T, Nakamura Y. DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics. 2018;34:1037–9.
Tita ATN, Andrews WW. Diagnosis and management of clinical chorioamnionitis. Clin Perinatol. 2010;37:339.
Kim CJ, Romero R, Chaemsaithong P, Chaiyasit N, Yoon BH, Kim YM. Acute chorioamnionitis and funisitis: definition, pathologic features, and clinical significance. Am J Obstet Gynecol. 2015;213(4 Suppl):S29–52.
Van Tyne D, Gilmore MS. Friend turned foe: evolution of enterococcal virulence and antibiotic resistance. Annu Rev Microbiol. 2014;68:337.
Gilmore MS, Lebreton F, van Schaik W. Genomic transition of enterococci from gut commensals to leading causes of multidrug-resistant hospital infection in the antibiotic era. Curr Opin Microbiol. 2013;16:10.
Dubin K, Pamer EG. Enterococci and their interactions with the intestinal microbiome. Microbiol Spectr. 2017;5:5.
Ramos S, Silva V, Dapkevicius M de LE, Igrejas G, Poeta P. Enterococci, from harmless bacteria to a pathogen. Microorganisms. 2020;8:1118.
Li M, Huang Z, Tao Z, Meng Y, Wen J, Zhang Q, et al. The role of upper and lower genital tract microbiota alterations in term chorionamnionitis: a prospective study. Front Microbiol. 2022;13:1069254.
Ncib K, Bahia W, Leban N, Mahdhi A, Trifa F, Mzoughi R, et al. Microbial diversity and pathogenic properties of microbiota associated with aerobic vaginitis in women with recurrent pregnancy loss. Diagnostics. 2022;12:2444.
Singh N, Pattnaik L, Panda SR, Jena P, Panda J. Fetomaternal outcomes in women affected with preterm premature rupture of membranes: an observational study from a tertiary care center in Eastern India. Cureus. 2022;14:e25533.
Javed A, Manzoor S. Comparative analysis of bacterial vaginosis microbiota among pregnant and non-pregnant females and isolation of phages against Enterococcus faecalis, Enterococcus faecium, and Shigella flexneri strains. Microb Pathog. 2020;149:104588.
Puranik V. Enterococci and Bacterial Infections. In: Elkady A, Sinha P, Hassan S, editors. Infections in Pregnancy: An Evidence-Based Approach (p. 115-120). Cambridge: Cambridge University Press; 2019. https://doi.org/10.1017/9781108650434.019.
Seliga-Siwecka JP, Kornacka MK. Neonatal outcome of preterm infants born to mothers with abnormal genital tract colonisation and chorioamnionitis: a cohort study. Early Hum Dev. 2013;89:271–5.
Zaghloul HAH, El Halfawy NM. Genomic insights into antibiotic-resistance and virulence genes of Enterococcus faecium strains from the gut of Apis mellifera. Microb Genom. 2022;8:mgen000896.
Mitchell SL, Simner PJ. Next-generation sequencing in clinical microbiology: are we there yet? Clin Lab Med. 2019;39:405–18.
Simar SR, Hanson BM, Arias CA. Techniques in bacterial strain typing: past, present, and future. Curr Opin Infect Dis. 2021;34:339.
Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol. 2012;30:693–700.
Koren S, Phillippy AM. One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr Opin Microbiol. 2015;23:110–20.
Chen Z, Erickson DL, Meng J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genomics. 2020;21:1–21.
Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13:e1005595.
Di Marco F, Spitaleri A, Battaglia S, Batignani V, Cabibbe AM, Cirillo DM. Advantages of long- and short-reads sequencing for the hybrid investigation of the Mycobacterium tuberculosis genome. Front Microbiol. 2023;14:186.
Chen H, Jiang T, Wu J, Sun Q, Zha Q, Jin S, et al. Genomic and phylogenetic analysis of a multidrug-resistant mcr-1-carrying Klebsiella pneumoniae recovered from a urinary tract infection in China. J Glob Antimicrob Resist. 2021;27:222–4.
Zheng W, Yue M, Zhang J, Ruan Z. Coexistence of two blaCTX-M-14 genes in a blaNDM-5-carrying multidrug-resistant Escherichia coli strain recovered from a bloodstream infection in China. J Glob Antimicrob Resist. 2021;26:11–4.
Kim SH, Cho SY, Kim HM, Huh K, Kang CI, Peck KR, et al. Sequence type 17 is a predictor of subsequent bacteremia in vancomycin-resistant Enterococcus faecium-colonized patients: a retrospective cohort study. Antimicrob Resist Infect Control. 2021;10:1–10.
Bourafa N, Abat C, Loucif L, Olaitan AO, Bentorki AA, Boutefnouchet N, et al. Identification of vancomycin-susceptible major clones of clinical Enterococcus from Algeria. J Glob Antimicrob Resist. 2016;6:78–83.
Lee T, Pang S, Abraham S, Coombs GW. Antimicrobial-resistant CC17 Enterococcus faecium: the past, the present and the future. J Glob Antimicrob Resist. 2019;16:36–47.
Lebreton F, van Schaik W, McGuire AM, Godfrey P, Griggs A, Mazumdar V, et al. Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. MBio. 2013;4:10.
Zhou X, Willems RJL, Friedrich AW, Rossen JWA, Bathoorn E. Enterococcus faecium: from microbiological insights to practical recommendations for infection control and diagnostics. Antimicrob Resist Infect Control. 2020;9:1–13.
Top J, Willems R, Van Der Velden S, Asbroek M, Bonten M. Emergence of clonal complex 17 Enterococcus faecium in the Netherlands. J Clin Microbiol. 2008;46:214–9.
Roper DI, Huyton T, Vagin A, Dodson G. The molecular basis of vancomycin resistance in clinically relevant Enterococci: crystal structure of D-alanyl-D-lactate ligase (VanA). Proc Natl Acad Sci U S A. 2000;97:8921–5.
Szakacs TA, Kalan L, McConnell MJ, Eshaghi A, Shahinas D, McGeer A, et al. Outbreak of vancomycin-susceptible Enterococcus faecium containing the wild-type vanA gene. J Clin Microbiol. 2014;52:1682–6.
Kohler P, Eshaghi A, Kim HC, Plevneshi A, Green K, Willey BM, et al. Prevalence of vancomycin-variable Enterococcus faecium (VVE) among vanA-positive sterile site isolates and patient factors associated with VVE bacteremia. PLoS ONE. 2018;13:e0193926.
Zhong Z, Kwok LY, Hou Q, Sun Y, Li W, Zhang H, et al. Comparative genomic analysis revealed great plasticity and environmental adaptation of the genomes of Enterococcus faecium. BMC Genomics. 2019;20:1.
Gibbs RS, Castillo MS, Rodgers PJ. Management of acute chorioamnionitis. Am J Obstet Gynecol. 1980;136:709–13.
Gibbs RS, Duff P. Progress in pathogenesis and management of clinical intraamniotic infection. Am J Obstet Gynecol. 1991;164(5 Pt 1):1317–26.
Conde-Agudelo A, Romero R, Jung EJ, Garcia Sánchez ÁJ. Management of clinical chorioamnionitis: an evidence-based approach. Am J Obstet Gynecol. 2020;223:848–69.
American College of Obstetricians and Gynecologists. Committee Opinion No. 712: Intrapartum management of intraamniotic infection. Obstet Gynecol. 2017;130:e95-101.
Fleiss N, Schwabenbauer K, Randis TM, Polin RA. What’s new in the management of neonatal early-onset sepsis? Arch Dis Child Fetal Neonatal Ed. 2023;108:10–4.
Jan AI, Ramanathan R, Cayabyab RG. Chorioamnionitis and management of asymptomatic infants ≥35 weeks without empiric antibiotics. Pediatrics. 2017;140:20162744.
Fuchs A, Bielicki J, Mathur S, Sharland M, Van Den Anker JN. Reviewing the WHO guidelines for antibiotic use for sepsis in neonates and children. Paediatr Int Child Health. 2018;38(Suppl 1):S3.
Subramanya SH, Amberpet R, Chaudhary D, Nayak N, Padukone S, Bairy I, et al. Neonatal sepsis due to glycopeptide resistant Enterococcus faecium from colonized maternal gut- rare case evidence. Antimicrob Resist Infect Control. 2019;8:29.
Trajkovska-Dokic E, Kaftandzieva A, Stojkovska S, Kuzmanovska A, Panovski N. Gastrointestinal colonization with vancomycin-resistant enterococci in hospitalized and outpatients. Open Access Maced J Med Sci. 2014;3:7–11.
Hannaoui I, Barguigua A, Serray B, El Mdaghri N, Timinouni M, Ait Chaoui A, et al. Intestinal carriage of vancomycin-resistant enterococci in a community setting in Casablanca. Morocco J Glob Antimicrob Resist. 2016;6:84–7.
Zhou X, García-Cobos S, Ruijs GJHM, Kampinga GA, Arends JP, Borst DM, et al. Epidemiology of extended-spectrum β-lactamase-producing E. coli and vancomycin-resistant enterococci in the Northern Dutch–German cross-border region. Front Microbiol. 2017;8:1914. https://doi.org/10.3389/fmicb.2017.01914.
Huang YS, Lai LC, Chen YA, Lin KY, Chou YH, Chen HC, et al. Colonization with multidrug-resistant organisms among healthy adults in the community setting: prevalence, risk factors, and composition of gut microbiome. Front Microbiol. 2020;11:1402.
Croughan S, O’Cronin D, O’Brien D, Roberts F, Underwood S, O’Connell J, et al. Vancomycin-resistant enterococci in patients attending for colonoscopy: an estimate of community prevalence. Ir Med J. 2022;115:649.
Kolar M, Pantucek R, Vagnerova I, Sauer P, Kesselova M, Cekanova L, et al. Prevalence of vancomycin-resistant enterococci in hospitalized patients and those living in the community in the Czech Republic. New Microbiol. 2006;29:121–5.
Moles L, Gómez M, Heilig H, Bustos G, Fuentes S, de Vos W, et al. Bacterial diversity in meconium of preterm neonates and evolution of their fecal microbiota during the first month of life. PLoS ONE. 2013;8:e66986.
Jiménez E, Marín ML, Martín R, Odriozola JM, Olivares M, Xaus J, et al. Is meconium from healthy newborns actually sterile? Res Microbiol. 2008;159:187–93.
Gosalbes MJ, Llop S, Vallès Y, Moya A, Ballester F, Francino MP. Meconium microbiota types dominated by lactic acid or enteric bacteria are differentially associated with maternal eczema and respiratory problems in infants. Clin Exp Allergy. 2013;43:198–211.
Doyle RM, Harris K, Kamiza S, Harjunmaa U, Ashorn U, Nkhoma M, et al. Bacterial communities found in placental tissues are associated with severe chorioamnionitis and adverse birth outcomes. PLoS ONE. 2017;12:e0180167.
Collins SM, Hacek DM, Degen LA, Wright MO, Noskin GA, Peterson LR. Contamination of the clinical microbiology laboratory with vancomycin-resistant enterococci and multidrug- resistant enterobacteriaceae: implications for hospital and laboratory workers. J Clin Microbiol. 2001;39:3772.
Katz KC, McGeer A, Low DE, Willey BM. Laboratory contamination of specimens with quality control strains of vancomycin-resistant enterococci in Ontario. J Clin Microbiol. 2002;40:2686–8.
Duckro AN, Blom DW, Lyle EA, Weinstein RA, Hayden MK. Transfer of vancomycin-resistant enterococci via health care worker hands. Arch Intern Med. 2005;165:302–7.
De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–9.
Guerrero-Araya E, Muñoz M, Rodríguez C, Paredes-Sabja D. FastMLST: a multi-core tool for multilocus sequence typing of draft genome assemblies. Bioinform Biol Insights. 2021;15:11779322211059238.
Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44:W16–21.
Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol. 2014;52:1501.
Johansson MHK, Bortolaia V, Tansirichaiya S, Aarestrup FM, Roberts AP, Petersen TN. Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J Antimicrob Chemother. 2021;76:101–9.
Alcock BP, Huynh W, Chalil R, Smith KW, Raphenya AR, Wlodarski MA, et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023;51:D690–9.
Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018;46:W246–51.
Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019;47:D687–92.
Tetzschner AMM, Johnson JR, Johnston BD, Lund O, Scheutz F. In silico genotyping of escherichia coli isolates for extraintestinal virulence genes by use of whole-genome sequencing data. J Clin Microbiol. 2020;58:10.
Feng Y, Zou S, Chen H, Yu Y, Ruan Z. BacWGSTdb 2.0: a one-stop repository for bacterial whole-genome sequence typing and source tracking. Nucleic Acids Res. 2021;49(D1):D644–D650. https://doi.org/10.1093/nar/gkaa821.
Alikhan N-F, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–21.
Acknowledgements
The authors are grateful to Maureen McGerty, M.A., for the critical readings of the manuscript and editorial support.
Funding
This research paper is supported by Specific League Funds from Mahidol University, Ramathibodi Funding Research RF-65048 and RF-65100 and Faculty of Medicine Ramathibodi Hospital Funding ID 3158, Mahidol University (Decentralized funding for CNMI, RF_65090).
This research was also supported, in part, by the Perinatology Research Branch, Division of Obstetrics and Maternal–Fetal Medicine, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, US Department of Health and Human Services (NICHD/NIH/DHHS); and, in part, by federal funds from NICHD/NIH/DHHS (Contract No. HHSN275201300006C). Dr. Roberto Romero has contributed to this work as part of his official duties as an employee of the United States Federal Government.
Author information
Authors and Affiliations
Contributions
P.P., R.R. and P.C. initiated the project, conceptualized, designed and conceived the study; P.M., P.V., T.K., P.N. and P.S. collected samples and conducted Microbiological work; T.W. and P.J. conducted sequencing and bioinformatic analysis; AS performed histopathological interpretation; P.P., R.R. and P.C. drafted the manuscript; P.P., R.R., T.W., P.J., I.T. and P.C. revised the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocols of this study were approved by the Institutional Research Board of the Faculty of Medicine, Ramathibodi Hospital, Mahidol University (COA.MURA2021/254 and COA.MURA 2022/675). The protocol was in accordance with the Declaration of Helsinki. Informed consent was obtained from all subjects.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
The list of point mutation of pbp5 gene, gyrA gene and parC gene identified in the genome of Entercoccus faecoum strain RAOG174. Supplementary Figure 1. A maximum likelihood phylogenetic tree with 1,000 bootstrap repeats constructed by using a whole genome sequence of the Enterococcus faecium strain RAOG174 (as highlighted in red). Only bootstrap support >75% was shown on a tree. Supplementary Figure 2. Comparative genomic visualization of the Enterococcus faecium (E. faecium) strain RAOG174 constructed with the Blast Ring Image Generator. Pink ring represents the E. faecium strain SRR24. Green ring indicates the E. faecium strain NBRC100486. Dark blue ring shows the E. faecium strain RAOG174. Black boxes represent the Insert Sequence (IS). Red boxes represent the intact phage regions.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pongchaikul, P., Romero, R., Mongkolsuk, P. et al. Genomic analysis of Enterococcus faecium strain RAOG174 associated with acute chorioamnionitis carried antibiotic resistance gene: is it time for precise microbiological identification for appropriate antibiotic use?. BMC Genomics 24, 405 (2023). https://doi.org/10.1186/s12864-023-09511-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09511-1