
Abstract
Background
Entomopathogenic fungi such as Beauveria bassiana are promising biological agents for control of malaria mosquitoes. Indeed, infection with B. bassiana reduces the lifespan of mosquitoes in the laboratory and in the field. Natural isolates of B. bassiana show up to 10-fold differences in virulence between the most and the least virulent isolate. In this study, we sequenced the genomes of five isolates representing the extremes of low/high virulence and three RNA libraries, and applied a genome comparison approach to uncover genetic mechanisms underpinning virulence.
Results
A high-quality, near-complete genome assembly was achieved for the highly virulent isolate Bb8028, which was compared to the assemblies of the four other isolates. Whole genome analysis showed a high level of genetic diversity between the five isolates (2.85–16.8 SNPs/kb), which grouped into two distinct phylogenetic clusters. Mating type gene analysis revealed the presence of either the MAT1–1–1 or the MAT1–2–1 gene. Moreover, a putative new MAT gene (MAT1-2–8) was detected in the MAT1–2 locus. Comparative genome analysis revealed that Bb8028 contains 163 genes exclusive for this isolate. These unique genes have a tendency to cluster in the genome and to be often located near the telomeres. Among the genes unique to Bb8028 are a Non-Ribosomal Peptide Synthetase (NRPS) secondary metabolite gene cluster, a polyketide synthase (PKS) gene, and five genes with homology to bacterial toxins. A survey of candidate virulence genes for B. bassiana is presented.
Conclusions
Our results indicate several genes and molecular processes that may underpin virulence towards mosquitoes. Thus, the genome sequences of five isolates of B. bassiana provide a better understanding of the natural variation in virulence and will offer a major resource for future research on this important biological control agent.
Similar content being viewed by others


Background
Beauveria bassiana is a cosmopolitan entomopathogenic fungus (EPF) with a broad host range covering most insect orders (e.g., Coleoptera, Diptera, Hemiptera, Lepidoptera) and also arthropods (subclass Acari) [1, 2]. B. bassiana is currently used commercially as a biological control agent for different arthropod pests in China such as the pine caterpillar (Thaumetopoea pityocampa), corn borer (Ostrinia furnacalis), peach aphid (Myzus persicae), and pine sawyer (Bursaphelenchus xylophilus) [3]. B. bassiana is also being developed as a biological control tool for vectors of human diseases, such as Anopheles mosquitoes that transmit malaria [4, 5], Aedes aegypti mosquitoes that are vectors of Dengue, Chikungunya and Zika virus [6, 7], the Triatoma infestans bug, the Chagas disease vector [8], and Ixodes ricinus ticks that transmit Lyme disease [9].
Infection by B. bassiana starts when conidia adhere to the host cuticle via hydrophobic interactions and subsequently germinate. A germ tube is formed that is able to penetrate the cuticle through a combination of mechanical pressure and cuticle degrading enzymes. Once the fungal structures reach the haemocoel, the fungus switches to growth as single-celled blastospores, which are capable of evading the insect immune system, exploit the haemolymph for nutrition, and secrete toxins that eventually kill the host. Once the host is dead, the fungus breaches the cuticle from the inside outwards, allowing the formation of conidia that upon dispersal start new infections [10].
A good understanding of the life history of B. bassiana as well as the genetic mechanisms of virulence is crucial to develop it as an effective and sustainable biological control agent. Substantial progress has been made in the genetic characterization of the mechanisms involved in the infection cycle of B. bassiana inside insects. Many of the recent findings regarding genes involved in virulence were possible due to the development of reliable protocols that allow genetic manipulation [11] together with the availability of a draft genome sequence [12]. For instance, it has been found that B. bassiana expresses two hydrophobin genes, hyd1 and hyd2, that are believed to be involved in surface hydrophobicity, adhesion, virulence and composition of the rodlet layer. A Δhyd1 knock-out mutant showed decreased virulence and spore hydrophobicity but unchanged surface adhesion, while a Δhyd2 mutant showed reduced hydrophobicity and surface adhesion but unchanged virulence [13]. Similarly, a cytochrome P450 gene, Bbcyp52x1, was shown to function in enzymatic breakdown of lipids in the waxy layer. A ΔBbcyp52x1 knock-out mutant showed reduced virulence on Galleria mellonella upon topical application of conidia, though not upon intra-hemocoel injection [14].
Despite all efforts made to understand virulence in B. bassiana (for a review, see [15]), many of the genes that were functionally analysed thus far are involved in general biological processes (e.g., conidiation, stress response) that pleiotropically affect virulence. This indicates that virulence is tightly connected to the biology and life history of Beauveria. In order to increase the efficacy and specificity of Beauveria against arthropods, we need to identify the genes exclusively implicated in entomopathogenic virulence. Therefore, strategies must be employed that allow de novo identification of genes and mechanisms specifically related to virulence. Our previous identification of ample natural genetic variation for virulence in B. bassiana isolates [16] opens the way for a comparative genomic approach. Such an approach offers a powerful opportunity to collect comprehensive information of genetic variation and identification of variants of virulence and their evolutionary dynamics. Importantly, knock-out mutant approaches are crucial and will play an important role in verifying the action of the candidate genes that emerge from comparative genomics of natural isolates that differ in virulence.
The aim of this study was to improve our knowledge in understanding the key factors involved in virulence of B. bassiana by a comparative genomics study. B. bassiana is well known for having a broad genetic, host, and virulence diversity. As a consequence, genome structure and DNA sequence variation between any two isolates could be related to many factors in the biology of this fungus (including neutral evolution/genetic drift). Therefore, we sequenced five isolates of contrasting virulence, selected from a panel of 29 B. bassiana isolates of which the natural variation in virulence had been previously characterized [16]. We assembled a gapless near-complete genome for the most virulent B. bassiana isolate, Bb8028, which provides the community a highly suitable reference genome to be used for comparison with other isolates.
Methods
Fungal isolates and maintenance
Beauveria bassiana isolates ARSEF 1520 (Bb1520), ARSEF 2597 (Bb2597), ARSEF 4305 (Bb4305), ARSEF 5078 (Bb5078) and ARSEF 8028 (Bb8028) were obtained from the USDA-ARS Collection of Entomopathogenic Fungal Cultures (ARSEF). These isolates were selected based on their previously characterized virulence against malaria mosquitoes. Isolates Bb2597 and Bb8028 were previously shown to display high virulence, Bb1520 has intermediate virulence, while Bb4305 and Bb5078 have low virulence [16]. Isolates were grown on Sabouraud Dextrose Agar with 1% yeast extract (SDAY) at 27 °C before DNA extraction and sequencing.
Genome and transcriptome sequencing
DNA was isolated from freeze-dried mycelium using a CTAB-based method [17]. All isolates were sequenced with paired-end libraries of 180 bp insert size, and with a read length of 2x100 bp. Isolates Bb1520, Bb2597, and Bb8028 were further sequenced with a mate-pair library of 10 kb (Bb1520, Bb2597) or 6 kb (Bb8028). Isolate Bb8028 was selected as a reference based on the quality of the assembly, therefore additional sequencing focussed on this isolate and was performed using two PacBio SMRT cells with an insert size of 20 kb. The paired-end and mate-pair sequencing were performed using Illumina HiSeq2000 technology at the Beijing Genome Institute (BGI, Hongkong, China). PacBio sequencing was performed using the RS II instrument at Keygene N.V. (Wageningen, the Netherlands).
To obtain the transcriptome, RNA from Bb8028 was isolated from three different sources: (1) 5-day old mycelia grown on rice medium; (2) proto fruiting bodies (stromata like-structures) obtained from a cross between Bb8028 and Bb5078 co-cultivated for 2 months on rice medium as described [18]; and (3) infected mosquitoes that had just died. Rice medium was composed of 60 g of brown rice and 10 g of dried silkworm pupae, and 100 ml of distilled water. RNA was isolated using a RNAeasy Plant mini Kit (Qiagen) following manufacturer’s recommendations and sequenced using a paired-end library of 180 bp insert size on an Illumina HiSeq2000 platform at BGI.
Genome assembly and annotation
For isolates Bb4305 and Bb5078, de novo assembly was performed using Celera assembler [19] with default settings except k-mer size, which was set to 15. Isolates Bb1520, Bb2597, and Bb8028 were assembled using Allpaths-LG [20] with default settings. Additionally, PacBio data was used to improve the assembly of Bb8028 using PBJelly [21] and quiver tools [22], and erroneously merged contigs were manually corrected. RNA-Seq libraries were assembled de novo with Oases [23] and Trinity [24]. Also, the RNA-Seq reads were mapped to the Bb8028 assembly using Tophat [25]. Completeness of the genome assemblies was assessed by the Benchmarking Universal Single-Copy Orthologs (BUSCO) v1.1b1 software tool [26]. RepeatMasker [27] was used to search for homologs of known repeats using the library from Repbase [28], and previously unknown repeats were predicted de novo with RepeatScout [29]. Local Collinear Blocks (LCBs) were identifed using progressiveMauve (build date Feb 25, 2015), with default settings. Parsing and inversion detection was performed using a custom python script. Genome annotation was performed using the MAKER (v.2.31.8) pipeline [30] with AUGUSTUS [31], SNAP [32], and GeneMark-ES [33] as ab initio gene predictors. The assembled RNA-Seq reads and all fungal proteins available in the Uniprot database were used as evidence for gene prediction. Furthermore, the predicted proteins were manually inspected and curated. The predicted proteins were functionally annotated using BLASTp [34] against the non-redundant database of the National Center of Biotechnology Information (NCBI) and InterProScan [35]. The BLASTp and InterproScan results were imported into Blast2GO version 3.2 [36] and Gene Ontology (GO) terms were predicted. Overrepresentation of GO terms was analysed using Fisher’s test as embedded in Blast2GO. Secreted proteins were predicted by SignalP 4.1 [37].
Pan-genome analysis
The pan-genomes and core genomes at the gene level were estimated using OrthoMCL [38] implemented in GET_HOMOLOGUES-EST [39] with e-value 1e−5 and 75% coverage. Besides the five newly sequenced genomes presented here, the previously reported genome of B. bassiana ARSEF 2860 (Bb2860) [12] was also included in the analysis.
Phylogenetic analysis
The phylogenetic relationships were determined with the software REALPHY [40] between all isolates of B. bassiana, including the previously sequenced isolate Bb2860 [12], and using C. militaris [41] as the outgroup of the tree. All genomes were mapped using Bowtie2 [42] on the genome of Bb8028 as a reference. After the alignment, a maximum likelihood phylogenetic tree was inferred with RAxML [43] using the generalised time reversible (GTR) plus GAMMA nucleotide substitution model and 100 rapid bootstraps.
Variant analysis
To identify single-nucleotide polymorphisms (SNPs), Freebayes (v0.9.21), a Bayesian method based program, was used [44] with a minimum alteration fraction of 0.90. All sequences were mapped with BWA [45] to the genome of isolate Bb8028. To minimize false positives, SNPs called at positions with low coverage (<10x) were not considered. SNP density and transition/transversion ratios were calculated using VCFtools [46].
Secondary metabolite analysis
Putative gene clusters that are predicted to be involved in biosynthesis of secondary metabolites were identified using antiSMASH (antibiotics and Secondary Metabolite Analysis SHell) version 3.0.4 [47].
Mating type gene analysis
The presence of mating type loci in all isolates of B. bassiana (including Bb2860) were identified by homology to characterized MAT genes of other filamentous fungi using BLASTp. The mating type alleles MAT1–1–1 and MAT1–2–1 were also determined by PCR with the primer pairs MAT112.F4/ MAT111.R5 and MAT2.F4/ MAT2.R5 [48], respectively.
Selection analysis
To detect regions under positive selection, protein-coding DNA alignments based on the orthologous genes were generated with ParaAT. Ratios of non-synonymous (Ka) to synonymous (Ks) nucleotide substitution rates per site were estimated for each alignment using KaKs_Calculator 2.0 [49]. A sliding window of 100 bp was used with a gamma distribution (gMYN) model.
Results
A high-quality Beauveria bassiana reference genome
Five B. bassiana isolates were sequenced that were previously phenotyped for virulence against malaria mosquitoes. Isolate Bb8028, which is consistently the most virulent isolate in our collection [16] was selected to serve as a high quality reference genome for the dataset. This isolate was sequenced with both short and long-reads, single molecule sequencing technology, while two isolates were sequenced with short reads on both short and long insert libraries (Bb1520 and Bb2597), and two isolates were sequenced with short reads only on short insert libraries (Bb4305 and Bb5078). The final assembly sizes were in the range of 34.45–38.83 Mb, slightly larger (33.7 Mb) than the assembly of the previously sequenced isolate of B. bassiana, Bb2860 [12]. The final assemblies resulted in 12 to 3,952 contigs (Table 1), and the five isolates were predicted to encode between 10,210 and 10,831 protein-coding genes. The genome assembly of isolate Bb8028 had only 12 contigs with an N50 contig length of 5.69 Mb, and there were no gaps present. The 12 contigs covering the genome of Bb8028 represent four full chromosomes (with telomeric repeats at both ends) and 5 chromosome ends with a single telomeric repeat.
The three highest quality assemblies (Bb8028, Bb1520 and Bb2597) were used to explore structural rearrangements between these three genomes. In total, 695 locally collinear blocks (LCBs) were shared between two or three genomes. We identified 18 cases of clear inversions, where three adjacent LCBs were shared between the three genomes and in one genome the middle block was inverted with respect to the others. Fifteen of these inversions occurred in strain Bb1520 (Bb8028 and Bb2597 remain collinear), while two inversions occurred in Bb8028 and one in Bb2597. The size of inverted regions ranged from ~1 kb to 220 kb. Some LCBs demonstrated significant size differences between the genomes (ranging from 3 to 65 kb), suggesting insertions or deletions. Two inversions in isolate Bb1520 occurred within the coding sequence of a predicted gene (in the Bb8028 reference genome), thereby truncating the coding sequence of that gene in Bb1520. The vast majority of inversions occurred outside coding sequences and were judged to have no impact on gene structure or regulatory regions.
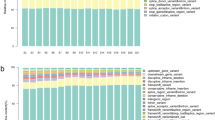
The completeness of all five genomes was assessed by checking for the representation of a set of universal single-copy orthologs for fungi [26], and this analysis indicated that each genome had at least 96% of the total BUSCOs (Additional File 1), suggesting a high quality of all the assemblies. The pan-genome is here defined as the combination of all the genes that are present in the genomes of all isolates of a particular species, and it includes the core genes, dispensable genes, and isolate-specific genes [50, 51]. A pan-genome analysis for B. bassiana for the five isolates sequenced in this study in combination with the previously sequenced isolate Bb2860 [12], indicated that the core genome of B. bassiana consists of 7,341 orthologous gene clusters (Fig. 1a). On the other hand, the pan-genome consists of 13,068 orthologous gene clusters (Fig. 1b).

Pan-genome analysis of B. bassiana isolates. a Estimation of the B. bassiana core genome, in which the number of shared genes is plotted as a function of the number of isolates sequentially added. b Estimation of B. bassiana pan-genome size, in which the numbers of all genes are plotted as a function of the number of isolates sequentially added
High levels of genetic diversity as revealed by SNPs and phylogenetic analysis
The phylogenetic relationships between isolates of B. bassiana were determined using the whole genome. Two distinct clusters were formed (Fig. 2), designated 1 and 2. Cluster 1 contains three isolates (Bb2597, Bb5078 and Bb8028), while the other three isolates (Bb1520, Bb4305 and Bb2860) are grouped in cluster 2. The grouping did not reflect a geographical distribution: the two isolates originating from USA were assigned to different clusters, as well as the two isolates of European origin. In addition, the two clusters did not reflect distinct virulence levels, as the low virulence isolate (Bb5078) grouped with the two high virulence isolates in cluster 1.

Phylogenetic tree based on whole genome assemblies of the different isolates of B. bassiana, with C. militaris as the outgroup to root the tree. On the right, the different clusters found are highlighted
All the read data that were available in this study were mapped to Bb8028 as a reference in order to detect SNPs. Within cluster 1, there were 2.85 and 4.8 SNPs per kb for isolates Bb5078 and Bb2597, respectively. In contrast, the genetic variation in isolates from cluster 2 against Bb8028 was four-fold higher, with an average of 16.6 and 16.8 SNPs per kb, for Bb1520 and Bb4305, respectively. Similarly, the genetic identity to respect to Bb8028 was higher in Bb5078 (90.4%) and Bb2597 (89.7%), and lower in Bb4305 (88.4%) and Bb1520 (87.9%). We searched for SNPs in coding sequences of a set of 54 genes that have been functionally analysed by knock-out mutagenesis (reviewed in [15]) between the five B. bassiana isolates sequenced in this study. There are two genes that contain a SNP with potentially high impact: in the α-glucoside transporter gene Bbagt1 in a single isolate, and in the thioredoxin gene trx3 in two isolates. The remaining 52 genes do not display any high impact SNPs.
A new mating type gene
Analysis of orthologous relationships between the genes of all isolates of B. bassiana in this study identified the presence of several mating type loci (Fig. 3). We identified two MAT genes, MAT1–1–1 and MAT1–1–2, in Bb2597 and Bb5078. The MAT1–1–2 gene was not previously identified in the published genome of Bb2860 [12], but based on BLAST hits, genes from Bb2597 and Bb5078 have homology to the MAT1–1–2 gene in C. militaris [41]. Further alignment of the putative MAT1–1–2 gene from Bb2597 and Bb5078 to scaffold BBA_S00027 from Bb2860 confirmed that this gene is also present in Bb2860 but was not characterized [12]. The MAT1-2 locus was identified by homology to the HMG-box domain in Bb1520, Bb4305 and Bb8028. A putative new MAT gene was found next to the MAT1–2–1 gene. BLASTp analysis and sequence alignments shows that it has partial homology to the newly described MAT1–2–8 gene in Villosiclava virens [52]. PCR confirmed presence of either MAT1–1–1 or MAT1–2–1 in each isolate (Additional File 2).

Graphical presentation of the synteny of mating type genes and their flanking region in B. bassiana genomes. Genes in the same colour show orthologous relationships. The dashed circle represents a gene that has not previously been identified in the genome of ARSEF 2860
The draft genome of B. bassiana isolate Bb2860 was reported to lack the spo11 gene, a meiosis-specific topoisomerase essential for meiotic recombination, and this observation was used as an explanation for the infrequent occurrence of the sexual cycle of B. bassiana [12]. The spo11 gene was however present in the genomes of all five B. bassiana isolates analysed in this study, and the conclusion about its absence in B. bassiana [12], based on a draft genome assembly, was thus erroneous.
Isolate-specific genes from Bb8028 include candidate virulence genes
Isolate Bb8028 was previously characterized as the most virulent towards malaria mosquitoes [16]. Therefore, we hypothesised that the high virulence could be due to the presence of genes unique to Bb8028 that are related to fungal virulence. In order to detect regions uniquely present in Bb8028, all reads from the four other isolates were mapped to the genome of Bb8028 (parsed in bins of 10 kb in size) and regions with very low or no read coverage were detected (Fig. 4). A total sequence length of 0.71 Mb (2% of the genome of Bb8028) displayed very low coverage in the four other isolates, these regions encompassed 390 genes. To independently verify whether these genes were indeed unique, raw reads of the four other isolates were then mapped to individual genes of Bb8028. This approach identified 252 genes from Bb8028 with low or no coverage in all four other isolates. Comparison of the two datasets yielded 163 genes that were identified by both methods (Additional File 3), which we therefore considered to be unique to isolate Bb8028. The location of these Bb8028 genes on the eight largest contigs was analysed. Interestingly, the genes showed a tendency to cluster together, especially close to telomeres (most notably at the ends of contigs 3, 5 and 6, and at the beginning of contigs 7, 8 and 12; Additional File 3). The unique Bb8028 genes were functionally annotated to evaluate their potential role in pathogenicity. Of the 163 genes, 57 (35%) have homology to genes in the Pathogen Host Interaction Database (PHI-base) [53], 47 (25.2%) were hypothetical proteins, and 27 (16.6%) were predicted to contain signal peptides. Both the number of genes classified as hypothetical proteins and those predicted to be secreted were higher than expected when compared to the complete gene content (15%, χ 2 = 12.02, d.f. = 1, p = 0.000527; and 10.4%, χ 2 = 5.77, d.f. = 1, p = 0.01629, respectively). By contrast, the number of genes homologous to genes represented in PHI-base follows the expectation based on the whole gene set (29.4%, χ 2 = 2.1223, d.f. = 1, p = 0.1452). An overrepresentation of gene ontology terms was not detected in the 163 genes that are unique to Bb8028.

Circular visualization of the alignment of genome sequencing reads of four isolates of Beauveria bassiana with respect to the Bb8028 reference assembly. The eight largest contigs are numbered 1 to 8 along the perimeter of the outer circle. The tracks represent the location of predicted genes for Bb8028 (a); mean read coverage (per 10 kb) for isolates Bb1520 (b), Bb2597 (c), Bb4305 (d) and Bb5078 (e); and location of the unique genes for Bb8028 (f). In tracks (b) to (e), colours represent the percentage of coverage against the Bb8028 reference assembly: red: 0–45%, orange: 45–63%, yellow: 63–77%, green: 77–90%, and blue: 90–100%)
Secondary metabolites play a role in fitness and virulence of fungal (entomo)pathogens [54]. A Non-Ribosomal Peptide Synthetase (NRPS) gene cluster is present in the unique gene set of Bb8028 at the start of contig 8. The NRPS gene (BB8028_0008g00080) has homology (52–54%) to several Metarhizium species, with the difference that BB8028_0008g00080 has a single adenylation domain and is 1059 amino acids in length, while its Metarhizium homologs possess two adenylation domains and encode proteins almost double in size. In addition, Bb8028 contains a unique polyketide synthase gene (BB8028_0001g06000.1).
Five genes unique to Bb8028 (thus lacking in the four other B. bassiana genomes) encode proteins possessing bacterial enterotoxin-like domains. Four of these contain a heat-labile enterotoxin domain (IPR001144) while one protein displays 54% similarity to the Vibrio cholera enterotoxin alpha chain domain (IPR008992). Outside the kingdom Eubacteria, these enterotoxin genes have homology almost exclusively to Beauveria, Cordyceps and Metarhizium species, all of which are entomopathogenic fungi (data not shown). Bb8028 also possesses 11 peptidase genes (from various types) that have no orthologs in any of the other four B. bassiana isolates. Furthermore, two genes encoding the enzyme cyanamide hydratase are uniquely present in Bb8028. Cyanamide hydratase converts cyanamide into urea, and has been studied in relation to fungal survival in the soil since cyanamide is used as a fertilizer and is also synthesized by vetch species [55, 56]. Bb8028 also contains 13 unique genes involved in transport processes, of which five belong to the major facilitator (MFS) superfamily, four to the family of ATP-binding cassette (ABC) transporters, and the remaining four are dedicated siderophore and dicarboxylate transporters, as well as a sodium/proton antiporter. ABC transporters can be involved in (multidrug) resistance to fungicides, as well as the secretion of antibiotics and toxins [57]. Recently, five ABC transporters were characterized in B. bassiana and found to be implicated in fungicide resistance, oxidative stress tolerance, and in virulence [58].
Finally, among the gene set unique to Bb8028 are also genes encoding enzymes putatively involved in the modification of chromatin proteins, which may modulate changes in gene expression [59]. Specifically, these genes encode three arginine deiminases and a putative histone arginine N-methyltransferase.
Although a detailed analysis of differential gene expression could not be done with the RNA data available due to the lack of sample replication, an exploratory examination was done to see whether unique genes of Bb8028 were highly expressed. Specifically, we looked for genes that had at least (1) double the expression (in Fragments Per Kilobase of transcript per Million mapped reads (FPKM)) in the library isolated from infected mosquitoes compared against the library from fruiting bodies or mycelia, and, (2) a value of at least one FPKM. Of the 163 genes, 15 genes fulfilled these criteria. Notably, three of the five genes annotated as a toxin were among these 15 genes (Additional File 4). Also one of the arginine deiminase genes and one of the cyanamide hydratase genes displayed a high expression in the mosquito library as compared to the other two libraries.
No clear pattern of positive selection in genes of Bb8028
Genes with a signature of positive selection may contribute to the adaptation to a changing environment and could be related to virulence. Therefore, we investigated whether genes were under positive selection in Bb8028. Ka/Ks ratios were calculated in pairwise comparisons between orthologs in Bb8028 and each of the four other isolates, separately. We detected 10, 5, 11, 10 genes of Bb8028 with Ka/Ks > 1 when compared to their orthologs in Bb1520, Bb2597, Bb4305, Bb5078, respectively. In total, 18 genes are considered to be under selection in Bb8028, but intriguingly no gene was shared among the four pairwise comparisons (Additional File 5). Among the genes under positive selection are two calmodulin-dependent protein kinase genes and an ABC transporter gene, while most of the other genes were of unknown function.
Discussion
In this study, we provide and compare the genomes of five isolates of B. bassiana that were previously characterized in a large-scale analysis of virulence against the malaria mosquito (An. coluzzii) [16]. These isolates have different levels of virulence, with Bb8028 being the most virulent isolate, followed by Bb2597, Bb1520, Bb4305, and finally Bb5078. Comparative genomics at the intraspecific level is an approach to determine the genetic basis of phenotypic variations detected between different isolates. The genome of B. bassiana was previously compared to other entomopathogenic fungi [12], and in order to avoid a duplication of this previous work, we focused on comparison between B. bassiana isolates, with an emphasis on Bb8028. A pan-genome analysis indicated that the core genome of B. bassiana consists of 7,341 orthologous gene clusters, while the pan-genome consists of 13,068 orthologous gene clusters. These numbers suggest that B. bassiana has an open pan-genome, as may be expected for a species that can typically colonize multiple environments [51].
The isolates analysed have a high level of diversity with 467,292 SNPs between Bb4305 and Bb8028, and 87,119 SNPs between Bb5078 and Bb8028. Previous studies have suggested that B. bassiana is not monophyletic, but rather is composed of several clusters that are morphologically identical [48, 60, 61]. One study [61] described seven phylogenetic clusters in B. bassiana that consisted of isolates which did not show any geographic distribution but had common climate characteristics, while a different study [48] described isolates that were distributed in five clusters in a particular hedgerow habitat, however, genetic distances were not determined on a genome-wide scale in these studies. A comparative genomic study of seven isolates of the plant pathogenic fungus Penicillium expansum revealed a genetic diversity between 1.3 and 7.1 SNPs/kb [62], while between two isolates of Aspergillus niger it was 7.8 SNPs/kb [63]. In both these cases, the genetic diversity was lower than observed in this study (2.9–16.8 SNPs/kb). The finding that B. bassiana possesses a high genetic diversity seems to be in agreement with the earlier suggestions that B. bassiana is not monophyletic [48, 60, 61] and warrants further studies to elucidate the species phylogeny on a whole genome scale.
B. bassiana normally reproduces asexually, and for a long time it was believed to be exclusively clonal. Developmental and phylogenetic studies, however, have linked it to the teleomorph Cordyceps bassiana [64, 65]. In the five isolates studied, the MAT1-1 and MAT1-2 loci were identified based on homology to the typical fungal mating type genes, encoding the alpha domain protein and the high mobility group box domain, respectively. The presence of either of two distinct alleles of the MAT locus in each B. bassiana isolate indicates that the species is putatively heterothallic. A new MAT gene was detected next to the MAT1–2–1 gene which, based on homology to a newly described gene in Villosiclava virens [52], is referred to as MAT1–2–8 following the nomenclature proposed by Dyer et al. [66]. We attempted to sexually cross different isolates of B. bassiana in several combinations to generate fertile fruiting bodies. Based on the mating type, six combinations were possible (Additional File 6). Unfortunately, none of the crosses generated ascospores, but the cross between Bb8028 and Bb5078 generated fruiting body-like structures without ascospores; these structures were used to generate a transcriptome library. When taking into consideration the phylogenetic clusters found, we realized that only Bb8028 and B5078 belong to the same cluster and had the lowest number of SNPs/kb, compared to the other possible combinations. Since B. bassiana could be polyphyletic, and reports have found a single mating type in several populations [48], it is not surprising that B. bassiana is limited mainly to asexual reproduction. It is worthwhile to invest in a reproducible protocol for sexual crosses under laboratory conditions. The ability to achieve sexual reproduction will enable genome-wide association studies (GWAS) of quantitative virulence in the offspring of two isolates that markedly differ in virulence, for instance Bb8028 and Bb5078.
In this analysis, we identified a suite of genes that are unique to Bb8028. Among these are a PKS gene and a gene cluster comprising an NRPS gene with homology to a M. anisopliae gene annotated as tyrocidine synthase (KJK81377). Tyrocidine has antibiotic activity, and the peptide synthesized by the NRPS gene product could thus have antibiotic activity. The set of genes unique to Bb8028 also comprised five genes encoding bacterial-like toxins and 13 transporter genes. The B. bassiana genome contains many more bacterial-like toxins than most other fungi [12]. Interestingly, we found preliminary evidence that some of the bacterial-like toxins are highly expressed in fungal infection of mosquitoes. A recent study [67] analysed gene expression in B. bassiana ARSEF 2860 infecting Plutella xylostella larvae at three different time points after inoculation (24 h, 36 h, 48 h). In the latter study, an enrichment of genes with functions in antioxidant activity, peroxidase activity, and proteolysis was detected among the genes that are highly expressed in B. bassiana during infection of the host insect. In contrast, toxin genes were not reported to be highly expressed during infection of P. xylostella larvae [67]. The mortality rate of P. xylostella larvae infected by B. bassiana at 48 h was 33%, which is much lower than the mortality rate of almost 100% of infected mosquitoes at the time that we sampled RNA for library construction. The infection cycle of B. bassiana is a complex process which includes several developmental transitions that require an intricate genetic regulation [15]. This biological complexity may explain why no overlap is found between these studies. Furthermore, B. bassiana is capable of infecting a wide range of hosts that have different chemical and physical compositions, which most likely requires a tailored response for each host-pathogen interactions.
Conclusions
The availability of a near-complete genome assembly of Bb8028 provided with a manually curated gene set, adds significant value to the previously published draft genome of B. bassiana [12]. As argued by [68], a high quality assembly including proper assembly of centromeres, telomeres, and other repetitive elements provides the ability to characterize structural rearrangements and facilitates a better identification of secondary metabolite gene clusters. Therefore, a high quality genome is a key step to uncover the biological complexity of an organism, as our analyses showed. The Bb8028 genome assembly represents a significant resource for future research into the biology of this entomopathogenic fungus and its relevance as a biological control agent. Understanding the key factors involved in virulence of B. bassiana will improve our methods to use this fungus as an effective and sustainable biological control agent against, amongst others, vectors of human diseases.
References
Kirkland BH, Westwood GS, Keyhani NO. Pathogenicity of entomopathogenic fungi Beauveria bassiana and Metarhizium anisopliae to Ixodidae tick species Dermacentor variabilis, Rhipicephalus sanguineus, and Ixodes scapularis. J Med Entomol. 2004;41:705–11.
de Faria MR, Wraight SP. Mycoinsecticides and Mycoacaricides: A comprehensive list with worldwide coverage and international classification of formulation types. Biol Control. 2007;43:237–56.
Li Z, Alves SB, Roberts DW, Fan M, Delalibera I, Tang J, Lopes RB, Faria M, Rangel DEN. Biological control of insects in Brazil and China: history, current programs and reasons for their successes using entomopathogenic fungi. Biocontrol Sci Technol. 2010;20:117–36.
Blanford S, Chan BHK, Jenkins N, Sim D, Turner RJ, Read AF, Thomas MB. Fungal pathogen reduces potential for malaria transmission. Science. 2005;308:1638–41.
Mnyone LL, Lyimo IN, Lwetoijera DW, Mpingwa MW, Nchimbi N, Hancock PA, Russell TL, Kirby MJ, Takken W, Koenraadt CJM. Exploiting the behaviour of wild malaria vectors to achieve high infection with fungal biocontrol agents. Malar J. 2012;11:87.
Darbro JM, Johnson PH, Thomas MB, Ritchie SA, Kay BH, Ryan PA. Effects of Beauveria bassiana on survival, blood-feeding success, and fecundity of Aedes aegypti in laboratory and semi-field conditions. Am J Trop Med Hyg. 2012;86:656–64.
de Paula AR, Brito ES, Pereira CR, Carrera MP, Samuels RI. Susceptibility of adult Aedes aegypti (Diptera: Culicidae) to infection by Metarhizium anisopliae and Beauveria bassiana: prospects for Dengue vector control. Biocontrol Sci Technol. 2008;18:1017–25.
Forlani L, Pedrini N, Girotti JR, Mijailovsky SJ, Cardozo RM, Gentile AG, Hernández-Suárez CM, Rabinovich JE, Juárez MP. Biological control of the Chagas disease vector Triatoma infestans with the entomopathogenic Fungus Beauveria bassiana combined with an aggregation cue: field, laboratory and mathematical modeling assessment. PLoS Negl Trop Dis. 2015;9:e0003778.
Hartelt K, Wurst E, Collatz J, Zimmermann G, Kleespies RG, Oehme RM, Kimmig P, Steidle JLM, Mackenstedt U. Biological control of the tick Ixodes ricinus with entomopathogenic fungi and nematodes: Preliminary results from laboratory experiments. Int J Med Microbiol. 2008;298:314–20.
Boomsma JJ, Jensen AB, Meyling NV, Eilenberg J. Evolutionary interaction networks of insect pathogenic fungi. Annu Rev Entomol. 2014;59:467–85.
Fang W, Zhang Y, Yang X, Zheng X, Duan H, Li Y, Pei Y. Agrobacterium tumefaciens-mediated transformation of Beauveria bassiana using an herbicide resistance gene as a selection marker. J Invertebr Pathol. 2004;85:18–24.
Xiao G, Ying S-H, Zheng P, Wang Z-L, Zhang S, Xie X-Q, Shang Y, St Leger RJ, Zhao G-P, Wang C, Feng M-G. Genomic perspectives on the evolution of fungal entomopathogenicity in Beauveria bassiana. Sci Rep. 2012;2:483.
Zhang S, Xia YX, Kim B, Keyhani NO. Two hydrophobins are involved in fungal spore coat rodlet layer assembly and each play distinct roles in surface interactions, development and pathogenesis in the entomopathogenic fungus, Beauveria bassiana. Mol Microbiol. 2011;80:811–26.
Zhang S, Widemann E, Bernard G, Lesot A, Pinot F, Pedrini N, Keyhani NO. CYP52X1, representing new cytochrome P450 subfamily, displays fatty acid hydroxylase activity and contributes to virulence and growth on insect cuticular substrates in entomopathogenic fungus Beauveria bassiana. J Biol Chem. 2012;287:13477–86.
Valero-Jiménez CA, Wiegers H, Zwaan BJ, Koenraadt CJM, van Kan JAL. Genes involved in virulence of the entomopathogenic fungus Beauveria bassiana. J Invertebr Pathol. 2016;133:41–9.
Valero-Jiménez CA, Debets AJM, van Kan JAL, Schoustra SE, Takken W, Zwaan BJ, Koenraadt CJM. Natural variation in virulence of the entomopathogenic fungus Beauveria bassiana against malaria mosquitoes. Malar J. 2014;13:479.
Möller EM, Bahnweg G, Sandermann H, Geiger HH. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res. 1992;20:6115–6.
Lee J-O, Shrestha B, Sung G-H, Han S-K, Sung J-M. Successful Development of Cordyceps bassiana Stromata from Beauveria bassiana. Mycobiology. 2010;38:13.
Myers EW. A whole-genome assembly of Drosophila. Science. 2000;287(80-):2196–204.
Ribeiro FJ, Przybylski D, Yin S, Sharpe T, Gnerre S, Abouelleil A, Berlin AM, Montmayeur A, Shea TP, Walker BJ, Young SK, Russ C, Nusbaum C, MacCallum I, Jaffe DB. Finished bacterial genomes from shotgun sequence data. Genome Res. 2012;22:2270–7.
English AC, Richards S, Han Y, Wang M, Vee V, Qu J, Qin X, Muzny DM, Reid JG, Worley KC, Gibbs RA. Mind the gap: upgrading genomes with Pacific Biosciences RS Long-Read Sequencing Technology. PLoS One. 2012;7:1–12.
Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10:563–9.
Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–92.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2.
RepeatMasker Open-4.0. 2013–2015. http://repeatmasker.org.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7.
Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:351–8.
Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, Moore B, Holt C, Alvarado AS, Yandell M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18:188–96.
Stanke M, Schöffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics. 2006;7:62.
Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59.
Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18:1979–90.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–40.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–6.
Li L. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89.
Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol. 2013;79:7696–701.
Bertels F, Silander OK, Pachkov M, Rainey PB, van Nimwegen E. Automated reconstruction of whole genome phylogenies from short sequence reads. Mol Biol Evol. 2014;31:1077–88.
Zheng P, Xia Y, Xiao G, Xiong C, Hu X, Zhang S, Zheng H, Huang Y, Zhou Y, Wang S, Zhao G-P, Liu X, St Leger RJ, Wang C. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011;12:R116.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. ArXiv e-prints. 2012;1207(3907):9.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, Breitling R, Takano E, Medema MH. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–43.
Meyling NV, Lübeck M, Buckley EP, Eilenberg J, Rehner SA. Community composition, host range and genetic structure of the fungal entomopathogen Beauveria in adjoining agricultural and seminatural habitats. Mol Ecol. 2009;18:1282–93.
Wang D, Zhang Y, Zhang Z, Zhu J, Yu J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinforma. 2010;8:77–80.
Vernikos G, Medini D, Riley DR, Tettelin H. Ten years of pan-genome analyses. Curr Opin Microbiol. 2015;23:148–54.
Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev. 2005;15:589–94.
Yu J-J, Sun W-X, Yu M-N, Yin X-l, Meng X-K, Zhao J, Huang L, Huang L, Liu Y-F. Characterization of mating-type loci in rice false smut fungus Villosiclava virens. FEMS Microbiol Lett. 2015;362:1–9.
Winnenburg R, Baldwin TK, Urban M, Rawlings C, Köhler J, Hammond-Kosack KE. PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 2006;34(Database issue):D459–64.
Molnár I, Gibson DM, Krasnoff SB. Secondary metabolites from entomopathogenic Hypocrealean fungi. Nat Prod Rep. 2010;27:1241–75.
Li J, Biss M, Fu Y, Xu X, Moore SA, Xiao W. Two duplicated genes DDI2 and DDI3 in budding yeast encode a cyanamide hydratase and are induced by cyanamide. J Biol Chem. 2015;290:12664–75.
Maier-Greiner UH, Obermaier-Skrobranek BM, Estermaier LM, Kammerloher W, Freund C, Wülfing C, Burkert UI, Matern DH, Breuer M, Eulitz M. Isolation and properties of a nitrile hydratase from the soil fungus Myrothecium verrucaria that is highly specific for the fertilizer cyanamide and cloning of its gene. Proc Natl Acad Sci U S A. 1991;88:4260–4.
Del Sorbo G, Schoonbeek H, De Waard MA. Fungal transporters involved in efflux of natural toxic compounds and fungicides. Fungal Genet Biol. 2000;30:1–15.
Song T-T, Zhao J, Ying S-H, Feng M-G. Differential contributions of five ABC transporters to mutidrug resistance, antioxidion and virulence of Beauveria bassiana, an entomopathogenic fungus. PLoS One. 2013;8:e62179.
Brosch G, Loidl P, Graessle S. Histone modifications and chromatin dynamics: A focus on filamentous fungi. FEMS Microbiol Rev. 2008;32:409–39.
Rehner SA, Posada F, Buckley EP, Infante F, Castillo A, Vega FE. Phylogenetic origins of African and Neotropical Beauveria bassiana s.l. pathogens of the coffee berry borer, Hypothenemus hampei. J Invertebr Pathol. 2006;93:11–21.
Ghikas DV, Kouvelis VN, Typas MA. Phylogenetic and biogeographic implications inferred by mitochondrial intergenic region analyses and ITS1-5.8S-ITS2 of the entomopathogenic fungi Beauveria bassiana and B. brongniartii. BMC Microbiol. 2010;10:174.
Julca I, Droby S, Sela N, Marcet-Houben M, Gabaldón T. Contrasting genomic diversity in two closely related postharvest pathogens: Penicillium digitatum and Penicillium expansum. Genome Biol Evol. 2016;8:218–27.
Andersen MR, Salazar MP, Schaap PJ, Van De Vondervoort PJI, Culley D, Thykaer J, Frisvad JC, Nielsen KF, Albang R, Albermann K, Berka RM, Braus GH, Braus-Stromeyer SA, Corrochano LM, Dai Z, Van Dijck PWM, Hofmann G, Lasure LL, Magnuson JK, Menke H, Meijer M, Meijer SL, Nielsen JB, Nielsen ML, Van Ooyen AJJ, Pel HJ, Poulsen L, Samson RA, Stam H, Tsang A, et al. Comparative genomics of citric-acid-producing Aspergillus niger ATCC 1015 versus enzyme-producing CBS 513.88. Genome Res. 2011;21:885–97.
Rehner SA, Buckley E. A Beauveria phylogeny inferred from nuclear ITS and EF1-α sequences: evidence for cryptic diversification and links to Cordyceps teleomorphs. Mycologia. 2005;97:84–98.
Sung J-M, Lee J-O, Humber RA, Sung G-H, Shrestha B. Cordyceps bassiana and production of stromata in vitro showing Beauveria anamorph in Korea. Mycobiology. 2006;34:1.
Dyer PS, Inderbitzin P, Debuchy R. Mating-type structure, function, regulation and evolution in the Pezizomycotina. In: Wendland J, editor. The Mycota I: Growth, Differentiation and Sexuality. 3rd ed. Switzerland: Springer International Publisher; 2016.
Chu Z-J, Wang Y-J, Ying S-H, Wang X-W, Feng M-G. Genome-wide host-pathogen interaction unveiled by transcriptomic response of diamondback moth to fungal infection. PLoS One. 2016;11:e0152908.
Thomma BPHJ, Seidl MF, Shi-Kunne X, Cook DE, Bolton MD, van Kan JAL, Faino L. Mind the gap; seven reasons to close fragmented genome assemblies. Fungal Genet Biol. 2015;90:24–30.
Acknowledgements
CAVJ thanks Dr. Michael Seidl for advice and assistance with bioinformatics analysis.
Funding
This research was funded by the Graduate School Production Ecology & Resource Conservation, grant 12072. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and material
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accessions: JRHA00000000, JTCW00000000, JTCX00000000, JTCY00000000, and JTCZ00000000. The raw data for this study are deposited in NCBI Sequence Read Archive under BioProject number PRJNA260878. Bb8028: SRR3269141 (DNA reads Illumina, short insert library), SRR3269142 (DNA reads Illumina, long insert library), SRR3269778 (RNA reads mycelium), SRR3269779 (RNA reads “fruiting bodies”), SRR3269780 (RNA reads infected mosquitoes), SRR3269861 (DNA reads PacBio library), and SRR3269862 (DNA reads PacBio library). Bb1520 data: SRR3269145 (DNA reads Illumina, short insert library) and SRR3269146(DNA reads Illumina, long insert library). Bb2597: SRR3269425 (DNA reads Illumina, short insert library) and SRR3269550 (DNA reads Illumina, long insert library). Bb4305: SRR326914 (DNA reads Illumina, short insert library). Bb5078: SRR3269144 (DNA reads Illumina, short insert library). The genome of B. bassiana strain Bb8028 will be accessible from Ensembl Fungi (release October 2016).
Authors’ contributions
CAVJ, SS, BJZ, and JALvK conceived the study and participated in its design and coordination. CAVJ carried out the laboratory work. CAVJ, DSV and LF performed the bioinformatics analysis. CAVJ, BJZ, and JALvK wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Both B.J. Zwaan and J.A.L. van Kan are joint corresponding authors on the publication
Additional files
Additional file 1:
Summary of the genome quality benchmarks performed with BUSCO. (XLSX 10 kb)
Additional file 2:
PCR verification of the presence of MAT1–1–1 (odd numbers) or MAT1–2–1 (even numbers) in the genomes of different isolates of Beauveria bassiana. (1–2) Bb1520, (3–4) Bb2597, (5–6) Bb4305, (7–8) Bb5078 and (9–10) Bb8028. (JPG 62 kb)
Additional file 3:
Summary of the unique genes of Bb8028. (XLSX 23 kb)
Additional file 4:
Summary of the unique genes of Bb8028 that were highly expressed in the infected mosquito library. (XLSX 11 kb)
Additional file 5:
Summary of genes of Bb8028 that under positive selection. (XLSX 10 kb)
Additional file 6:
Additional_file_6.xlsx. Fruiting body formation from isolates of Beauveria bassiana with different mating types. (XLSX 9 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Valero-Jiménez, C.A., Faino, L., Spring in’t Veld, D. et al. Comparative genomics of Beauveria bassiana: uncovering signatures of virulence against mosquitoes. BMC Genomics 17, 986 (2016). https://doi.org/10.1186/s12864-016-3339-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-016-3339-1