
Abstract
Background
The Pacific white shrimp (Litopenaeus vannamei) is the world’s most prevalent cultured crustacean species. However, the supply of high-quality broodstocks is limited and baseline information related to its reproductive activity and molecular issues related to gonad development are scarce. In this study, we performed transcriptome sequencing on the gonads of adult male and female L. vannamei to identify sex-related genes.
Results
A total of 25.16 gigabases (Gb) of sequences were generated from four L. vannamei gonadal tissue libraries. After quality control, 24.11 Gb of clean reads were selected from the gonadal libraries. De-novo assembly of all the clean reads generated a total of 65,218 unigenes with a mean size of 1021 bp and a N50 of 2000 bp. A search of all-unigene against Nr, SwissProt, KEGG, COG and NT databases resulted in 26,482, 23,062, 20,659, 11,935 and 14,626 annotations, respectively, providing a total of 30,304 annotated unigenes. Among annotated unigenes, 12,320 unigenes were assigned to gene ontology categories and 20,659 unigenes were mapped to 258 KEGG pathways. By comparing the ovary and testis libraries, 19,279 testicular up-regulated and 3,529 ovarian up-regulated unigenes were identified. Enrichment analysis of differentially expressed unigenes resulted in 1060 significantly enriched GO terms and 34 significantly enriched KEGG pathways. Nine ovary-specific, 6 testis-specific, 45 testicular up-regulated and 39 ovarian up-regulated unigenes were then confirmed by semi-quantitative PCR and quantitative real-time PCR. In addition, using all-unigenes as a reference, a total of 13,233 simple sequence repeats (SSRs) were identified in 10,411 unigene sequences.
Conclusions
The present study depicts the first large-scale RNA sequencing of shrimp gonads. We have identified many important sex-related functional genes, GO terms and pathways, all of which will facilitate future research into the reproductive biology of shrimp. We expect that the SSRs detected in this study can then be used as genetic markers for germplasm evaluation of breeding and imported populations.
Similar content being viewed by others


Background
The Pacific white shrimp (Litopenaeus vannamei) is a species of Penaeus shrimp that are native to the eastern Pacific Ocean, from the Mexican state of Sonora as far south as northern Peru [1]. It has become the world’s most prevalent cultured crustacean species as a result of its fast growth, adaptability to a wide range of salt and temperature, strong disease resistance, and low demand for dietary protein [2]. By 2004, global production of L. vannamei approached 1,116,000 tons, and exceeded that of Penaeus monodon [3]. By 2010, production had reached 2,721,000 tons [4]. However, the very limited supply of high-quality broodstocks is contrasted with the heavy demand of shrimp larvae from large-scale cultivation, especially in non-native countries such as China. The employment of inferior-quality broodstocks could lead to an eventual loss in gametic and larval quality, and production would then decline. Studies aimed at improving reproductive performance would therefore be helpful for the industrial applications of L. vannamei. However, previous studies primarily focused on disease resistance mechanisms and culturing techniques of L. vannamei [5–9], and baseline information related to its reproductive activity and molecular aspects of gonadal development remain scarce. Thus, it is important to understand the regulatory mechanisms of reproductive phenotypes in this species.
The first step toward understanding molecular mechanisms of reproduction is to identify and characterize sex-related genes and regulatory pathways. Many efforts have been made to reveal sex-related genes, and many of these genes are cloned and characterized in shrimp; for example, M-phase phosphoprotein 6 (MPP6) [10], cell division cycle 2 (Cdc2) [11], cyclin A and cyclin B [12], gonad-inhibiting hormone (GIH) [13], mitogen-activating protein kinase 1 (MAPK1) [14], prostaglandin reductase 1 [15], and valosin-containing protein (VCP) [16] in Penaeus monodon; and sex-determiner transformer-2 (Tra-2) [17], activated protein kinase C1 (RACK1) [18], and cell apoptosis susceptibility (FcCAS) [19] in Chinese shrimp Fenneropenaeus chinensis. In L. vannamei, vasa-like, vitellin, gonadotropin-releasing hormone-like and a sex-related marker have been identified [20–23]. Additionally, research methods, such as suppression subtractive hybridization SSH [24, 25], proteomic analysis [26, 27], EST sequencing [28–30], and microarray [28, 31, 32], have also been applied to scientific studies of shrimp in order to reveal potential sex-related genes. However, because of the lack of genomic sequences, comprehensive identification of sex-related genes and construction of regulatory networks associated with shrimp gonadal development are lacking.
Newly developed next-generation high-throughput sequencing technology has become a powerful tool for identifying genes involved in gonadal development, sex determination and sex differentiation [33–37]; and for SNP/SSR marker discovery [38–40] in aquaculture species where the genomic sequences are not available. In the present study, we performed transcriptomic sequencing of the gonads of 13-month-old adult male and female L vannamei to identify sex-related genes. The gonadal transcriptomic data of one-day post-eyestalk- ablation females (Day1O) and six-day post-eyestalk ablation females (Day6O) were also used for de-novo assembly and annotation so as to identify more genes during ovarian maturation. Results from the transcriptomic analysis would be particularly important for better understanding the regulation of gonadal development between sexes in this economically important aquaculture species. In addition, real-time PCR verification of 104 sex differentially expressed genes herein validates the reliability of the transcriptomic analysis strategy, and emphasizes some candidate genes of interest involved in sex determination and gonadal development for further functional studies.
Methods
Ethics statement
All procedures involving the handling and treatment of shrimp used in this study were conducted with the approval of the Animal Care and Use Committee of the Guangxi Academy of Fishery Sciences, Nanning, China.
Experimental shrimp and sample collection
L. vannamei used in this study were reared at Fangchenggang aquaculture base, Guangxi Academy of Fishery Sciences, Nanning, China. The experimental shrimp were 13 months of age with a weight of 40–50 g. First, shrimp were anesthetized on ice for three minutes, then testes from male shrimp (Testis), and ovaries from the pre-eyestalk ablation female shrimp (PreO) were removed. The ovaries of one-day post-eyestalk ablation female shrimp (Day1O) and the six-day post-eyestalk ablation female shrimp (Day6O) were also isolated. The gonadal tissues (testis for male and ovary for female) were cut into cubes of approximately 0.5 × 0.5 × 0.5 cm in size and immediately immersed in liquid nitrogen overnight, and then stored at −80 °C until RNA extraction. Some pieces of gonad from each shrimp were fixed in 4 % PFA and sectioned at 5 μm for hematoxylin and eosin (HE) staining and observation by light microscopy.
RNA isolation, library preparation, and Illumina Hiseq2500 sequencing
Total RNA was isolated using TRIzol® Reagent (Invitrogen, CA, U.S.) according to the manufacturer’s instructions, and genomic DNA was removed using DNase I (Takara, Tokyo, Japan). Then RNA quality was determined with a 2100 Bioanalyser (Agilent, CA, U.S.) and quantified using a ND-2000 (NanoDrop Technologies, DE, U.S.). Three RNA samples from each group (Testis, PreO, Day1O, and Day6O) were pooled equally, and the mRNA-seq libraries were then prepared using 5 μg of pooled total RNA and the TruSeqTM RNA sample preparation Kit (Illumina, San Diego, CA). First, mRNA was isolated with oligo(dT) beads and then fragmented to 100–400 bp by fragmentation buffer. Second, double-stranded cDNA was synthesized using a SuperScript double-stranded cDNA synthesis kit (Invitrogen, CA) with random hexamer primers (Illumina). Then the synthesized cDNA was subjected to end-repair, phosphorylation, and ‘A’ base addition according to Illumina’s library construction protocol. The libraries were amplified by PCR for 15 cycles using Phusion DNA polymerase (NEB), and then target cDNA fragments of 200–300 bp were selected using 2 % Low Range Ultra Agarose gel. After quantification by TBS380, paired-end sequencing of 101 bp reads was performed for the four cDNA libraries in one lane on an Illumina HiSeq2500 high-throughput sequencer.
De-novo assembly and annotation
The raw paired-end reads were trimmed and quality controlled by filter fq (BGI internal software) to remove reads with adaptors, reads with unknown nucleotides larger than 5 % and low-quality reads (the rate of reads in which the quality value ≦ 10 was more than 20 %). Then the clean data from samples (Testis, PreO, Day1O, Day6O) were used to perform de-novo assembly with Trinity (https://github.com/trinityrnaseq/trinityrnaseq/wiki; version Trinityrnaseq_r2013-02-25) and with min_kmer_cov set to 2 and all other parameters set to default [41]. The longest- assembled sequences per gene model were called contigs. Then the reads were mapped back to the contigs, as with paired-end reads we are able to detect contigs from the same transcript as well as the distances between these contigs. Finally, we retrieved sequences without Ns and these could not be extended at either end. Such sequences were thereby defined as Unigenes. TGICL (http://sourceforge.net/projects/tgicl/files/tgicl%20v2.1/; v2.1) [42] was used to further assemble all the unigenes from different samples to form a single set of non-redundant unigenes (called all-unigenes). The all-unigenes displaying >70 % sequence identities were grouped into a cluster, in which the prefix is CL, and the cluster ID is locater after. And the others were singletons whose the prefix was Unigene.
The completeness of the transcriptome assembly was tested by Core Eukaryotic Genes Mapping Approach (CEGMA) software (cegma_v2.4.010312, using the default parameters), by comparing known 248 Core Eukaryotic Genes (CEGs) and the transcripts assembled by Trinity. CEGMA was developed to identify a subset of 248 highly conserved core eukaryotic genes (CEGs) deriving from six diverse model organisms in eukaryotic genomes [43].
All non-redundant unigene sequences were searched against protein databases (Nr, SwissProt, KEGG, COG.) using blastx (evalue < 0.00001) and the nucleotide database NT (e-value < 0.00001) by blastn (evalue < 0.00001) (http://blast.ncbi.nlm.nih.gov/Blast.cgi; v2.2.26 + x64-linux). Protein function information was predicted from annotation of the most similar protein available in the databases.
BLAST2GO (http://www.blast2go.com/b2ghome; v2.5.0, release 2012-08-01) [44] program was used to retrieve GO annotations of unigenes for describing biological processes, molecular functions and cellular components. Metabolic pathway analysis was performed using online KEGG Automatic Annotation Server (http://www.genome.jp/; BGI internal version, Release 63.0) [45].
Identification of sex-specifically expressed and differentially expressed genes
SOAPaligner/soap2 (http://soap.genomics.org.cn/) was used to map the reads to the assembled transcriptome. Unique mapped reads including paired-end reads for which only one part matched, were used to calculate the level of gene expression using the fragments per kb per million fragments method (FPKM) [46]. The method edgeR was used to identify differentially expressed genes (DEGs) between two samples [47]. The threshold for the P-value was determined by the false-discovery rate (FDR). Unigenes with FDR ≤ 0.001 and ratio of FPKMs of the two samples larger than 2 (genes for which FPKM < 1 were filtered) were considered to be differentially expressed genes in this study.
In addition, functional-enrichment analysis was performed to identify which GO terms and metabolic pathways were significantly enriched in DEGs. Hypergenometric test and 0.05 cutoff P adjusted were used for analysis of the enrichment of functional terms. GO functional enrichment and KEGG pathway analysis were carried out using Goatools (https://github.com/tanghaibao/Goatools) and KOBAS (http://kobas.cbi.pku.edu.cn/home.do) [48].
Simple sequence repeat (SSR) detection
SSRs were detected among the unigenes using MISA software (MIcroSAtellite; http://pgrc.ipk-gatersleben.de/misa/misa.html; version 1.0). Six types of SSRs were investigated: mono-, di-, tri-, quad-, penta-, and hexa-nucleotide repeats; and we kept the SSRs in which the lengths at both ends of the Unigene were more than 150 bp for primer design by Primer 3 (http://www.onlinedown.net/soft/51549.htm; Release 2.3.4).
Semi-quantitative and quantitative real-time PCR validation
Quantitative and semi-quantitative real-time PCR were used to verify sex differentially expressed genes as identified from the gonadal transcriptome.
The sequences of 47 up-regulated testicular and 57 up-regulated ovarian unigenes were chosen for primer design using Array Designer 4 (http://premierbiosoft.com/dnamicroarray/index.html). The β-actin (BQF: 5′-GTGTGACGACGAAGTAGC-3′, BQR: 5′-GATACCTCGCTTGCTCTG-3′) was used as a reference gene. Total RNAs were reverse-transcribed with Goscript™ reverse transcription system (Promega, U.S.) according to the manufacturer’s instructions.
Primers for Testis-specific (FPKM of PreO = 0 and FDR ≦ 0.001) and PerO-specific (FPKM of Testis = 0 and FDR ≦ 0.001) unigenes were first amplified by semi-quantitative PCR using equivalent cDNAs from testis and ovary of pre-eyestalk ablation shrimp as templates. PCR products were analyzed with gel electrophoresis using 1.2 % agarose gel. When a gene was found to be expressed only in testis, the gene was considered to be a testis-specific gene, while a gene that was expressed only in PreO was considered to be an ovary-specific gene. For the genes that were expressed in both ovary and testis, quantitative real-time PCR was further conducted to verify whether they were differentially expressed in ovaries and testes. The primers for other DEGs were also verified by quantitative real-time PCR.
The quantitative real-time PCR was performed in the Applied Biosystems 7500 fast real-time system using THUNDERBIRD qPCR Mix (TOYOBO, Japan) as recommended by the manufacturer. The original cDNAs were diluted 100-fold for the target gene and β-actin amplification, and the PCR cycle for both the target genes and the β-actin was as follows: 3 min at 94 °C; then 36 cycles of 15 s at 94 °C, 30 s at 56 °C and 30 s at 72 °C; followed by 10 min at 72 °C. The PCRs used to detect all the target genes and β-actin reference gene were performed with three biologic replicates. The specificity of the amplification was assessed by a melting curve analysis to exclude primers with nonspecific amplification peaks. The relative expression level of target genes was calculated with the 2−ΔΔCT method [49]. The Student’s t-test was conducted using SPSS 23.0 (http://www-01.ibm.com/software/analytics/spss/), and significant differences were determined at a P-value < 0.05 (two-tailed test).
Results
Sequence analysis and assembly
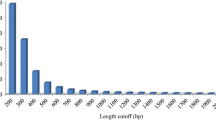
A total of 25.16 gigabases (Gb) of sequences were generated from four L. vannamei gonadal tissue libraries (The raw reads data can be obtained from the NCBI Short Read Archive [SRA] under accession number SRA SRP059164.) After quality control with filter_fq, 63,782,344, 60,300,542, 58,195,380, and 61,048,948 clean reads were retrieved from the testis, PreO, Day1O and Day6O library, respectively. De-novo assembly of all the clean reads from four libraries generated a total of 65,218 unigenes with a mean size of 1021 bp and N50 of 2,000 bp (Table 1) (The assembled transcriptome can be obtained from the NCBI Transcriptome Shotgun Assembly (TSA) Database under accession number GDUV00000000). The length distribution of the unigenes obtained is illustrated in Fig. 1. The completeness of the transcriptome assembly was tested by CEGMA software by comparing known 248 Core Eukaryotic Genes (CEGs) and the transcripts assembled by Trinity. As a result, 234 out of 248 (94.35 %) CEGs were deemed to be complete proteins in the transcriptome, and 245 out of 248 (98.79 %) CEGs were found in the transcriptome including some partial proteins (Table 2), indicates a high level of completeness of the transcriptome.

Length distribution of all-unigenes. X-axis, size distribution of unigenes; Y-axis, number of unigenes in different length ranges
Sequence annotation
All-unigene sequences were searched against Nr, SwissProt, KEGG, COG and NT databases, which returned 26,482 (40.61), 23,062 (35.36), 20,659 (31.68), 11,935 (18.30), and 14,626 (22.43 %) matches, respectively; providing a final total of 30,304 annotated unigenes (46.47 %) (Table 3).
The 20,659 unigenes with a KO annotation were mapped to 258 pathways (Additional file 1: Table S1). The top 3 pathways were metabolic pathways (1,517 unigenes), RNA transport (562 unigenes), and regulation of actin cytoskeleton (501 unigenes). Importantly, the main biological pathways involved in germ cell meiosis during gonadal development were cell cycle (ko04110, 233 unigenes), DNA replication (ko03030, 61 unigenes), mismatch repair (ko03430, 41 unigenes), base excision repair (ko03410, 73 unigenes), oocyte meiosis (ko04114, 141 unigenes) and homologous recombination (ko03440, 41). The primary pathways involved in oogenesis, spermatogenesis and gonadal maturation were MAPK signaling pathway (ko04010, 309 unigenes), GnRH signaling pathway (ko04912, 118 unigenes), progesterone-mediated oocyte maturation (ko04914, 153 unigenes), focal adhesion (ko04510, 412 unigenes), calcium signaling pathway (ko04020, 229 unigenes), ubiquitin mediated proteolysis (ko04120, 344 unigenes) and wnt signaling pathway (ko04310, 228 unigenes). Further investigation of these pathways would be expected to reveal the regulatory mechanisms governing reproductive processes in L. vannamei.
Sex-biased gene identification and enrichment analysis
Comparison of gene expression levels in testis and different ovarian stages revealed similar numbers of differentially expressed genes (testis vs. PreO, 22,808 DEGs; testis vs. Day1O, 22,168 DEGs; testis vs. Day6O, 22187 DEGs), and over 84 % of the DEGs between testis and different ovarian stages are always the same (Fig. 2); and unigenes of testis and PreO libraries were selected for further analysis. Among 45,998 unigenes expressed in testis and ovary with lengths > 200 bp and FPKM > 1, 22,808 (45.6 %) showed sex-biased expression (Additional file 1: Table S2); 3,529 unigenes were up-regulated in the ovary and 19,279 were up-regulated in the testis (Fig. 2). Interestingly, the number of male-biased genes was much greater than that for female-biased genes, and the testicular up-regulated genes showed a greater average in fold-change than did the female-biased transcripts (Calculating from data in Additional file 1: Table S2, the average FC for male-biased and female-biased genes was 106.9 and 21.1, respectively).

Statistical evaluation of differentially expressed genes between testis and ovaries that were obtained at different points in time. a: Venn diagram of DEGs that showed more expression in the testis than in the ovaries; b: Venn diagram of DEGs that showed more expression in the ovaries than in the testis
Gene ontology (GO) annotation was performed to classify sex-biased genes (Fig. 3). Results also showed a much higher number of male-biased genes than female-biased genes. Most of the GO terms showed significantly higher counts for male-biased genes. At the molecular function level, there were significantly more DEGs in catalytic activity and binding GO terms than in other terms. The cell part and cell GO terms had greatest DEG counts at the cellular component level. At the biological process level, cellular processes, metabolic processes, and single-organism processes had the top counts. By enrichment analysis, there were 49, 53, and 312 enriched GO-terms for male-biased DEGs, and 111, 155, and 380 enriched GO-terms for female-biased DEGs, in the cellular component, the molecular function and the biology process categories, respectively (Additional file 1: Table S3). The most enriched GO-terms for male-biased DEGs included DNA-directed RNA polymerase in the cellular component category, RNA polymerase activity in the molecular function category and isoprenoid biosynthetic and metabolic process in the biology process category (Table 4). As for female-biased DEGs, the most enriched GO-terms at cellular component level was mitochondrion and ribosome, the most enriched GO-terms associated with molecular function were aminoacyl-tRNA ligase activity, and the most enriched biology process were translation, cellular biosynthetic process and organic substance biosynthetic process (Table 4).

Distribution of DEGs among GO terms in biological processes, molecular functions, and cellular components
Enriched pathways (Table 4) associated with male-biased DEGs were mRNA surveillance pathway, RNA transport, ABC transporters, terpenoid backbone biosynthesis etc. And enriched pathways for female-biased DEGs were oxidative phosphorylation, metabolic pathways, aminoacyl-tRNA biosynthesis, steroid biosynthesis, ribosome biogenesis in eukaryotes, DNA replication, vasopressin-regulated water re-absorption, neuroactive ligand-receptor interaction, cell cycle, nucleotide excision repair etc.
According to the enrichment analysis results, transcription was the most enriched activity in testis and translation and mitochondrion synthesis were the most enriched activities in ovary. Further studies on DEGs associated with the GO terms and pathways are needed to reveal the different molecular mechanisms that apparently exist between ovarian and testicular developmental processes.
Real-time PCR confirmation of sex-biased genes
One hundred and four DEGs with different levels of FPKM and FDR values were selected for RT-PCR validation to determine the veracity of the transcriptomic analysis. In male-biased genes, 6 of 14 testis-specific genes showed a specific testicular expression pattern by semi-quantitative PCR (Fig. 4), and the other 8 genes were significantly up-regulated in the testis as assessed with quantitative real-time PCR (Table 5). For the 33 male-biased DEGs, 32 showed the significantly up-regulated expression in testis, and only one gene was up-regulated in ovary (Table 5). In the female-biased genes selected, 9 of 14 PreO-specific genes in the ovary showed a specific expression pattern by semi-quantitative PCR (Fig. 5), and the other 5 genes were significantly up-regulated in ovary (Table 6). For the 43 female-biased DEGs, 33 showed up-regulated ovarian expression, 5 showed up-regulated testicular expression and 5 did not show expression differences between ovary and testis (Table 6). In total, 9 ovary-specific, 6 testis-specific, 45 testicular up-regulated and 39 ovarian up-regulated unigenes were obtained by real-time PCR confirmation.

Testis-specific genes. M, DNA marker DL2000; T, testis; O, ovary. TS are the series numbers for testis-specific genes from bioinformatics analysis

Ovary-specific genes. M, DNA marker DL2000; T, testis; O, ovary. OS are the series numbers for ovary-specific genes from bioinformatics analysis
SSRs in the gonadal transcriptome of L. vannamei SSR detection was performed with the MicroSAtellite (MISA) software using all-unigenes as references. A total of 13,233 SSRs were identified in 10,411 unigene sequences, with 2,192 (21.05 %) unigenes containing more than one SSR. Among the different types of SSRs, the di-nucleotide repeats were the most abundant, accounting for 41.83 %, followed by the tri-nucleotide repeats (28.25 %), mono-nucleotide repeats (24.79 %), quad-nucleotide repeats (2.86 %), penta-nucleotide repeats (1.23 %) and hexa-nucleotide repeats (1.04 %) (Fig. 6). The distributions of SSRs in unigenes and primers for SSRs are shown in Additional file 1: Table S6.

Distribution of putative SSRs in the transcriptome of the L. vannamei gonad. X-axis, distribution of SSR types; Y-axis, number of different SSR types
Discussion
L. vannamei is one of the most important aquaculture species and has attracted the attention of many researchers. A high-throughput RNA sequencing strategy has already been used in studying the mechanisms of viral resistance [50–53] and nitrite adaptation [54], as well as in revealing gene expression patterns in muscle, hepatopancreas, gills and pleopods of normal shrimp [55, 56]. However, data regarding its reproductive or sex phenotypes is lacking. Herein, we performed RNA-Seq on the gonads of adult males and females in an attempt to unravel sex-related genes. The reason we chose adult individuals for gonadal transcriptomic analysis was that we focused primarily on genes involved in gonadal development and gametogenesis. Moreover, these cell types showed more diversity in adult gonads; for example, oogonia/spermatogonia, and primary and secondary oocytes/spermatocytes are all present in adult gonads (Additional file 1: Figure S1). This stage selection was appropriate since we identified in our assembled sequences a great majority of genes involved in gonadal development, oogenesis, and spermatogenesis, as well as some sex-determining genes previously reported.
Reference gonadal transcriptome of L. vannamei
De-novo assembly of the sequencing data from four L. vannamei gonadal cDNA libraries resulted in 65,218 unigenes with an average length of 1021 bp. Approximately half of the unigenes (46.47 %) had significant matches against existing sequences and 40.65 % were annotated using Gene Ontology terms. The L. vannamei sequences possessed top matches with D. pulex sequences, since this latter species is also a crustacean and its entire genome has been sequenced [57]. KEGG pathway analysis showed that, the main biological pathways involved in gonadal development and gametogenesis were obtained, which will facilitate the further in-depth analysis of the relationships between different genes in the transcriptome of gonads. However, there were far fewer pathways involved in spermatogenesis than in oogenesis, and more female-biased genes (75.6 %) than male-biased genes (37.9 %) showed NR hits. This could be due to the fact that researchers have mainly focused on the reproductive phenotypes of female crustaceans because of their economic importance to aquaculture [15, 26]; thus, genetic information for female crustaceans has been much richer than for males.
Sex-determining genes
The transcription factor, sex-determining region of the Y-chromosome (Sry), is the sex-determining gene in mammalians. The correct expression of Sry triggers testicular development, while decreased or delayed Sry expression leads to testicular defects [58]. Sry acts by activating the Sox9 gene [59], and then, downstream genes of Sox9 promote testis development. Once expression of sox9 or its downstream genes is impeded, the gonad can switch to ovarian development [60]. Doublesex (Dsx) is a male sex-determining gene that plays an important role in controlling sexual dimorphism in invertebrate organisms such as nematodes, insects and Daphnia magna, as well as in vertebrates [61–63]. Fem-1 is also a male sex-determining gene required for somatic and germline development in the testis of C. elegans [64, 65]. Herein, we identified all male sex-determining genes mentioned above in L. vannamei, including a testicular up-regulated Sry (Unigene7231_All), sox9 (CL86.Contig2_All, Unigene2458_All), Dsx (Unigene35364_All) and seven fem-1 or fem-1-like genes (Unigene24042_All, Unigene9434_Al, Unigene9463_All, Unigene24047_All, Unigene11407_All, Unigene2036_All, Unigene15025_All).
Foxl2 (Forkhead box l2) encodes a conserved transcription factor and is a sex determiner in female vertebrates. It is preferentially expressed in the ovary and involved in ovarian differentiation and maintenance by repression of testis-specific genes [66, 67]. Although foxl2 orthologs have been cloned in some invertebrates, its role in sex determination or differentiation remains unclear [68]. We found foxl2 (Unigene24037_All) in our gonadal transcriptomic data, and intriguingly, it shows up-regulated expression in testis.
The identification of known sex-determining genes in the present study proved that transcriptomic sequencing is a powerful tool in mining sex-determining genes. The exact role of these genes in sex determination of L. vannamei remains to be examined.
Identification of genes involved in germline determination and development
Gonadal tissue is composed of germ cells and somatic cells. Germ cells are the only cells that can undergo both meiosis and mitosis, and finally differentiate into gametes (spermatozoon and oocyte); and sperm-oocyte binding in sexually reproducing organisms constitutes the beginning of the next generation. To maintain germ cell fate and regulate its development and differentiation, gene expression patterns in germ cells need to be regulated at the transcriptional and post-transcriptional levels. Moreover, it is also important to maintain the niche formed by somatic cells so as to maintain the characteristics of germ cells. Transcriptional regulation of gene expression is widely used in early differentiation stages of germ cells. In mouse, interaction of blimp1 (B-lymphocyte-induced maturation protein 1) with Prmt (histone methyltransferase) represses chromatin transcription and prevents trans-differentiation from germ cells to somatic cells. In the blimp1-knockout mouse, germ cells cannot form [69–72]. In the present study, a blimp1 (Unigene15201_All), three Prmt (PRMT2: Unigene7507_All, PRMT3: CL2301.Contig2_All, CL2301.Contig1_All, PRMT6: Unigene14757_All), and a chromatin target of Prmt1 protein genes (Unigene4147_All) were identified, indicating a similar regulation of germ cell fate at the transcriptional level in shrimp.
The regulation of RNA translation, however, is more common and important in germ cells. A battery of evolutionally conserved RNAs is essential for germ cell proliferation, survival and differentiation; some of these genes are called germ cell markers, because they exist throughout the germ cell life cycle. Vasa, first isolated in Drosophila, is expressed throughout germ cell developmental stages in many invertebrates and vertebrates species [73, 74]. As a molecular marker, vasa was used to track germ cell specification, migration and differentiation [75–77]; and loss of function of vasa led to germ cell specification failure and defects in germ cell development in Drosophila, mouse and nematode [78–81]. Vasa were hypothesized to function by inhibiting expression of genes that are responsible for somatic differentiation in germ cells [82]. This hypothesis was further supported by studies of nanos, a target of vasa, which can act together with pumilio to repress mitosis, transcription and translation during the development of the germline [83–85]. The DAZ (Deleted in Azoospermia) family is another well-known gene family that consists of three genes specifically expressed in germ cells: BOULE, DAZ-Like (DAZL) and DAZ. DAZ and DAZL are expressed throughout almost the entire life cycles of germ cells, and are essential for germ cell determination, differentiation and maturation; while BOULE is mainly involved in germ cell meiosis [86–89]. In the present study, vasa (Unigene1169_All), nanos (Unigene22266_All), pumilio (CL285.Contig16_All), boule (Unigene1947_All) and three DAZ-associated protein (daz interacting protein 1, Unigene13632_All; DAZ-associated protein 1-like, Unigene20_All; DAZ-associated protein 2, Unigene4428_All) were identified, offering abundant genetic information for further study of germline determination and development.
miRNAs are also involved in RNA regulation of germ cells. Knockout of the Dicer gene prevented splicing of miRNAs--the regulator of germ cell-specific genes-- leading to the termination of germ cell development [90]. Additionally, the PIWI-mediated piRNA pathway regulates the expression of germ cell determination genes, such as vasa and oskar. The PIWI-null mutant fly (Drosophila) and mouse exhibited a deficiency in germ cell formation and differentiation [91, 92]. The existence of Dicer (Unigene17990_All) and two piwi (piwi 1, Unigene399_All; piwi 2, Unigene21063_All) in our transcriptome made miRNA-mediated regulation in L. vannamei possible.
The gonadal cell niche surrounding germ cells is also important for the preservation of germ cell fate [93]. For example, Notch signaling and its downstream genes in nematode [94], BMPs (bone morphogenetic proteins) in Drosophila [95] and GDNF (glial cell line-derived neurotrophic factor, GDNF) in mammals are produced by gonadal somatic cells [96] such as Sertoli cells, and are the known pathway or factors that might control meiotic timing and germ cell numbers. We retrieved two notch genes (Notch, Unigene14535_All; Notch2, Unigene14705_All); two BMP genes (BMP1, Unigene14770_All; BMP7, Unigene12011_All); and two GDNF associated genes (GDNF-inducible zinc finger protein 1, Unigene4392_All; GDNF family receptor alpha-3, Unigene7144_All) in the gonadal transcriptome of L. vannamei. Moreover, 101 sequences for genes in the Wnt signaling pathway were also obtained. This information will further benefit the study of the mechanisms of interaction between germ cells and somatic cells in L. vannamei.
Sex-biased genes involved in spermatogenesis and oogenesis are identified
Except for similar cell lines in both male and female gonads, there are remarkable differences between the ovary and testis that involve morphology, cell types and biologic processes; and also gene expression patterns and molecular regulatory mechanisms. A large number of gonadal differentially expressed genes have been identified in the present transcriptome, and many more genes were over-expressed in the testis (19,279) compared to the ovary (3,529). This male-biased gene expression pattern in the gonads has been observed in Drosophila [97, 98], Caenorhabditis elegans [99], fishes [100, 101] and mammals [102], as well as in the green mud crab (Scylla paramamosain) [36], a crustacean species. This phenomenon may be explained by the previous assumption that male development is regulated by activating a series of testis-specific genes and/or by repressing genes vital for ovarian development [103, 104].
The most important biologic processes in the testis and ovary are spermatogenesis and oogenesis, respectively. As expected, testis-specific and differentially expressed genes include members with functions pertinent to spermatogenic stages. For example, the male-biased gene spermatogonial stem-cell renewal factor (CL2114.Contig2_All), MLH1 (CL2662.Contig1_All, CL2662.Contig2_All) and RHAU (Unigene22544_All) are essential for early stages of spermatogenesis, including spermatogonial maintenance and differentiation; and knockout of RHAU or MLH1 resulted in deficiencies in spermatogonial or spermatocytic differentiation in the mouse [105–107]. Genes involved in later stages of spermatogenesis include Spermatogenesis-associated proteins (Spata2, Unigene8309_All; Spata5 (CL1808.Contig1_All, CL1808.Contig2_All); Spata20, Unigene9757_All), spermatogenesis regulator (Unigene14897_All); sperm associated antigens ciliary and flagellar proteins such as Sperm-associated antigen (Spag2, Unigene7744_All; Spag7, Unigene482_All); Sperm-specific protein PHI-2B/PHI-3 (Unigene12515_All); Sperm protamine P2 (Unigene20056_All); Spermatid-specific protein T1 (Unigene12459_All, Unigene17251_All); Round spermatid basic protein 1 (Unigene6458_AlL); major sperm protein (Unigene9914_All); and Motile sperm domain-containing protein 2 (Unigene9914_All). In addition, the testis-biased genes testis-specific serine proteases (TESSP) (Unigene17312_All, Unigene2408_All, Unigene25019_All) are involved in germ cell survival during meiosis [108, 109], and T-complex testis-specific protein 1(t-complex 11) (Unigene4999_All) might function in insemination processes so as to stimulate sperm capacitation and inhibit the acrosome reaction [110].
Among the female-biased sequences, we focused on genes involved in oogenic processes. Vitellogenesis is the central process in oogenesis. In crustaceans, vitellogenin--the precursor of yolk protein--is synthesized in the hepatopancreas and ovary [111, 112]. The synthesis of vitellogenin in oviparous vertebrates is regulated by the E2-ER-Hsp90-Vtg pathway, in which estrogen receptor (ER) and heat shock protein 90 (Hsp90) mediate the enhancement of vitellogenin transcription by estrogen or estrogen-like hormones [113, 114]. In our transcriptomic data, we identified vitellogenin (CL1767.Contig3_All), vitellogenin receptor (VgR) (Unigene2102_All), estrogen receptor (CL2390.Contig2_All, Unigene9144_All), estrogen receptor-binding protein (Unigene2782_All) and HSP90 (Unigene15703_All) genes, indicating that an E2-ER-Hsp90-Vtg pathway exists in crustaceans. Further experiments should focus on whether and how E2, ER and Hsp90 regulate the synthesis of vitellogenin in shrimp.
The second meiotic division and oocyte maturation constitute another vital process in oogenesis. MPF (maturation-promoting factor), mainly composed of CDK1 (encoded product of the Cdc2 gene) and Cyclin B proteins, is a primary regulator of this process. MPF can be activated by G2 to reach the M phase transition, and the active MPF promotes rapid maturation of oocytes [115]. In the present study, we identified G2/mitotic-specific cyclin-A (Unigene10629_All), cyclin-B (Unigene4127_All, Unigene1805_All), cyclin-B3 (CL352.Contig2_All), cyclin-F (Unigene11044_All); and the components of MPF, Cdc2 (Unigene3813_All) and Cyclin B (CL2390.Contig3_All). The identification of these genes will facilitate further study on the artificial induction of oocyte maturation in L. vannamei. In addition, oocyte quality is hypothesized to be improved by high levels of the expression of superoxide dismutase genes, which could neutralize reactive oxygen species and protect the embryo during its development [116]. Herein, we found four superoxide dismutase homologs (Unigene14142_All, Unigene419_All, Unigene6388_All, Unigene8984_All) that are predominantly expressed in the ovary, indicating a similar maternal protection of offspring by shrimp; and antioxidant defense techniques may be useful in improving the overall quality of shrimp larvae.
Verification of sex-biased genes
Among 104 differentially expressed gonadal unigenes, 9 genes specifically expressed in ovary and 6 specifically expressed in testis were verified by semi-quantitative RT-PCR; while of the remaining DEGs tested, 45 testis-predominant unigenes and 39 ovary-predominant unigenes were verified by quantitative real-time PCR analysis. These gonadal specific and differentially expressed genes must play important roles in certain parts of gonadal development in L. vannamei. However, there are some differences observed between transcriptomic analysis and qRT-PCR data concerning fold-changes in gene expression in testis and ovary, and one male-biased DEG showed ovarian up-regulated expression and 5 female-biased DEGs showed testis-predominant expression. This is most likely due to the differences that often existing between the bioinformatics analysis of next-generation sequencing technology and the actual experimental analysis [117]; and biologic replicates (which are more valuable and accurate for detecting differently expressed genes), were not available during the course of the present study [118].
Discovery of SSR markers
Simple sequence repeats (SSRs) are single-locus markers with high allelic variation and are widely applicable to molecular genetics studies, including research involving genetic diversity assessment, comparative genomics, gene flow characterization, and genetic linkage mapping [119]. In L.vannamei, a number of microsatellite sequences have been reported for genetic map construction and quantitative trait locus (QTL) detection [120–122], and next-generation sequencing has facilitated the discovery of a relatively large set of SSRs in the hepatopancreas of the shrimp by the present team [53, 123]. The SSRs identified in this study can serve as genetic markers for quantifying genetic diversity of germplasm within breeding and imported populations, identifying paternity of the breeding families and managing broodstocks, which are the major issues for the selectively breeding and farming of introduced species. They can also be used in QTL mapping and marker-assisted select ion (MAS) in L. vannamei and other phylogenetically similar shrimp species. This may promote genetic gain to traits of interest for aquaculture, such as reproduction, sex determination, growth, and tolerance against environmental stress.
Conclusions
The present study encompasses the first large-scale RNA sequencing of shrimp gonads. We have identified many important sex-related functional genes, GO terms and pathways, all of which will facilitate future research into the reproductive biology of shrimp. The SSRs detected in this study can be used as genetic markers for germplasm evaluation of breeding and imported populations.
Abbreviations
- cDNA:
-
complementary DNA
- Day1O:
-
ovary of one-day post-eyestalk ablation female shrimp
- Day6O:
-
ovary of six-day post-eyestalk ablation female shrimp
- DEGs:
-
differentially expressed genes
- FDR:
-
false discovery rate
- FPKM:
-
fragments per kilo base per million
- GO:
-
gene ontology
- NR:
-
NCBI non-redundant protein database
- PreO:
-
Ovary of pre-eyestalk ablation female shrimp
References
Penaeus vannamei (Boone, 1931). Species Fact Sheets. Food and Agriculture Organization. Retrieved June 8, 2011.
Seafood Red list. Greenpeace. Retrieved 6 August, 2012.
Penaeus vannamei (Boone, 1931). Cultured Aquatic Species Information Programme. Food and Agriculture Organization. Retrieved June 8, 2011.
Penaeus vannamei (Boone, 1931). Species Fact Sheet. Food and Agriculture Organization. Retrieved June 2012.
Bartholomay LC, Loy DS, Dustin Loy J, Harris DL. Nucleic-acid based antivirals: augmenting RNA interference to ‘vaccinate’Litopenaeus vannamei. J Invertebr Pathol. 2012;110(2):261–6.
Flegel TW. Historic emergence, impact and current status of shrimp pathogens in Asia. J Invertebr Pathol. 2012;110(2):166–73.
Li F, Xiang J. Recent advances in researches on the innate immunity of shrimp in China. Dev Comp Immunol. 2013;39(1–2):11–26.
Lightner DV. Virus diseases of farmed shrimp in the Western Hemisphere (the Americas): a review. J Invertebr Pathol. 2011;106(1):110–30.
Lightner DV, Redman RM, Pantoja CR, Tang KF, Noble BL, Schofield P, et al. Historic emergence, impact and current status of shrimp pathogens in the Americas. J Invertebr Pathol. 2012;110(2):174–83.
Zhou J, Qiu L, Jiang S, Zhou F, Huang J, Yang L, et al. Molecular cloning and mRNA expression of M-phase phosphoprotein 6 gene in black tiger shrimp (Penaeus monodon). Mol Biol Rep. 2013;40(2):1301–6.
Phinyo M, Visudtiphole V, Roytrakul S, Phaonakrop N, Jarayabhand P, Klinbunga S. Characterization and expression of cell division cycle 2 (Cdc2) mRNA and protein during ovarian development of the giant tiger shrimp Penaeus monodon. Gen Comp Endocrinol. 2013;193(1):103–11.
Visudtiphole V, Klinbunga S, Kirtikara K. Molecular characterization and expression profiles of cyclin A and cyclin B during ovarian development of the giant tiger shrimp Penaeus monodon. Comp Biochem Physiol A Mol Integr Physiol. 2009;152(4):535–43.
Treerattrakool S, Panyim S, Chan SM, Withyachumnarnkul B, Udomkit A. Molecular characterization of gonad-inhibiting hormone of Penaeus monodon and elucidation of its inhibitory role in vitellogenin expression by RNA interference. FEBS J. 2008;275(5):970–80.
Ponza P, Yocawibun P, Sittikankaew K, Hiransuchalert R, Yamano K, Klinbunga S. Molecular cloning and expression analysis of the Mitogen-activating protein kinase 1 (MAPK1) gene and protein during ovarian development of the giant tiger shrimp Penaeus monodon. Mol Reprod Dev. 2011;78(5):347–60.
Wimuttisuk W, Tobwor P, Deenarn P, Danwisetkanjana K, Pinkaew D, Kirtikara K, et al. Insights into the prostanoid pathway in the ovary development of the penaeid shrimp Penaeus monodon. PLoS One. 2013;8(10), e76934.
Talakhun W, Khamnamtong B, Nounurai P, Klinbunga S, Menasveta P. Characterization, expression and localization of valosin-containing protein in ovaries of the giant tiger shrimp Penaeus monodon. Gene. 2014;533(1):188–98.
Li S, Li F, Wen R, Xiang J. Identification and characterization of the sex-determiner transformer-2 homologue in Chinese shrimp, Fenneropenaeus chinensis. Sex Dev. 2012;6(5):267–78.
Ren Q, Zhou J, Zhao XF, Wang JX. Molecular cloning and characterization of a receptor for activated protein kinase C1 (RACK1) from Chinese white shrimp; Fenneropenaeus chinensis. Dev Comp Immunol. 2011;35(6):629–34.
Wen R, Li F, Xie Y, Li S, Xiang J. A homolog of the cell apoptosis susceptibility gene involved in ovary development of Chinese shrimp Fenneropenaeus chinensis. Biol Reprod. 2011;86(1):1–7.
Aflalo ED, Bakhrat A, Raviv S, Harari D, Sagi A, Abdu U. Characterization of a vasa-like gene from the pacific white shrimp Litopenaeus vannamei and its expression during oogenesis. Mol Reprod Dev. 2007;74(2):172–7.
Garcia-Orozco KD, Vargas-Albores F, Sotelo-Mundo RR, Yepiz-Plascencia G. Molecular characterization of vitellin from the ovaries of the white shrimp Penaeus (Litopenaeus) vannamei. Comp Biochem Physiol B Biochem Mol Biol. 2002;133(3):361–9.
Tinikul Y, Poljaroen J, Nuurai P, Anuracpreeda P, Chotwiwatthanakun C, Phoungpetchara I, et al. Existence and distribution of gonadotropin-releasing hormone-like peptides in the central nervous system and ovary of the Pacific white shrimp, Litopenaeus vannamei. Cell Tissue Res. 2011;343(3):579–93.
Zhang LYC, Zhang Y, Li L, Zhang X, Zhang Q, Xiang J. A genetic linkage map of Pacific white shrimp (Litopenaeus vannamei): sex-linked microsatellite markers and high recombination rates. Genetica. 2006;131(1):37–49.
Leelatanawit R, Klinbunga S, Aoki T, Hirono I, Valyasevi R, Menasveta P. Suppression subtractive hybridization (SSH) for isolation and characterization of genes related to testicular development in the giant tiger shrimp Penaeus monodon. BMB Rep. 2008;41(11):796–802.
Xie Y, Li F, Wang B, Li S, Wang D, Jiang H, et al. Screening of genes related to ovary development in Chinese shrimp Fenneropenaeus chinensis by suppression subtractive hybridization. Comp Biochem Physiol Part D Genomics Proteomics. 2010;5(2):98–104.
Cui J, Wu LT, Chu KH. Comparative proteomic profiling during ovarian development of the shrimp Metapenaeus ensis. Mol Biol Rep. 2014;41(1):519–28.
Talakhun W, Phaonakrop N, Roytrakul S, Klinbunga S, Menasveta P, Khamnamtong B. Proteomic analysis of ovarian proteins and characterization of thymosin-beta and RAC-GTPase activating protein 1 of the giant tiger shrimp Penaeus monodon. Comp Biochem Physiol Part D Genomics Proteomics. 2014;11:9–19.
Wongsurawat T, Leelatanawit R, Thamniemdee N, Uawisetwathana U, Karoonuthaisiri N, Menasveta P, et al. Identification of testis-relevant genes using in silico analysis from testis ESTs and cDNA microarray in the black tiger shrimp (Penaeus monodon). BMC Mol Biol. 2010;11(1):55.
Leelatanawit R, Sittikankeaw K, Yocawibun P, Klinbunga S, Roytrakul S, Aoki T, et al. Identification, characterization and expression of sex-related genes in testes of the giant tiger shrimp Penaeus monodon. Comp Biochem Physiol A Mol Integr Physiol. 2009;152(1):66–76.
Preechaphol R, Leelatanawit R, Sittikankeaw K, Klinbunga S, Khamnamtong B, Puanglarp N, et al. Expressed sequence tag analysis for identification and characterization of sex-related genes in the giant tiger shrimp Penaeus monodon. J Biochem Mol Biol. 2007;40(4):501–10.
Leelatanawit R, Klanchui A, Uawisetwathana U, Karoonuthaisiri N. Validation of reference genes for real-time PCR of reproductive system in the black tiger shrimp. PLoS One. 2012;7(12), e52677.
Leelatanawit R, Uawisetwathana U, Klinbunga S, Karoonuthaisiri N. A cDNA microarray, UniShrimpChip, for identification of genes relevant to testicular development in the black tiger shrimp (Penaeus monodon). BMC Mol Biol. 2011;12:15.
He L, Jiang H, Cao D, Liu L, Hu S, Wang Q. Comparative transcriptome analysis of the accessory sex gland and testis from the Chinese mitten crab (Eriocheir sinensis). PLoS One. 2013;8(1):e53915.
Meng XL, Liu P, Jia FL, Li J, Gao BQ. De novo Transcriptome Analysis of Portunus trituberculatus Ovary and Testis by RNA-Seq: Identification of Genes Involved in Gonadal Development. PLoS One. 2015;10(6):e0128659.
Song YN, Shi LL, Liu ZQ, Qiu GF. Global analysis of the ovarian microRNA transcriptome: implication for miR-2 and miR-133 regulation of oocyte meiosis in the Chinese mitten crab, Eriocheir sinensis (Crustacea:Decapoda). BMC Genomics. 2014;15:547.
Gao J, Wang X, Zou Z, Jia X, Wang Y, Zhang Z. Transcriptome analysis of the differences in gene expression between testis and ovary in green mud crab (Scylla paramamosain). BMC Genomics. 2014;15:585.
Jiang H, Xing Z, Lu W, Qian Z, Yu H, Li J. Transcriptome analysis of red swamp crawfish Procambarus clarkii reveals genes involved in gonadal development. PLoS One. 2014;9(8), e105122.
Chen X, Mei J, Wu J, Jing J, Ma W, Zhang J, et al. A comprehensive transcriptome provides candidate genes for Sex determination/differentiation and SSR/SNP markers in yellow catfish. Mar Biotechnol (NY). 2015;17(2):190–8.
Valenzuela-Miranda D, Gallardo-Escarate C, Valenzuela-Munoz V, Farlora R, Gajardo G. Sex-dependent transcriptome analysis and single nucleotide polymorphism (SNP) discovery in the brine shrimp Artemia franciscana. Mar Genomics. 2014;18 PB:151–4.
Yu Y, Wei J, Zhang X, Liu J, Liu C, Li F, et al. SNP discovery in the transcriptome of white Pacific shrimp Litopenaeus vannamei by next generation sequencing. PLoS One. 2014;9(1), e87218.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19(5):651–2.
Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23(9):1061–7.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8.
Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat Protoc. 2013;8(9):1765–86.
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(Web Server issue):W316–22.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25(4):402–8.
Chen X, Zeng D, Xie D, Zhao Y, Yang C, Li Y, et al. Transcriptome analysis of Litopenaeus vannamei in response to white spot syndrome virus infection. PLoS One. 2013;8(8), e73218.
Sookruksawong S, Sun F, Liu Z, Tassanakajon A. RNA-Seq analysis reveals genes associated with resistance to Taura syndrome virus (TSV) in the Pacific white shrimp Litopenaeus vannamei. Dev Comp Immunol. 2013;41(4):523–33.
Xue S, Liu Y, Zhang Y, Sun Y, Geng X, Sun J. Sequencing and de novo analysis of the hemocytes transcriptome in Litopenaeus vannamei response to white spot syndrome virus infection. PLoS One. 2013;8(10), e76718.
Zeng D, Chen X, Xie D, Zhao Y, Yang C, Li Y, et al. Transcriptome analysis of Pacific white shrimp (Litopenaeus vannamei) hepatopancreas in response to Taura syndrome Virus (TSV) experimental infection. PLoS One. 2013;8(2), e57515.
Guo H, Ye CX, Wang AL, Xian JA, Liao SA, Miao YT, et al. Trascriptome analysis of the Paci fi c white shrimp Litopenaeus vannamei exposed to nitrite by RNA-seq. Fish Shellfish Immunol. 2013;35(6):2008–16.
Li C, Weng S, Chen Y, Yu X, Lu L, Zhang H, et al. Analysis of Litopenaeus vannamei transcriptome using the next-generation DNA sequencing technique. PLoS One. 2012;7(10), e47442.
Ghaffari N, Sanchez-Flores A, Doan R, Garcia-Orozco KD, Chen PL, Ochoa-Leyva A, et al. Novel transcriptome assembly and improved annotation of the whiteleg shrimp (Litopenaeus vannamei), a dominant crustacean in global seafood mariculture. Sci Rep. 2014;4:7081.
Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331(6017):555–61.
Larney C, Bailey TL, Koopman P. Switching on sex: transcriptional regulation of the testis-determining gene Sry. Development. 2014;141:2195–205.
Sekido R, Lovell-Badge R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature. 2008;453(7197):930–4.
Warr N, Greenfield A. The molecular and cellular basis of gonadal sex reversal in mice and humans. Wiley Interdiscip Rev Dev Biol. 2012;1(4):559–77.
Burtis KC, Baker BS. Drosophila doublesex gene controls somatic sexual differentiation by producing alternatively spliced mRNAs encoding related sex-specific polypeptides. Cell. 1989;56(6):997–1010.
Kato Y, Kobayashi K, Watanabe H, Iguchi T. Environmental sex determination in the branchiopod crustacean Daphnia magna: deep conservation of a Doublesex gene in the sex-determining pathway. PLoS Genet. 2011;7(3), e1001345.
Raymond CS, Murphy MW, O’Sullivan MG, Bardwell VJ, Zarkower D. Dmrt1, a gene related to worm and fly sexual regulators, is required for mammalian testis differentiation. Genes Dev. 2000;14(20):2587–95.
Doniach T, Hodgkin J. A sex-determining gene, fem-1, required for both male and hermaphrodite development in Caenorhabditis elegans. Dev Biol. 1984;106(1):223–35.
Hodgkin J. Sex determination in the nematode C. elegans: analysis of tra-3 suppressors and characterization of fem genes. Genetics. 1986;114(1):15–52.
Georges A, Auguste A, Bessiere L, Vanet A, Todeschini AL, Veitia RA. FOXL2: a central transcription factor of the ovary. J Mol Endocrinol. 2013;52(1):R17–33.
Pisarska MD, Barlow G, Kuo FT. Minireview: roles of the forkhead transcription factor FOXL2 in granulosa cell biology and pathology. Endocrinology. 2011;152(4):1199–208.
Teaniniuraitemoana V, Huvet A, Levy P, Klopp C, Lhuillier E, Gaertner-Mazouni N, et al. Gonad transcriptome analysis of pearl oyster Pinctada margaritifera: identification of potential sex differentiation and sex determining genes. BMC Genomics. 2014;15:491.
Ohinata Y, Payer B, O’Carroll D, Ancelin K, Ono Y, Sano M, et al. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature. 2005;436(7048):207–13.
Ancelin K, Lange UC, Hajkova P, Schneider R, Bannister AJ, Kouzarides T, et al. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat Cell Biol. 2006;8(6):623–30.
Ohinata Y, Ohta H, Shigeta M, Yamanaka K, Wakayama T, Saitou M. A signaling principle for the specification of the germ cell lineage in mice. Cell. 2009;137(3):571–84.
Vincent SD, Dunn NR, Sciammas R, Shapiro-Shalef M, Davis MM, Calame K, et al. The zinc finger transcriptional repressor Blimp1/Prdm1 is dispensable for early axis formation but is required for specification of primordial germ cells in the mouse. Development. 2005;132:1315–25.
Raz E. The function and regulation of vasa-like genes in germ-cell development. Genome Biol. 2000;1(3):reviews1017.
Xu H, Gui J, Hong Y. Differential expression of vasa RNA and protein during spermatogenesis and oogenesis in the gibel carp (Carassius auratus gibelio), a bisexually and gynogenetically reproducing vertebrate. Dev Dyn. 2005;233(3):872–82.
Gallardo T, Shirley L, John G, Castrillon DH. Generation of a germ cell-specific mouse transgenic Cre line, Vasa-Cre. Genesis. 2007;45(6):413–7.
Wessel GM, Fresques T, Kiyomoto M, Yajima M, Zazueta V. Origin and development of the germ line in sea stars. Genesis. 2014;52(5):367–677.
Xu H, Lim M, Dwarakanath M, Hong Y. Vasa identifies germ cells and critical stages of oogenesis in the Asian seabass. Int J Biol Sci. 2014;10(2):225–35.
Roussell DL, Bennett KL. glh-1, a germ-line putative RNA helicase from Caenorhabditis, has four zinc fingers. Proc Natl Acad Sci U S A. 1993;90(20):9300–4.
Styhler S, Nakamura A, Swan A, Suter B, Lasko P. vasa is required for GURKEN accumulation in the oocyte, and is involved in oocyte differentiation and germline cyst development. Development. 1998;125(9):1569–78.
Tanaka SS, Toyooka Y, Akasu R, Katoh-Fukui Y, Nakahara Y, Suzuki R, et al. The mouse homolog of Drosophila Vasa is required for the development of male germ cells. Genes Dev. 2000;14(7):841–53.
Li M, Hong N, Xu H, Yi M, Li C, Gui J, et al. Medaka vasa is required for migration but not survival of primordial germ cells. Mech Dev. 2009;126(5):366–81.
Seydoux G, Strome S. Launching the germline in Caenorhabditis elegans: regulation of gene expression in early germ cells. Development. 1999;126(15):3275–83.
Deshpande G, Calhoun G, Yanowitz JL, Schedl PD. Novel functions of nanos in downregulating mitosis and transcription during the development of the Drosophila germline. Cell. 1999;99(3):271–81.
Forbes A, Lehmann R. Nanos and Pumilio have critical roles in the development and function of Drosophila germline stem cells. Development. 1998;125(4):679–90.
Parisi M, Lin H. Translational repression: a duet of Nanos and Pumilio. Curr Biol. 2000;10(2):R81–3.
Li M, Shen Q, Xu H, Wong FM, Cui J, Li Z, et al. Differential conservation and divergence of fertility genes boule and dazl in the rainbow trout. PLoS One. 2011;6(1), e15910.
Peng JX, Xie JL, Zhou L, Hong YH, Gui JF. Evolutionary conservation of Dazl genomic organization and its continuous and dynamic distribution throughout germline development in gynogenetic gibel carp. J Exp Zool B Mol Dev Evol. 2009;312B(8):855–71.
Yen PH. Putative biological functions of the DAZ family. Int J Androl. 2004;27(3):125–9.
Xu H, Li M, Gui J, Hong Y. Cloning and expression of medaka dazl during embryogenesis and gametogenesis. Gene Expr Patterns. 2007;7(3):332–8.
Stadler BM, Ruohola-Baker H. Small RNAs: keeping stem cells in line. Cell. 2008;132(4):563–6.
Kuramochi-Miyagawa S, Kimura T, Ijiri TW, Isobe T, Asada N, Fujita Y, et al. Mili, a mammalian member of piwi family gene, is essential for spermatogenesis. Development. 2004;131(4):839–49.
Megosh HB, Cox DN, Campbell C, Lin H. The role of PIWI and the miRNA machinery in Drosophila germline determination. Curr Biol. 2006;16(19):1884–94.
Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132(4):598–611.
Kershner A, Crittenden SL, Friend K, Sorensen EB, Porter DF, Kimble J. Germline stem cells and their regulation in the nematode Caenorhabditis elegans. Adv Exp Med Biol. 2013;786:29–46.
Chen S, Wang S, Xie T. Restricting self-renewal signals within the stem cell niche: multiple levels of control. Curr Opin Genet Dev. 2011;21(6):684–9.
Meng X, Lindahl M, Hyvonen ME, Parvinen M, de Rooij DG, Hess MW, et al. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 2000;287(5457):1489–93.
Parisi M, Nuttall R, Naiman D, Bouffard G, Malley J, Andrews J, et al. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science. 2003;299(5607):697–700.
Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature. 2007;450(7167):233–7.
Jiang M, Ryu J, Kiraly M, Duke K, Reinke V, Kim SK. Genome-wide analysis of developmental and sex-regulated gene expression profiles in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2001;98(1):218–23.
Small CM, Carney GE, Mo Q, Vannucci M, Jones AG. A microarray analysis of sex- and gonad-biased gene expression in the zebrafish: evidence for masculinization of the transcriptome. BMC Genomics. 2009;10:579.
Tao W, Yuan J, Zhou L, Sun L, Sun Y, Yang S, et al. Characterization of gonadal transcriptomes from Nile tilapia (Oreochromis niloticus) reveals differentially expressed genes. PLoS One. 2013;8(5), e63604.
Rinn JL, Rozowsky JS, Laurenzi IJ, Petersen PH, Zou K, Zhong W, et al. Major molecular differences between mammalian sexes are involved in drug metabolism and renal function. Dev Cell. 2004;6(6):791–800.
Swain A, Lovell-Badge R. Mammalian sex determination: a molecular drama. Genes Dev. 1999;13(7):755–67.
Tilmann C, Capel B. Cellular and molecular pathways regulating mammalian sex determination. Recent Prog Horm Res. 2002;57:1–18.
Gao X, Ma W, Nie J, Zhang C, Zhang J, Yao G, et al. A G-quadruplex DNA structure resolvase, RHAU, is essential for spermatogonia differentiation. Cell Death Dis. 2015;6, e1610.
Eaker S, Cobb J, Pyle A, Handel MA. Meiotic prophase abnormalities and metaphase cell death in MLH1-deficient mouse spermatocytes: insights into regulation of spermatogenic progress. Dev Biol. 2002;249(1):85–95.
Kanatsu-Shinohara M, Shinohara T. Spermatogonial stem cell self-renewal and development. Annu Rev Cell Dev Biol. 2013;29:163–87.
Yoneda R, Kimura AP. A testis-specific serine protease, Prss41/Tessp-1, is necessary for the progression of meiosis during murine in vitro spermatogenesis. Biochem Biophys Res Commun. 2013;441(1):120–5.
Yoneda R, Takahashi T, Matsui H, Takano N, Hasebe Y, Ogiwara K, et al. Three testis-specific paralogous serine proteases play different roles in murine spermatogenesis and are involved in germ cell survival during meiosis. Biol Reprod. 2013;88(5):118,111–114.
Fraser LR, Hosseini R, Hanyalogou A, Talmor A, Dudley RK. TCP-11, the product of a mouse t-complex gene, plays a role in stimulation of capacitation and inhibition of the spontaneous acrosome reaction. Mol Reprod Dev. 1997;48(3):375–82.
Xie S, Sun L, Liu F, Dong B. Molecular characterization and mRNA transcript profile of vitellogenin in Chinese shrimp, Fenneropenaeus chinensis. Mol Biol Rep. 2009;36(2):389–97.
Phiriyangkul P, Utarabhand P. Molecular characterization of a cDNA encoding vitellogenin in the banana shrimp, Penaeus (Litopenaeus) merguiensis and sites of vitellogenin mRNA expression. Mol Reprod Dev. 2006;73(4):410–23.
Fliss AE, Benzeno S, Rao J, Caplan AJ. Control of estrogen receptor ligand binding by Hsp90. J Steroid Biochem Mol Biol. 2000;72(5):223–30.
Coccia E, De Lisa E, Di Cristo C, Di Cosmo A, Paolucci M. Effects of estradiol and progesterone on the reproduction of the freshwater crayfish Cherax albidus. Biol Bull. 2010;218(1):36–47.
Kishimoto T. Activation of MPF at meiosis reinitiation in starfish oocytes. Dev Biol. 1999;214(1):1–8.
Kably Ambe A, Ruiz Anguas J, Carballo Mondragon E, Corona De Lau C, Karchmer Krivitsky S. [Correlation between follicle levels of superoxide dismutase and oocyte quality, fertilization rates and embryo development]. Ginecol Obstet Mex. 2004;72:335–44.
Su ZŁP, Li S, Thierry-Mieg J, Thierry-Mieg D, Shi W, Wang C, et al. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the sequencing quality control consortium. Nat Biotechnol. 2014;32(9):903–14.
Liu Y, Zhou J, White KP. RNA-seq differential expression studies: more sequence or more replication? Bioinformatics. 2014;30(3):301–4.
Li YC, Korol AB, Fahima T, Beiles A, Nevo E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol. 2002;11(12):2453–65.
Ball AO, Leonard S, Chapman RW, Xu Z, Zuniga G, Alcivar-Warren A, et al. Charaterization of (GT)n microsatellites from native white shrimp (Penaeus setiferus). Mol Ecol. 1998;7(9):1251–3.
Andriantahina F, Liu X, Huang H. Genetic Map construction and Quantitative Trait Locus (QTL) detection of growth-related traits in litopenaeus vannamei for selective breeding applications. PLoS One. 2013;8:9.
Meehan D. High frequency and large number of polymorphic microsatellites in cultured shrimp. Penaeus (Litopenaeus) vannamei [Crustacea:Decapoda]. 2003;5(4):311–30.
Santos CA, Blanck DV, de Freitas PD. RNA-seq as a powerful tool for penaeid shrimp genetic progress. Front Genet. 2014;5:298.
Acknowledgments
This work was supported by the National High Technology Research and Development Program (863 Program) of China (2012AA10A404), the special funds for Ba Gui Scholars project (Grant No. BGXZ-NMBDX-05), and the China Agriculture Research System (Grant No. CARS-47). We would also like to thank LetPub (www.letpub.com) for linguistic assistance during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CXH and PJX conceived the study, participated in its design and coordination and wrote the manuscript. WPY and PJX directed sampling and dissections of shrimps to form the library of gonadic tissue. WPY and CXL conducted the RT-PCR experiments and data analysis, ZDG and LM performed RNA extraction, cDNA library construction and sequencing. ZB and ZYZ performed the bioinformatic analysis, assembly, annotation, and SSR discovery. All authors read and approved the final manuscript.
Jinxia Peng and Pinyuan Wei contributed equally to this work.
Additional file
Additional file 1: Figure S1.
Histological features of the testis and pre-eyestalk ablation shrimp ovary. Both gonads are in early development stages and contain mostly spermatogonia, spermatocytes, oogonia and oocytes. Table S1. List of KEGG pathways for gonadally expressed genes. Table S2. List of differentially expressed genes in male and female gonads. (FDR < 0.001). Table S3. Enriched GO terms and pathways for sex-biased genes. Table S4. Primers and verification information of male-biased genes. Table S5. Primers and verification information of female-biased genes. Table S6. The distributions of SSRs in unigenes and primers for SSRs. (ZIP 3741 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Peng, J., Wei, P., Zhang, B. et al. Gonadal transcriptomic analysis and differentially expressed genes in the testis and ovary of the Pacific white shrimp (Litopenaeus vannamei). BMC Genomics 16, 1006 (2015). https://doi.org/10.1186/s12864-015-2219-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-015-2219-4