
Abstract
Background
Microbes infecting cystic fibrosis patients’ respiratory tract are important in determining patients’ functional status. Representatives of Burkholderiales order are the most dangerous. The goal of our investigation was to reveal the diversity of Burkholderiales, define of their proportion in the microbiome of various parts of respiratory tract and determine the pathogenicity of the main representatives.
Results
In more than 500 cystic fibrosis patients, representing all Federal Regions of Russia, 34.0% were infected by Burkholderia cepacia complex (Bcc), 21.0% by Achromobacter spp. and 12.0% by Lautropia mirabilis. B. cenocepacia was the most numerous species among the Bcc (93.0%), and A. ruhlandii was the most numerous among Achromobacter spp. (58.0%). The most abundant genotype in Bcc was sequence type (ST) 709, and in Achromobacter spp. it was ST36. These STs constitute Russian epidemic strains. Whole genome sequencing of strains A. ruhlandii SCCH3:Ach33–1365 ST36 and B. cenocepacia GIMC4560:Bcn122 ST709 revealed huge resistomes and many virulence factors, which may explain the difficulties in eradicating these strains. An experience of less dangerous B. cenocepcia ST710 elimination was described. Massively parallel sequencing of 16S rDNA amplicons, including V1-V4 hypervariable regions, was used to definite “healthy” microbiome characteristics. Analysis of maxillary sinus lavage of 7 patients revealed infection with Proteobacteria of the same ST as pathogens from sputum, suggesting that the maxillary sinus is a source of infection in cystic fibrosis patients.
Conclusions
Characterization of the Russian epidemic bacterial strains in the sputum and sinuses of cystic fibrosis patients have better defined the importance of Burkholderiales bacteria. This information may aid in the development of effective approaches for treatment of this disease.
Similar content being viewed by others


Background
Microbes infecting the cystic fibrosis (CF) patients’ respiratory tracts adversely affect the patients’ functional status. Among more than 200 bacterial species that may be involved in this disease [1], Burkholderiales order representatives are the most dangerous: well-known representatives are Burkholderia cepacia complex (Bcc) or emerging Achromobacter spp., Pandoraea spp., Lautropia mirabilis, Variovorax spp. All of them are multidrug resistant, transmissible and biofilm forming, which makes them difficult to eradicate [2]. Cepacia syndrome was described by Isles [3], who have noted that patients infected with Pseudomonas cepacia (the name of Bcc in 1970–1980 years) have greater impairment of pulmonary function than those with P. aeruginosa [3]. Timely and accurate bacteria identification is key to infection containment in CF. Molecular-genetic methods are more effective for Burkholderiales, which are closely related, difficult to cultivate and complicated in taxonomic affiliation. Thus Lipuma [1] named Burkholderiales “a challenge for conventional microbiology”. Since MultiLocus Sequence Typing (MLST) became the routine in Russian, in 2012, examination of CF patients’ samples by this method as in classical, so in express version is underway [4, 5], first for Bcc, then for Achromobacter spp. [6]. Then revealing new Burkholderiales genera has led to the necessity of developing new in-home schemes [7]. Diversity of species and genotypes was estimated for Bcc and Achromobacter spp., but these characteristics must be under permanent surveillance because of constantly increasing numbers of CF patients in the monitorable group. Russian epidemic strains B. cenocepacia ST709 and A. ruhlandii ST36 required the closer attention, so we began investigating strains by whole genome sequencing (WGS). Identification of individual pathogens may still be important for choosing treatment strategies in CF, but the entire microbial community of the respiratory tract can act as pathogens [8], so there is need to examine for diversity. The pioneers in culture-independent techniques for respiratory tract samples, Rogers et al. [9], demonstrated the power of bacterial diversity analysis through use of 16S rDNA sequencing. Next-generation sequencing of 16S rDNA then became the gold standard in bacterial diversity analysis [10], and changes in microbial community diversity in various stages of disease were identified [11]. Thus, we have decided to apply this technique in chronically Bcc-infected patients to help appreciate the process of microbial community recovery and to search for additional sources of bacterial infection in CF patients.
Methods
Biosamples and bacterial strains
Materials for investigation included sputum and tracheal aspirate from more than 500 patients’ cohorts, representing all Federal Regions of Russia, and from maxillary sinus lavage of 7 adult patients. The patients’ ages ranged from 4 months to 67 years (mean, 18.8 years; median, 19.0 years). The samples were collected from patients attending these CF Centers: Research Institute of Pulmonology; National Scientific and Practical Center of Children’s Health; Russian Children’s Clinical Hospital; and Regional CF Centers.
Approval of the Biomedical Ethics Committee of Research Center was obtained in May 2012 (Proceedings Record N1 d.d. 05.17.2012). Adult subjects or parents of minor subjects provided written informed consent.
Ten patients (P1-P10) were included in the microbiome analysis (Additional file 1: Table S4). The patients’ ages ranged from 23 to 36 years. Patients were subdivided into 3 groups according to the lung disease status, which was controlled by the percentage of predicted forced expiratory volume in 1 s (FEV1,%) [12, 13]: a) CF patients mild lung disease (FEV1% > 70%); b) CF patients with a moderate lung disease (70 < FEV1% > 40); c) CF patients with a severe lung disease (FEV1% < 40).
B. cepacia and B. gladioli strains of new STs were isolated from the sputum samples at the laboratory of biologically active nanostructure by Dr. R.S. Ovchinnikov.
Strain SCCH3:Ach33–1365, kindly provided by Dr. A.V. Lasareva of the National Scientific and Practical Center of Children’s Health, and strain GIMC4560:Bcn122, kindly provided by the laboratory of molecular epidemiology of our Research Center, were used for the whole genome sequencing (WGS).
DNA isolation
DNA was isolated according to the protocol of the Maxwell 16 Tissue DNA Purification Kit for Maxwell MDX Instrument (Promega). Preparation of genomic DNA for WGS was performed according to the protocols [14].
Bacteria genotyping
Bacterial identification and genotyping technology were conducted according to two approaches: 1) detailed classical MLST protocols for Bcc and Achromobacter spp. completed by 16S rDNA target and in-home elaborated protocols for other emerging Burkholderiales; and 2) two/three-locus express protocols for prompt screening.
The MLST protocol for Bcc allows the differentiation of all Bcc species and their genotypes or identification of STs [15]. The MLST protocol for Achromobacter spp. is suitable for all Achromobacter species representatives [16]. The additive target nrdA765 allows the immediate determination of strain species. The protocols, alleles’ sequences and software for data analysis are available on the PubMLST site [17]. For detection of Pandoraea spp. we used gltB and recA targets from the Bcc MLST scheme. Gene gyrB was suitable for revealing Variovorax paradoxus. For Lautropia mirabilis we used gltB target from Bcc MLST scheme, and home-elaborated selective target - phbB gene, coding 3-ketoacyl-ACP reductase. The amplification was performed with primers phbB_LauF 5’-GCGCATCGCTTACGTGACTG-3′, phbB_LauR 5’-GGATGGTGGACACGATGCGA-3′ and program: 95 °C—3 min, (95 °C—30 s, 60 °C—40 s, 72 °C—1 min) × 35, 72 °C—5 min. The product size was 608 bp. 16S rDNA gene fragments were amplified according to Voronina et al. [7].
Express protocol for the samples analysis included three targets: gltB, gyrB and 16S rDNA and was performed according to Voronina et al. [5].
Polymerase chain reaction (PCR) products were sequenced according to the protocol of BigDye Terminator 3.1 Cycle Sequencing kit for the Genetic Analyzer 3500 Applied Biosystems. The electrophoretic DNA separation was performed in 50-cm capillaries with POP7 polymer.
New MLST alleles and STs were managed by curators of Bcc and Achromobacter spp. MLST Databases and submitted with the following ids: 1149–1155, 1189–1267, 1501, 1443, 2029–2030 for Bcc and 605–629, 773–774 for Achromobacter spp. Sequences of Pandoraea, Variovorax and Lautropia genes were deposited in GenBank under the accession numbers: KM410934, KM410935; KJ848780; KT345191 - KT345194. Achromobacter spp. gltB gene sequences from express protocols had accession numbers KC817498-KC817502, KF290958, KF290959, KF297891, KF963246- KF963250, KJ364657, KJ439616, KJ941209, KM262752, KM262753, KP027420, KP765437- KP765444, KP940471.
Lung microbiome analysis
Lung microbiome was analyzed by massively parallel sequencing of 16S rDNA amplicons based on the MiSeq Illumina platform. 16S rDNA amplicons, which included V1-V4 hypervariable regions (753 bp), were used for the preparation of paired-end libraries according to the Nextera XT DNA Library Prep Kit protocol, then sequenced by the MiSeq Reagent Kit v3 (600 cycles) according to the supplied protocol. The Microbial Genomics Module of CLC Genomic Workbench v.8.5.1 was used with default settings to perform Operational Taxonomic Unit clustering. Greengenes database v13_5 [18] was used as reference with 97% threshold.
Whole genome sequencing
WGS with 454 Roche technology was performed for Russian epidemic strains A. ruhlandii SCCH3:Ach33–1365 ST36 (AruhST36) and B. cenocepacia GIMC4560:Bcn122 ST709 (BcenST709). Shotgun and paired-end libraries were prepared. Paired-end libraries were built according to the 3 kb protocol. The sequencing procedure was performed with a GS Junior Titanium Sequencing Kit, and a GS Junior+ Series XL+ Kit, according to the manufacturer’s guidelines. Genome assembly was performed with 454 Sequencing System Software v.2.7 and v.3.0 (Roche).
Reference genomes
For assembling genomes, the following references presented in the NCBI Genome database were used: A. xylosoxidans strain MN001 (CP012046.1) and A. denitrificans strain USDA-ARS-USMARC-56712 (CP013923.1) as the main references; A. xylosoxidans C54 (GL636060.1); A. xylosoxidans A8 (HE798385.1) and A. xylosoxidans strain FDAARGOS_150 (CP014028) as the additional references. The main reference for three chromosomes of BcenST709 was B. cenocepacia J2315 (NC_011000.1- NC_011002.1). The additional references for genome assembling were B. cenocepacia strain 895 (CP015036.1 - NZ_CP015038.1); B. cenocepacia strain MSMB384WGS (NZ_CP013450.1 - CP013452.1); B. cenocepacia strain ST32 (NZ_CP011917.1 - CP011919.1); B. cenocepacia PC184 cont1.51 (AAKX01000051.1); B. ubonensis strain MSMB2109WGS (NZ_LPHO01000048.1); B. cepacia strain DDS 7H-2 chromosome 3 (CP007785.1).
Bioinformatic analyses
The software Rapid Annotations Subsystems Technology and SEED [19, 20] were used for genome annotation. Complementary analysis was made with help of conserved domains search services: KEGG [21], KEGG OC [22], COGs [23]; protein subcellular localization prediction software: TMHMM Server v.2.0 [24], SignalP 4.1 Server [25], PSORTb version 3.0.2 [26], InterPro server [27, 28]. Prophage sequences were revealed with help of PHAST (PHAge Search Tool) [29, 30]. WGS data are available in GenBank: Accession Numbers are CP017433 (A. ruhlandii SCCH3:Ach33–1365) and CP020599 - CP020601 (B. cenocepacia GIMC4560:Bcn122 chr1–3).
Type III secretion system and effectors prediction
Sequence of type III secretion system (T3SS) gene cluster (Accession Number AY028431) was used for searching the analogs in the BcenST709 genome. Sequences of the proteins annotated in T3SS were the references for identifying T3SS components in the AruhST36 proteome. EffectiveDB server was used for prediction of proteins secreting by T3SS (effectors) [31, 32]. Coiled-coil domains of effectors were identified with COILS server [33, 34]. The potential phosphorylation sites of effectors were predicted by NetPhos 3.1 Server [35, 36].
Results
Diversity of the Burkholderiales representatives in Russian CF patients’ respiratory tracts
Burkholderiales species are relevant for Russian CF patients. In an analyzed cohort 34.0% of patients were infected by Bcc, 21.0% by Achromobacter spp., and 12.0% by Lautropia mirabilis. Pandoraea pnomenusa was seldom found [37]. Two patients had this bacterium in the respiratory tract, one together with B. mulivorans and Lautropia mirabilis. Variovorax paradoxus was detected in the samples of three patients who have congenital lung malformation and in one nurse from the pulmonary department.
Bcc was represented by 6 species (Fig. 1), the most abundant being B. cenocepacia which constituted 93.0% of all Bcc cases. Among 19 identified Bcc genotypes (Table 1) ST709 was constituted 77.0%. Despite the high B. cenocepacia prevalence had not changed since the beginning of the control period, in 2012 [4], the prevalence of ST709 decreased somewhat [7], because the proportion of ST208 increased to 9.0%. The diversity of Bcc species and ST increased in 2016–2017 years, when B. gladioli, B. cepacia and B. stabilis strains were isolated. Interestingly, two B. cepacia strains (ST438 and ST1083) were found in the sputum of one patient: the newly registered ST1083 was a double-locus variant of ST438 (gyrB and lepA genes).

Diversity of Burkholderia cepacia complex bacteria among the study CF patients
Among Achromobacter species A. ruhlandii was the most abundant (58.0%) (Fig. 2). A. ruhlandii ST36 was the most abundant Achromobacter (35.7%), which was more abundant than all A. xylosoxidans genotypes (34.7%) (Table 1). The next most abundant A. ruhlandii genotype was ST261 (14.0%).

Diversity of Achromobacter spp. bacteria among the study CF patients
Retrospective analysis of CF patients revealed the replacement of ST261 by ST36 and nosocomial outbreak of A. ruhlandii ST36 infection in the late 1990s in Russian Children’s Clinical Hospital [6]. The bacterium continues circulating among CF patients, among which the youngest infected patient is 3 years old.
B. cepacia ST709 and A. ruhlandii ST36 are transmissible, are most abundant among Burkholderiales strains, decrease the respiratory function of patients, and have spread into all Russian Federal Regions, except the Far Eastern Region. Thus, these bacteria have been designated Russian epidemic strains.
B. cepacia ST709 is the most dangerous: 75% of patients, who have died since beginning of the control, in 2012, were infected by ST709.
We have asked, how often CF patients are co-infected with multiple Burkholderiales representatives. The answer seems to be, seldom: only 1.7% of patients from our cohort were co-infected. The following combinations were identified in patients of various ages: B. gladioli ST903 and A. ruhlandii ST36 (patient 32 years old); B. cenocepacia ST709 and A. ruhlandii ST36 (29 years); A. xylosoxidans and L. mirabilis (19 years); B. cenocepacia ST709 and L. mirabilis (19 years); B. multivorans ST783, P. pnomenusa and L. mirabilis (19 years); B. cenocepacia ST709 and A. ruhlandii ST261 (17 years); B. cenocepacia ST710 and A. xylosoxidans ST182 (4 years). These co-infections have not correlated with respiratory tract function or nutritional status of the patients. It should be noted that in the first example, B. gladioli ST903 and A. ruhlandii ST36 changed B. multivorans ST711 after a long infection period.
Comparison of two epidemic strains genomes.
To try to understand the reason for the pathogenicity of the Russian epidemic strain B. cenocepacia ST709 and A. ruhlandii ST36 we performed WGS on them. The features of the epidemic strains’ genomes are presented in Table 2. The BcenST709 genome is bigger and more complicated; it consists of three chromosomes and contains nearly twice the amount of insertion sequence (IS) elements and prophages, two of which are intact (Table 2, Additional file 2: Table S1, Additional file 3: Table S2). In contrast, the clustered regularly interspaced short palindromic repeats (CRISPR), which are elements of bacterial immune systems, were predicted only in the AruhST36 genome. As a result, AruhST36 may be better protected from foreign genetic elements.
Multiple natural antibiotic resistance is a characteristic of Burkholderiales bacteria, which originate from the soil [1]. Huge resistomes of BcenST709 and AruhST36 include beta-lactamases in numbers approximately proportional to the genome size. As illustrated in Table 3, most BcenST709 beta-lactamases are on chromosome 2. Four classes of beta-lactamases were present in the proteomes of epidemic strains, and most of the proteins were from class C, followed by B, A and D, with one protein. An important characteristic of these proteins is resistance to beta-lactam inhibitors.
Beta-lactamases of class C are more active on cephalosporins than benzylpenicillin, are usually resistant to inhibition by clavulanic acid, and are active on cephamycins, such as cefoxitin [38]. Class B beta-lactamases (metallo-beta-lactamases) determine resistance to almost all beta-lactam antibiotics, including carbapenems [39]. Class A beta-lactamases are represented by two types: PER (Pseudomonas extended resistance) and PenA (penicillin-binding protein 2). The PER type encodes high resistance to cephalosporins (ceftazidime and gentamicin) and intermediate resistance to aminoglycosides (amikacin) [40]. The PenA type, first revealed in Burkholderia mallei, determines resistance to penicillins and ceftazidime [41].
Class D beta-lactamases are oxacillinases, but they can hydrolyze both benzyl penicillin and its derivatives (oxacillin, cloxacillin, amino- and carboxypenicillin) [42]. Oxacillinases are genera taxonomic markers in class Proteobacteria [43] and species-specific marker for Achromobacter spp. [44]. Allele OXA-258 is typical for A. ruhlandii, OXA-114 for A. xylosoxidans and A. marplatensis, and OXA-364 for A. dolens [6].
Aminoglycoside phosphotransferase and macrolide glycosyltransferase amplify the resistome of BcenST709, but only aminoglycoside acetyltransferases complete the resistome of AruhST36.
Active transport is one of the most important mechanisms that determine resistance of microbial cell to antimicrobial agents. We have found that almost 50% of AruhST36 proteins participate in transport of various substances (29.3% of proteome are inner membrane proteins, 21.3% have transmembrane domains and 2.4% are outer membrane proteins). Resistance-Nodulation-Division (RND) family transporters are widespread among gram-negative bacteria. RND are tripartite, which allows them to export drugs directly into the external medium, rather than into the periplasm [45]. The number of RND transporters in AruhST36 and BcenST709 are also proportionate to the genome size. The genes of RND transporters are organized in the operons in both genomes. For all operons, promoter regions have been characterized. But as demonstrated by analysis of the operons, not all RND pumps have complete number of components, so their effective function is questionable. For example, two AruhST36 RND operons (AruCF_0558−AruCF_0559; AruCF_1425−AruCF_1427) encode two components, whereas AruCF_5254−AruCF_5259 operon encodes only one component of the RND pumps. As illustrated in Table 3, nearly half of BcenST709 RND operons are incomplete, so two epidemic strains have equal number of RND pumps with complete function.
A second problem in defining RND transporter characteristics is estimating the numbers of transmembrane domains in the internal membrane components. According to Putman et al. [46], internal membrane components of RND pumps have 12 transmembrane domains, but only 7 RND pumps of AruhST36 meet this requirement: three internal membrane proteins had 11 and 10 transmembrane domains. However, it should be noted that fewer transmembrane domains were present in RND pumps components of Burkholderia cenocepacia J2315 also [47]. Thus, proteins with fewer transmembrane domains can be functional.
RND transporters provide resistance to a broad spectrum of substances. For example, three RND transporter operons of AruhST36 encode resistance to cobalt, zinc and cadmium, and two operons encode resistance to the antiseptic acriflavin. RND operon AxyABM provides resistance to cephalosporins (except cefepime), fluoroquinolones and chloramphenicol [48]. This type of RND pump was first described in A. xylosoxidans. RND efflux pump CmeABC is common among gram-negative bacteria, determines ciprofloxacine resistance, and acts synergistically with tet(O) gene to promote resistance to tetracycline [49]. The other RND-type operon, AxyXY-OprZ, can extrude cefepime, carbapenems, some fluoroquinolones, tetracyclines, and erythromycin from the epidemic strains’ cells. Also AxyXY-OprZ plays a role in the innate resistance to aztreonam [50].
The other way that microbial cells can attain antimicrobial resistance is through modification of lipopolysaccharides. In the case of polymyxin resistance formation, this modification occurs by the addition of 4-amino-4-deoxy-L-arabinose to lipid A, a process that is determined by the arnBCADTEF operon, as described in P. aeruginosa [51]. Search in AruhST36 genome for genes with the same function as in arnBCADTEF operon revealed 7 open reading frames (ORF) putatively participating in polymyxin resistance.
In description of bacterial resistance, genes encoding resistance to abiotic substances also must be considered. In the AruhST36 genome, three relevant genes are annotated: for albicidin resistance protein, pyrabactin resistance family protein and for ethidium bromide-methyl viologen resistance protein EmrE. These genes are evidence for soil and plant root systems as an early ecological niche of epidemic strains: albicidin is synthesized by the plant pathogenic bacterium Xanthomonas albilineans [52]; and the synthetic sulfonamide pyrabactin mimics abscisic acid, a naturally produced stress hormone in plants that inhibits cell growth.
Numerous capsular proteins, the part of virulence potential responsible for adhesion, are present in both genomes (Table 3).
The next participants in the adaptation and pathogenic function of epidemic strains are pili and flagella. However, flagella cannot be considered without type III secretion system (T3SS) injectisome because of their common origin [53]. The AruhST36 and BcenST709 genomes encode various pili: BcenST709 contains IVа and VIb type pili, and AruhST36 contains only VIb type pili.
The number of flagellar proteins is similar in the proteomes of epidemic strains (Table 3), but the number of chemotaxis proteins is different; only one methyl-accepting chemotaxis protein was revealed in BcenST709, but six chemotaxis proteins were detected in AruhST36.
The T3SSs of these strains are more different. First, the size of gene cluster is bigger in BcenST709 and has a unique sequence, but one gene (virB1) of AruhST36 T3SS is located separately. The number of ORF, coding by T3SS gene cluster, is bigger in the AruhST36 genome. The similarity of T3SS injectisome proteins of two epidemic strains is only 28–57%. The analysis based on the traditional ATPase of flagellum sequences suggested the membership of different T3SS families for AruhST36 and BcenST709 T3SSs: the first one belongs to the EscN/YscN/HrcN family, and the second one to the Hrc-Hrp2 family of T3SS. Troisfontaines et al. [53] suggested that the presence of the T3SS Ysc family is an indicator of an extracellular pathogen.
Our searching for effectors of T3SS included four steps (Table 3): predicting secreted proteins with the help of EffectiveDB server; correction of data based PSORTb information, choosing only coiled-coil domain containing proteins, and data-sorting according to MxxY motif in protein sequence (Additional file 3: Table S2). For example, two of AruhST36 proteins (AruCF_0917 and AruCF_5495), selected by this method, had 3 copies of MxxY motif and were involved in heme metabolism. All sorted proteins were candidate effectors that can disrupt key cellular processes of host innate immunity [54, 55].
Thus Russian epidemic strains have huge resistomes and adaptive potential to resist their eradication, and the question of whether resources are available to overcome other genotypes of Burkholderiales must be asked. However, the physicians of Russian Adult CF Center have experience in eradication of B. cenocepacia ST710. Lung microbiome analysis became the basis of evidence-based medicine since 2010s for confirmation of therapy efficacy in case of tuberculosis, asthma and other bronchopulmonary diseases. We used microbiome analysis for examination of Burkholderia eradication and for elaborating criteria of the health microbiome recovery.
Microbiome analysis in lung and maxillary sinus samples
Patient 10 (P10) was under complex control since 2012. The main infecting bacterium was Mycobacterium abscessus, and aggressive therapy was directed at it (Fig. 3). In the beginning of 2012, B. cenocepacia ST710 was revealed in the patient’s sputum. After one year of treatment, Bcc was not detected by MLST, but a small concentration of it was present one month later. In May 2014, we detected only a trace of Bcc with the help of recA gene amplification, which is the most sensitive among Bcc MLST targets. Since July 2014, growth of M. abscessus on special medium has not been detected. Therapy became less aggressive and shorter duration. The last six sputum samples (Fig. 3) have been checked by MLST, and by massively parallel sequencing of 16S rDNA V1-V4 hypervariable regions to analyze the steps of microbiome recovery.

Time frames of therapy and Bcc control of patient 10. First row – Bcc control time (blue arrow): ST710 – some MLST targets detection, no – absence of PCR products in MLST analysis, trace – trace Bcc quality, detected only by recA gene primers. Second row – years of control. Third row – therapy. Black arrow points the month of the therapy
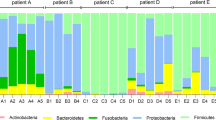
As depicted on Fig. 4, the proportion of Burkholderiacea in the bacterial community of sputum samples has changed from 0.0 to 18.0%. 16S rDNA is a six-copies gene in the Burkholderia genome and is a more sensitive target than are one-copy genes of the MLST scheme. However, the results of MLST correlate with those of massively parallel sequencing.

Betaproteobacteria families in patients’ microbiome based on 16S rDNA sequencing data. S – sputum, N – maxillary sinus lavage. The brackets combined the samples from each patient. The stars indicate the samples from the sinuses. The arrow indicates the sample obtained during the patient 10 treatment with Ivacaftor
Only trace amounts of Mycobacterium were present in samples P10S1 and P10S3 (0.149 and 0.257%), a finding which suggests that M. abscessus had been eradicated. According to Fig. 5, a “healthier” microbiome, corresponding to growth of Bacteroidetes, Firmicutes and Actinobacteria (but not Mycobacteria), has been established. Analysis of Proteobacteria phylum is illustrated in Fig. 6: the proportion of Epsilonproteobacteria in P10S3, P10S4, and P10S6 samples increased, and Neisseriales became the remarkable part of Betaproteobacteria class in the same samples (Fig. 4).

Microbial phyla in patients’ microbiome based on 16S rDNA sequencing data. S – sputum, N – maxillary sinus lavage. The brackets combined the samples from each patient. The stars indicate the samples from the sinuses. The arrow indicates the sample obtained during the patient 10 treatment with Ivacaftor

Proteobacteria classes in patients’ microbiome based on 16S rDNA sequencing data. S – sputum, N – maxillary sinus lavage. The brackets combined the samples from each patient. The stars indicate the samples from the sinuses. The arrow indicates the sample obtained during the patient 10 treatment with Ivacaftor
Thus, a central feature of a “healthier” microbiome in CF patients is that of increasing bacterial diversity, which is evident first at the phyla level.
Two last samples of P10 were associated with a new therapeutic approach in CF: treatment with Ivacaftor. Ivacaftor is a potentiator of the cystic fibrosis transmembrane conductance regulator (CFTR) and thus improves the transport of chloride through the ion channel. It was possible to use this treatment because P10 patient had N1303 K/G461E mutations in the cftr gene. The P10S7 sample was obtained days before start of therapy, and P10S8 was obtained after 4.5 months of therapy. As illustrated in Figs. 4, 5 and 6, the P10S7 sample had the “healthiest” microbiome. The P10S8 microbiome had representatives of various Proteobacteria classes and some families of Betaproteobacteria; in addition, trace amounts of Mycobacteria (0.158%) were present and B. cenocepacia ST710 was absent. These data are consistent with the view that Ivacaftor had improved the patient’s lung drainage. It must be noted the quick positive influence of Ivacoftor on the lung function of P10 patient (FEV1 increased from 50 to 67% during first 3 days of treatment, and slight increasing to 68% was observed during next 4.5 month, Additional file 1: Table S4). We consider this result as effect of the previous recovery of the microbiome diversity.
We compared P10 data with the results of chronically infected patients’ analyses. P1 was infected by P. aeruginosa; P2 by P. aeruginosa and Achromobacter spp.; P3 by B. multivorans ST835; P4 by B. multivorans ST712 and P. aeruginosa; P5 by P. aeruginosa and A. ruhlandii ST261; P6 by B. cenocepacia ST709 and P. aeruginosa; P7 by A. ruhlandii ST261; P8 by B. cenocepacia ST709 and P. aeruginosa. P9 was not chronically infected but had A. ruhlandii ST261 detected once.
In the group of 9 patients P9 had the “healthiest” microbiome and no bacteria in maxillary sinus lavage. The next stable patient was P3. Massively parallel sequencing (Fig. 5) suggested the prevalence of Firmicutes in P3S1 and P3S2, but demonstrated a small “healthy part” of microbiome, id est., Bacteroidetes and Actinobacteria. However, maxillary sinus lavage analysis of this patient (P3N1) demonstrated abundant Burkholderia, and MLST suggested B. cenocepacia ST835. The last 5 patients (P4N1-P8N1) had more than 50% of Proteobacteria in maxillary sinus lavage, and the main pathogen had the same ST as the pathogen from sputum; these data suggested the maxillary sinus can be an additive source of infection in CF patients. Comparing representatives of Firmicutes in sputum and sinus lavage samples we noted the prevalence of Streptococcaceae (up to 82%) in sputum microbiome, and presence less than 7% of Staphylococcaceae. However the proportion of Staphylococcaceae and in particular Staphylococcus aureus was increased only in maxillary sinus samples (up to 29%) of two patients.
Among all the samples we registered, Proteobacteria and Firmicutes were the most abundant phyla, reaching 100% and 93% of the microbiome, respectively. As a rule lower amounts of Proteobacteria were associated with higher proportions of Firmicutes (correlation coefficient was − 0.95), which probably reflects the competitive relationship between these phyla. In general, the presence of proteobacteria was associated with the suppression of other taxonomic groups of microorganisms and a decrease in microbial diversity. This, a moderate negative correlation was observed between the phyla Proteobacteria and Actinobacteria (− 0.65) and the phyla Proteobacteria and Bacteroidetes (− 0.67). This observation suggests that Proteobacteria is the most dangerous taxon in CF patients’ respiratory tracts.
Discussion
Burkholderia cepacia syndrome is famous, and B. cenocepacia clone of ST234 has intercontinental distribution; the Russian epidemic strain BcenST709 is a member of this clone. Another famous representative of this clone is B. cenocepacia J2315 (ST28). This strain was isolated from CF patient, infected by the most notorious epidemic B. cenocepacia lineage ET-12. It has been revealed that ET-12 comprised a group of strains that have been causing devastating infections in Canadian, UK and European CF populations [56]. There is less information about the distribution of A. ruhlandii. Of strains submitted to the Achromobacter MLST database, 17.6% belong to this species [17]. The most famous is Danish epidemic strain, DES, A. ruhlandii A83/DSM25711 [57]. We found only 4 incomplete genomes of A. ruhlandii in GenBank (Assembly GCA_001637085.1, GCA_001637095.1, GCA_001637105.1, GCA_001637115.1); all of them were submitted by scientists from Brazil (Universidade do Estado do Rio de Janeiro), the country with the highest Achromobacter infection rate among CF patients: 21.0%, with 23.9% among children [58].
However, some complete Achromobacter genomes submitted as A. xylosoxidans or A. denitrificans were determined to be A. ruhlandii on the base of Achromobacter PubMLST server analysis [17]. This result is evidence of the necessity of strain verification based on new species criteria. Four strains form the NCBI Genome database were the object of our analysis: Three strains were isolated from CF patients (A. xylosoxidans MN001, CP012046.1, USA; A. xylosoxidans FDAARGOS_162, CP014065.1, USA; and A. ruhlandii strain 7022, NZ_LVKN01000001:NZ_LVKN01000089, Brazil), and one strain (A. denitrificans USDA-ARS-USMARC-56712, CP013923.1, USA) was from a domestic cow with respiratory disease. All strains were identified as A. ruhlandii on the base of MLST analyses. Strains A. xylosoxidans MN001 and A. ruhlandii 7022 had the same ST36 as that of the Russian epidemic strain AruhST36. A. xylosoxidans FDAARGOS_162 differed in 4 positions of allelic profile, and A. denitrificans USDA-ARS-USMARC-56712 had a new ST with new alleles of two loci. We compared prophage profiles of ipso facto A. ruhlandii strains; one common prophage typical for all these strains was revealed (Fig. 7). The fewest prophages were in a strain isolated from domestic cow. All clinical strains had specific prophages as individual markers. Thus, we conclude there is intercontinental spread of A. ruhlandii also. It should be noted that strains from Russia, USA and Brazil, which had the same ST36, were isolated from CF patients. This fact suggested the danger of A. ruhlandii ST36.

Prophage diversity in A. ruchlandii genomes
Multidrug resistance of Russian epidemic strains is main problem of these stains eradication. Genomic data suggested the huge resistant potential of strains. We compared obtained data with resistance of well-known CF pathogens B. cenocepacia J2315 and A. xylosoxidans NH44784–1996. Holden et al. [59] revealed at least four β-lactamases encoded in the J2315 genome, including: two class A, one class C, and one class D. Genes coding these β-lactamases were located on the chromosomes 2 and 3. However in BcenST709 there was more number of β-lactamase genes, and these genes belonged to 4 different classes. Moreover 5 from 13 β-lactamase genes were located on the chromosome 1.
The number of β-lactamase genes in A. xylosoxidans NH44784–1996 genome was 12. It was 2 greater than in AruhST36 [60]. One gene - oxa-114, coding β-lactamase of class D, had species specific allele. In A. ruhlandii this gene had allele 258.
A characteristic of AruhST36 T3SS is additional evidence of its danger, id est., as mentioned above, the presence of T3SS family Ysc in AruhST36 is indicative of an extracellular pathogen [53]. NCBI draft Genome data indicate that the T3SS family Ysc is present in two more Achromobacter spp. strains: one strain (Achromobacter sp. 2789STDY5608607, NZ_CYSX01000006.1) was isolated from CF sputum, the other (A. xylosoxidans KCJK1737) from Bos taurus.
Moreover, this type of T3SS family is typical of another Betaproteobacteria - Bordetella species. In B. pertussis, B. parapertussis, and B. bronchiseptica a T3SS cluster was found and identified as locus bsc [61]. This cluster included 30 ORF encoding T3SS genes, BscN ATPase, type III secreted proteins, and putative chaperones. The T3SS allows B. bronchiseptica to avoid phagocytosis and reduce the proinflammatory response [62]. At least in B. bronchiseptica, the Bsc system favors long-term colonization of the respiratory tract epithelium and downregulation of the immune response [62, 63].
BcenST709 T3SS belongs to another family, id est., Hrc-Hrp2. NCBI BLAST analysis of the T3SS gene cluster revealed other B. cenocepacia genomes with similarity in this area (Additional file 4: Table S3). The largest coverage (97–99%) and similarity (99%) of the T3SS cluster were found in strains isolated mainly from CF patients (indicated at the top of the table). The B. cenocepacia strains isolated from the environment (except one, CP013452.1) have less coverage (76%) and similarity to the T3SS cluster (90%). This information suggests that spread of pathogenic B. cenocepacia strains occurs among CF patients.
However, Russian physicians have demonstrated the ability to eradicate less dangerous B. cenocepacia strain. Microbiome analysis suggested that massively parallel sequencing of 16S rDNA is a valid method for the appreciating therapeutic efficacy.
The case of patient 10 (P10) demonstrated that eradication of B. cenocepacia ST710 can result in recovery of microbiota diversity, approximating the microbiota of CF infants, as illustrated by Madan et al. [64]: genera of Firmicutes, Bacteriodetes and only Neisseria from Proteobacteria were present in the respiratory microbiota of patients from birth to 21 months [64]. Madan et al. analyzed oropharyngeal swabs, having believed that these traditional for infants samples [65] accurately reflected the microbial populations in the respiratory tract. In our investigation of the tracheal aspirate the most of the two phyla, Actinobacteria and Bacteroidetes, representatives are considered a “healthy part” of the respiratory tract, and can be as much as 20% the bacterial community of CF infants’ lungs [66].
Microbiome observations of patient P10 demonstrate positive dynamics, the first, due to complex antibiotic treatment: eradication of M. abscessus and B. cenocepacia ST710 and recovery of microbiota diversity. The second positive effect was that of treatment with Ivacaftor (P10S8), which likely improved lung drainage and additional emission of different Proteobacteria. Another healthier microbiome was observed in the respiratory tract of patient P9. It was marked by higher proportion of Firmicutes (69–79%), Actinobacteria (9%–14%), and Bacteroidetes (17%–3%), and a smaller proportion of Proteobacteria (4–1%). The microbiomes of P9 and P10 were characterized also by the presence of the poorly explored candidate phylum, TM7. Some data refer to a negative role of TM7, associated with human inflammatory mucosal diseases [67]. However, in this study we found only an association between TM7 and abundant Actinobacteria, which is an obligate epibiont of an Actinomyces odontolyticus [66].
An important finding of this study, which began as a search for a constant source of Proteobacteria in CF patients, was that the patients’ sinuses may harbor higher concentrations of dominant microorganism than do the lungs. Even the stable patient P3 had an abundance of Burkholderia in the maxillary sinus lavage. Thus, although adult CF patients with chronic infection may have periods of “healthier” microbiota, Burkholderia in the paranasal sinuses may be continual source of aggressive bacteria, entering the lungs.
The respiratory tract of CF patients is a dynamic ecosystem, and complete recovery of “healthy” microbiome is a long-term task. This task is complicated by various aspects of the CF patient’s physiology, as summarized in the review of Dickson et al. [68]. One component may be gastroesophageal dysfunction, which is common among CF patients and accelerates the migration of microbiota to the lungs. On the other hand, impaired mucociliary clearance inhibits the elimination of microbes. The lung destruction dramatically decreases the surface area of lungs and favors microbial growth; Burkholderiales are well adapted to the injured respiratory tract. Thus, effective treatment of these bacteria will require carefully matched therapy, perhaps with new drugs, taking into account WGS data and metabolic pathways of Burkholderiales.
Conclusions
Complicated and dynamic bacterial communities in the airway of CF patients have been revealed recently. The bacterial communities change over time, in response to disease progression and response to antibiotic therapies. Our data demonstrate the positive effects of Bcc eradication and the importance of controlling the microbiome of CF patients’ sinuses. Eradication of registered Russian epidemic strains is difficult because of the complicated huge genomes encoding numerous resistance and virulence factors. Reverse vaccinology, based on WGS data and bioinformatics analyses are leading to approaches for Burkholderiales treatment and the development of new vaccines. Moreover, a multi-omics approaches could reveal the potential of the microbiome and some features of bacteria-bacteria and bacteria-lung interactions. Host functional changes could be monitored as well as the metabolites originating from both the host and the microbiome. Thus, changes in the microbiome and the host may be simultaneously followed to provide a more comprehensive picture of the dynamic changes that occur during disease. Surveying respiratory tract samples of adult and, especially, infant CF patients must be longitudinal; such surveys permit temporal shifts in bacterial diversity during airway infection and reveal the predictors of lung disease states (inflammation, exacerbation, stable).
Abbreviations
- AruhST36:
-
Strain A. ruhlandii SCCH3:Ach33–1365 ST36
- Bcc:
-
Burkholderia cepacia complex
- BcenST709:
-
Strain B. cenocepacia GIMC4560:Bcn122 ST709
- CF:
-
Cystic fibrosis
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- Flp:
-
Fimbrial low-molecular weight protein
- IS:
-
Insertion sequence
- MLST:
-
MultiLocus Sequence Typing
- N1:
-
Maxillary sinus lavage number in microbiome analysis
- ORF:
-
Open reading frames
- P1:
-
Patient’s number in microbiome analysis
- PCR:
-
Polymerase chain reaction
- PenA:
-
Penicillin-binding protein 2
- PER:
-
Pseudomonas extended resistance
- RND:
-
Resistance-Nodulation-Division
- S1:
-
Sputum sample number in microbiome analysis
- ST:
-
Sequence types
- T3SS:
-
Type III secretion system
- WGS:
-
Whole genome sequencing
References
Lipuma JJ. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev. 2010;23(2):299–323. https://doi.org/10.1128/CMR.00068-09.
Mahenthiralingam E. Emerging cystic fibrosis pathogens and the microbiome. Paediatr Respir Rev. 2014;15(Suppl 1):13–5. https://doi.org/10.1016/j.prrv.2014.04.006.
Isles A, Maclusky I, Corey M, Gold R, Prober C, Fleming P, Levison H. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J Pediatr. 1984;104(2):206–10.
Voronina OL, Chernukha MY, Shaginyan IA, Kunda MS, Avetisyan LR, Orlova AA, Lunin VG, Avakyan LV, Kapranov NI, Amelina EL, Chuchalin AG, Gintsburg AL. Characterization Of genotypes for Burkholderia cepacia complex strains isolated from patients in hospitals of the Russian federation. Mol Gen Mikrobiol Virusol. 2013;28(2):22–9.
Voronina OL, Kunda MS, Aksenova EI, Orlova AA, Chernukha MY, Lunin VG, Amelina EL, Chuchalin AG, Gintsburg AL. The express diagnostic of microorganisms affecting respiratory tract of patients with mucoviscidosis. Klin Lab Diagn. 2013;11:53–8. Article in Russian
Voronina OL, Kunda MS, Ryzhova NN, Aksenova EI, Semenov AN, Lasareva AV, Semykin SY, Amelina EL, Simonova OI, Krasovsky SA, Lunin VG, Chuchalin AG, Baranov AA, Gintsburg AL. Diversity and hazard of respiratory infection of Achromobacter spp. in cystic fibrosis patients. Pulmonology. 2015;25(4):389–401. https://doi.org/10.18093/0869-0189-2015-25-4-389-401. Article in Russian
Voronina OL, Kunda MS, Ryzhova NN, Aksenova EI, Semenov AN, Lasareva AV, Amelina EL, Chuchalin AG, Lunin VG, Gintsburg AL. The variability of the order Burkholderiales representatives in the healthcare units. Biomed Res Int. 2015;2015:680210. https://doi.org/10.1155/2015/68021.
Klepac-Ceraj V, Lemon KP, Martin TR, Allgaier M, Kembel SW, Knapp AA, Lory S, Brodie EL, Lynch SV, Bohannan BJ, Green JL, Maurer BA, Kolter R. Relationship between cystic fibrosis respiratory tract bacterial communities and age, genotype, antibiotics and Pseudomonas Aeruginosa. Environ Microbiol. 2010;12(5):1293–303. https://doi.org/10.1111/j.1462-2920.2010.02173.x.
Rogers GB, Hart CA, Mason JR, Hughes M, Walshaw MJ, Bruce KD. Bacterial diversity in cases of lung infection in cystic fibrosis patients: 16S ribosomal DNA (rDNA) length heterogeneity PCR and 16S rDNA terminal restriction fragment length polymorphism profiling. J Clin Microbiol. 2003;41(8):3548–58.
Flight WG, Smith A, Paisey C, Marchesi JR, Bull MJ, Norville PJ, Mutton KJ, Webb AK, Bright-Thomas RJ, Jones AM, Mahenthiralingam E. Rapid detection of emerging pathogens and loss of microbial diversity associated with severe lung disease in cystic fibrosis. J Clin Microbiol. 2015;53(7):2022–9. https://doi.org/10.1128/JCM.00432-15.
Goddard AF, Staudinger BJ, Dowd SE, Joshi-Datar A, Wolcott RD, Aitken ML, Fligner CL, Singh PK. Direct sampling of cystic fibrosis lungs indicates that DNA-based analyses of upper-airway specimens can misrepresent lung microbiota. Proc Natl Acad Sci U S A. 2012;109(34):13769–74. https://doi.org/10.1073/pnas.1107435109.
Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, Enright PL, Hankinson JL, Ip MSM, Zheng J, Stocks J, The Ers Global Lung Function Initiative. ERS TASK FORCE REPORT. Multi-ethnic reference values for spirometry for the 3–95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40:1324–43.
Quanjer P. H., Stanojevic S., Cole T. J., Stocks J. Implementing GLI-2012 regression equations. Version 19 July 2015. http://www.ers-education.org/guidelines/global-lung-function-initiative/gli-2012-explained.aspx.
Current Protocols in Molecular Biology. Kate Wilson Unit 2.4 Preparation of genomic DNA from bacteria. Wiley Online Library, 2001. DOI: https://doi.org/10.1002/0471142727.mb0204s56.
Spilker T, Baldwin A, Bumford A, Dowson CG, Mahenthiralingam E, LiPuma JJ. Expanded multilocus sequence typing for Burkholderia species. J Clin Microbiol. 2009;47(8):2607–10. https://doi.org/10.1128/JCM.00770-09.
Spilker T, Vandamme P, Lipuma JJ. A multilocus sequence typing scheme implies population structure and reveals several putative novel achromobacter species. J Clin Microbiol. 2012;50:3010–5. https://doi.org/10.1128/JCM.00814-12.
The Main Site PubMLST. The Site hosted at The Department of Zoology, University of Oxford, UK. http://pubmlst.org. Accessed 06 Jan 2017.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–72.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. https://doi.org/10.1186/1471-2164-9-75.
Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–702. https://doi.org/10.1093/nar/gki866.
KEGG: Kyoto Encyclopedia of Genes and Genomes. The site of Kanehisa Laboratories. URL: http://www.genome.jp/kegg/, 06.2017.
KEGG OC (KEGG Ortholog Cluster). URL: http://www.genome.jp/tools/oc/, Accessed 06 Jan 2017.
COGs, Phylogenetic classification of proteins encoded in complete genomes. URL: http://www.ncbi.nlm.nih.gov/COG/, 06.2017.
Center for Biological Sequence Analysis Server. URL http://www.cbs.dtu.dk/services/TMHMM/, Accessed 06 Jan 2017. Hosted in Technical University of Denmark.
Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane region. Nat Methods. 2011;8:785–6. https://doi.org/10.1038/nmeth.1701.
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FSL. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26(13):1608–15.
Mitchell A, Chang HY, Daugherty L, Fraser M, Hunter S, Lopez R, et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015; https://doi.org/10.1093/nar/gku1243.
Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–40. https://doi.org/10.1093/bioinformatics/btu031.
Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39(Web Server issue):W347–52. https://doi.org/10.1093/nar/gkr485.
The PHAST website is maintained by Dept. of Biological Sciences, University of Alberta, Edmonton, AB, Canada. URL: http://phast.wishartlab.com/contact.html. Accessed 06 Jan 2017.
The EffectiveDB website is maintained by University of Vienna, Austria, URL: http://www.effectors.org/. Accessed 06 Jan 2017.
Eichinger V, Nussbaumer T, Platzer A, Jehl MA, Arnold R, Rattei T. EffectiveDB--updates and novel features for a better annotation of bacterial secreted proteins and type III, IV, VI secretion systems. Nucleic Acids Res. 2016;44(D1):D669–74. https://doi.org/10.1093/nar/gkv1269.
The COILS website is maintained by SIB Swiss Institute of Bioinformatics, URL: http://www.ch.embnet.org/software/COILS_form.html. Accessed 06 Jan 2017.
Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252(5009):1162–4.
The NetPhos 3.1 Server is maintained by Center for biological sequence analysis of Technical University of Denmark, URL: http://www.cbs.dtu.dk/services/NetPhos/. Accessed 06 Jan 2017.
Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4(6):1633–49.
Simonova OI, Voronina OL, Yu. V. G, Amelina EL, Burkina NI, Lasareva AV, Kunda MS, Ryzhova NN, Chernevich VP. Features of the treatment of the cystic fibrosis patient with mixt microbial respiratory infection, including Pandoraea pnomenusa. Rus Pediatr J. 2016;19(2):113–22. https://doi.org/10.18821/1560-9561-2016-19(2)-113-122. Article in Russian
Bush K, Jacoby GA. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother. 2010;54(3):969–76. https://doi.org/10.1128/AAC.01009-09.
Wommer S, Rival S, Heinz U, Galleni M, Frere J-M, Franceschini N, Amicosante G, Rasmussen B, Bauer R, Adolph H-W. Substrate-activated zinc binding of Metallo-beta-lactamases. J Biol Chem. 2002;277(27):24142–7. https://doi.org/10.1074/jbc.M202467200.
Vahaboglu H, Oztürk R, Aygün G, Coşkunkan F, Yaman A, Kaygusuz A, Leblebicioglu H, Balik I, Aydin K, Otkun M. Widespread detection of PER-1-type extended-spectrum beta-lactamases among nosocomial Acinetobacter and Pseudomonas Aeruginosa isolates in Turkey: a nationwide multicenter study. Antimicrob Agents Chemother. 1997;41(10):2265–9.
C T, Moore RA, Baker P, Woods DE. Burkholderia pseudomallei class a beta-lactamase mutations that confer selective resistance against ceftazidime or clavulanic acid inhibition. Antimicrob Agents Chemother. 2003;47(7):2082–7.
Poirel L, Naas T, Nordmann P. Diversity, epidemiology, and genetics of class D beta-lactamases. Antimicrob Agents Chemother. 2010;54(1):24–38. https://doi.org/10.1128/AAC.01512-08.
Doi Y, Poirel L, Paterson DL, Nordmann P. Characterization of a naturally occurring class D beta-lactamase from Achromobacter xylosoxidans. Antimicrob Agents Chemother. 2008;52(6):1952–6.
Papalia M, Almuzara M, Cejas D, Traglia G, Ramírez MS, Galanternik L, Vay C, Gutkind G, Radice M. OXA-258 from Achromobacter ruhlandii: a species-specific marker. J Clin Microbiol. 2013;51(5):1602–5. https://doi.org/10.1128/JCM.03043-12.
Nikaido H, Takatsuka Y. Mechanisms of RND multidrug efflux pumps. Biochim Biophys Acta. 2009;1794(5):769–81. https://doi.org/10.1016/j.bbapap.2008.10.004.
Putman M, van Veen HW, Konings WN. Molecular properties of bacterial multidrug transporters. Microbiol Mol Biol Rev. 2000;64(4):672–93.
Guglierame P, Pasca MR, de Rossi E, Buroni S, Arrigo P, Manina G, Riccardi G. Efflux pump genes of the resistance-nodulation-division family in Burkholderia cenocepacia genome. BMC Microbiol. 2006;6:66. https://doi.org/10.1186/1471-2180-6-66.
Bador J, Amoureux L, Duez J-M, Drabowicz A, Siebor E, Llanes C, Neuwirth C. First description of an RND-type multidrug efflux pump in Achromobacter xylosoxidans, AxyABM. Antimicrob Agents Chemother. 2011;55(10):4912–4. https://doi.org/10.1128/AAC.00341-11.
Lin J, Michel LO, Zhang Q. CmeABC functions as a multidrug efflux system in Campylobacter jejuni. Antimicrob Agents Chemother. 2002;46(7):2124–31.
Bador J, Amoureux L, Blanc E, Neuwirth C. Innate aminoglycoside resistance of Achromobacter xylosoxidans is due to AxyXY-OprZ, an RND-type multidrug efflux pump. Antimicrob Agents Chemother. 2013;57(1):603–5. https://doi.org/10.1128/AAC.01243-12.
Barrow K, Kwon DH. Alterations in two-component regulatory systems of phoPQ and pmrAB are associated with polymyxin B resistance in clinical isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2009;53(12):5150–4. https://doi.org/10.1128/AAC.00893-09.
Kretz J, Kerwat D, Schubert V, Gratz S, Pesic A, Semsary S, Cociancich S, Royer M, Sussmuth RD. Total synthesis of albicidin: a lead structure from Xanthomonas albilineans for potent antibacterial gyrase inhibitors. Angew Chem Int Ed Eng. 2015;54(6):1969–73. https://doi.org/10.1002/anie.201409584.
Troisfontaines P, Cornelis GR. Type III secretion: more systems than you think. Physiology (Bethesda). 2005;20:326–39.
Ruckdeschel K, Deuretzbacher A, Haase R. Crosstalk of signalling processes of innate immunity with Yersinia Yop effector functions. Immunobiology. 2008;213(3–4):261–9. https://doi.org/10.1016/j.imbio.2007.11.001.
Adiga R, Karunasagar I, Karunasagar I. Bioinformatics based analysis of type III secretion system effector protein of Vibrio Vulnificus. Adv Appl Sci Res. 2010;1(3):222–8.
Mahenthiralingam E, Urban TA, Goldberg JB. The multifarious, multireplicon Burkholderia cepacia complex. Nat Rev Microbiol. 2005;3(2):144–56.
Ridderberg W, Wang M, Nørskov-Lauritsen N. Multilocus sequence analysis of isolates of Achromobacter from patients with cystic fibrosis reveals infecting species other than Achromobacter xylosoxidans. J Clin Microbiol. 2012;50(8):2688–94. https://doi.org/10.1128/JCM.00728-12.
Pereira RH, Carvalho-Assef AP, Albano RM, Folescu TW, Jones MC, Leão RS, Marques EA. Achromobacter xylosoxidans: characterization of strains in Brazilian cystic fibrosis patients. J Clin Microbiol. 2011;49(10):3649–51. https://doi.org/10.1128/JCM.05283-11.
Holden MT, Seth-Smith HM, Crossman LC, Sebaihia M, Bentley SD, Cerdeño-Tárraga AM, Thomson NR, Bason N, Quail MA, Sharp S, Cherevach I, Churcher C, Goodhead I, Hauser H, Holroyd N, Mungall K, Scott P, Walker D, White B, Rose H, Iversen P, Mil-Homens D, Rocha EP, Fialho AM, Baldwin A, Dowson C, Barrell BG, Govan JR, Vandamme P, Hart CA, Mahenthiralingam E, Parkhill J. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J Bacteriol. 2009;191(1):261–77. https://doi.org/10.1128/JB.01230-08.
Jakobsen TH, Hansen MA, Jensen PØ, Hansen L, Riber L, Cockburn A, Kolpen M, Rønne Hansen C, Ridderberg W, Eickhardt S, Hansen M, Kerpedjiev P, Alhede M, Qvortrup K, Burmølle M, Moser C, Kühl M, Ciofu O, Givskov M, Sørensen SJ, Høiby N, Bjarnsholt T. Complete genome sequence of the cystic fibrosis pathogen Achromobacter xylosoxidans NH44784-1996 complies with important pathogenic phenotypes. PLoS One. 2013;8(7):e68484. https://doi.org/10.1371/journal.pone.0068484.
Fauconnier A, Veithen A, Gueirard P, Antoine R, Wacheul L, Locht C, Bollen A, Godfroid E. Characterization of the type III secretion locus of Bordetella pertussis. Int J Med Microbiol. 2001;290(8):693–705.
Yuk MH, Harvill ET, Cotter PA, Miller JF. Modulation of host immune responses, induction of apoptosis and inhibition of NF-kappa B activation by the Bordetella type III secretion system. Mol Microbiol. 2000;35:991–1004.
Nogawa H, Kuwae A, Matsuzawa T, Abe A. The type III secreted protein BopD in Bordetella bronchiseptica is complexed with BopB for pore formation on the host plasma membrane. J Bacteriol. 2004;186:3806–13.
Madan JC, Koestler DC, Stanton BA, Davidson L, Moulton LA, Housman ML, Moore JH, Guill MF, Morrison HG, Sogin ML, Hampton TH, Karagas MR, Palumbo PE, Foster JA, Hibberd PL, O'Toole GA. Serial analysis of the gut and respiratory microbiome in cystic fibrosis in infancy: interaction between intestinal and respiratory tracts and impact of nutritional exposures. MBio. 2012; 3(4). pii: e00251–12. doi: https://doi.org/10.1128/mBio.00251-12
Rosenfeld M, Emerson J, Accurso F, Armstrong D, Castile R, Grimwood K, Hiatt P, McCoy K, McNamara S, Ramsey B, Wagener J. Diagnostic accuracy of oropharyngeal cultures in infants and young children with cystic fibrosis. Pediatr Pulmonol. 1999;28(5):321–8.
Voronina O, Ryzhova N, Kunda M, Amelina E, Chuchalin A, Gintsburg A. Airways bacterial community composition in assessing the results of antibacterial therapy. 40th European Cystic Fibrosis Conference, Seville, Spain, 7–10 June 2017 – e-Poster. J Cyst Fibros. 2017;16S1:–S92.
He X, JS ML, Edlund A, Yooseph S, Hall AP, Liu SY, Dorrestein PC, Esquenazi E, Hunter RC, Cheng G, Nelson KE, Lux R, Shi W. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. PNAS. 2015;112(1):244–9. www.pnas.org/cgi/doi/10.1073/pnas.1419038112
Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The microbiome and the respiratory tract. Annu Rev Physiol. 2016;78:481–504. https://doi.org/10.1146/annurev-physiol-021115-105238.
Acknowledgements
We acknowledge otolaryngologist Shumkova Galina L. for sinus puncture preparation, microbiologist Ovchinnikov Roman S. for new Burkholderia species strain cultivation, senior researcher Botikov Andrey G. for technical support of MiSeq equipment.
Funding
This work was supported by the grant 16–04-00445 of the Russian foundation for basic research “The study of genomes fluidity and determination of possible adaptation mechanisms of the epidemiologically significant Bukholderia cenocepacia ST709 and Achromobacter ruhlandii ST36 isolates, aggressively depressing the cystic fibrosis patients’ respiratory function” and by the State Order to the N.F. Gamaleya National Research Center for Epidemiology and Microbiology. The publication costs for this article were funded also by the grant 16–04-00445.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
About this supplement
This article has been published as part of BMC Genomics Volume 19 Supplement 3, 2018: Selected articles from Belyaev Conference 2017: genomics. The full contents of the supplement are available online at https://bmcgenomics.biomedcentral.com/articles/supplements/volume-19-supplement-3.
Author information
Authors and Affiliations
Contributions
OV, MK, EA, NR, NS conceived and designed the experiments, sequenced, assembled and annotated the genomes of strains, and microbiome data; OV supervised the study and contributed to manuscript writing; MK performed resistome analysis and assisted in the preparation of figures and text; EA participated in data interpretation; AS contributed to bioinformatic analysis, NR was responsible for the microbiome analysis and data presentation, and contributed to manuscript writing; EA and AC were responsible for patient care, the selection of patients recruited in this study, the generation and interpretation of clinical data; AG performed general supervision of the research group. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval of the Biomedical Ethics Committee of Research Center was obtained in May 2012 (Proceedings Record N1 d.d. 05.17.2012). Adult subjects or parents of minor subjects provided written informed consent.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S4.
Characteristics of the patients, included in the microbiome analysis. FEV1,% - percentage of predicted forced expiratory volume in 1 s; SD - substantial decline; S – stable. (XLSX 13 kb)
Additional file 2: Table S1.
Genome features and virulence factors of A. ruhlandii ST36. (XLSX 59 kb)
Additional file 3: Table S2.
Genome features and virulence factors of B. cenocepacia ST709. (XLSX 61 kb)
Additional file 4: Table S3
B. cenocepacia strains with similar T3SS gene cluster. (XLSX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Voronina, O.L., Kunda, M.S., Ryzhova, N.N. et al. On Burkholderiales order microorganisms and cystic fibrosis in Russia. BMC Genomics 19 (Suppl 3), 74 (2018). https://doi.org/10.1186/s12864-018-4472-9
Published:
DOI: https://doi.org/10.1186/s12864-018-4472-9