
Abstract
Recent progress in understanding the silencing of transposable elements in the model plant Arabidopsis has revealed an interplay between DNA methylation, histone methylation and small interfering RNAs. DNA and histone methylation are not always sufficient to maintain silencing, and RNA-based reinforcement can be needed to maintain as well as initiate it.
Similar content being viewed by others


Throughout evolution, genomes have been invaded by 'selfish' DNA elements that use them as vehicles for self-propagation. In order to defend themselves against these genomic parasites, genomes need something akin to an immune system - a mechanism that can distinguish self from non-self at the nucleic-acid level and inactivate the non-self sequences. In the last few years, studies of DNA methylation, post-translational histone modifications and RNA silencing in the plant Arabidopsis thaliana and other organisms have begun to reveal what appears to be just such an integrated genome defense system.
An essential property of eukaryotic cells is the ability to establish heritable patterns of gene silencing without alterations in DNA sequence. Methylation of cytosine nucleotides, usually within CG dinucleotides, is the most common form of covalent DNA modification in the eukaryotic kingdom, and most eukaryotes use it to propagate epigenetic control [1]. For example, DNA methylation plays an important role in imprinting (silencing of genes specifically on the basis of their origin in one or other parent) and in mammalian X-chromosome inactivation. But in many organisms, a more widespread role of methylation appears to be in silencing of parasitic DNA sequences. DNA methylation is predominantly found at repetitive sequences that are descended from transposable elements and viruses, and it marks them for transcriptional inactivity [2].
Another feature of gene silencing is covalent modification of histones, especially methylation of lysine 9 of histone H3 (H3 K9) [3]. Methyltransferases that include a SET domain (named after three members of the family, Su(var)3-9, Enhancer-of-zeste and Trithorax) have been identified as catalyzing this modification; mutations in the Drosophila Su(var)3-9 gene are dominant suppressors of heterochromatin-induced silencing. Another gene with such a suppressor mutant phenotype encodes Heterochromatin-associated Protein 1 (HP1), which contains a chromodomain that specifically binds methylated H3 K9 [4].
In Neurospora and Arabidopsis, a reduction in H3 k9 methylation leads to a reduction in DNA methylation [5–7]. A question then arises as to what determines the substrates for H3 k9 methylation throughout the genome. Recent findings have implicated small interfering RNAs (siRNAs) in this process. Members of gene families involved in posttranscriptional silencing by siRNAs (the Dicer, RNA dependent RNA polymerase (RdRP) and Argonaute (Ago) families) have been shown to play important roles in transcriptional gene silencing in plants, animals and fungi [8–11]. Mutations in these genes lead to loss of H3 K9 methylation in a number of organisms and to loss of DNA methylation in Arabidopsis. A complex that mediates transcriptional RNA silencing in Schizosaccharomyces pombe contains a protein of the Argonaute family and a chromodomain protein, further reinforcing the connection between siRNAs and H3 K9 methylation [12].
Genome-scale observation of the features of silencing
Many of the advances in understanding gene silencing have been made in plants, largely using model reporter systems in which changes in silencing can be sensitively detected. For example, screens for genes that relieve the silencing of the Arabidopsis SUP and PAI loci that is induced by inverted repeats led to the discovery of three components of epigenetic processes: the CHROMOMETHYLASE3 (CMT3) DNA methyltransferase; an H3 K9 methyltransferase (KYP, also called SUVH4); and an Argonaute family member (AGO4) [6–8, 13, 14]. These studies revealed that mobile elements are among the targets for DNA methylation by CMT3, an observation confirmed by microarray analysis of DNA methylation patterns in mutant cmt3- plants [15]. Recently, the correlations between DNA methylation, H3 K9 methylation and siRNAs were shown to extend over a large contiguous portion of the Arabidopsis genome [16]. These features were primarily associated with mobile elements, suggesting that multiple silencing mechanisms are used for controlling genomic parasites. When a mutation in the ATP-dependent chromatin remodeling protein DDM1 was introduced, methylation of both DNA and H3 K9 was sharply decreased at mobile elements, with concomitant increases in transcription of these elements. Even the siRNAs were decreased in abundance in a ddm1- mutant background. Interdependent mechanisms involving chromatin remodeling, DNA methylation, histone methylation and siRNAs therefore maintain mobile elements in a silent state in Arabidopsis.
What is responsible for this interdependence? One possibility is that DDM1 facilitates synthesis of siRNAs, which would trigger downstream silencing events. But the loss of siRNA-mediated silencing components has no obvious effects on the silencing of extensive genomic regions [11, 17]. An alternative possibility is that multiple components of epigenetic silencing act at the same place at the same time. In this way, the elimination of the ATP-dependent remodeler that provides access to chromatin would have effects on multiple silencing components. For example, if DDM1 were to act at the replication fork to promote DNA methylation and chromatin assembly, then a concerted process of silencing would be disrupted in ddm1- mutant plants. Indeed, there are intriguing connections between various epigenetic-silencing components and the machinery for DNA replication and replication-coupled chromatin assembly. Where silent regions are extensive, replication-coupled mechanisms might suffice, but where they are small, targeting by siRNAs would be required to reinforce silencing. Below, we explore these concepts in light of recent evidence.
Active and passive maintenance of methylation
Most DNA methylation in both plants and animals is on CG dinucleotides [1]. A CG methylated on one strand but not the other (hemi-methylated), which results from replication of fully methylated DNA, serves as a substrate for a maintenance methyltransferase that restores the site to a fully methylated state. The enzyme responsible for maintenance of CG methylation, called DNA methyltransferase 1 (Dnmt1), was first cloned in mice and is associated with DNA replication foci during the S (synthesis) phase of the cell cycle [1]. An orthologous Arabidopsis enzyme called MET1 is similarly required for maintenance of CG methylation [18].
The Dnmt1 subfamily of cytosine DNA methyltransferases should be capable of maintaining methylation passively, so that the only signal required for a methylation pattern to be propagated is the initial methylation itself. Some of the best evidence for this comes from experiments in plants after induction of RNA-dependent DNA methylation [19]. CG methylation and transcriptional silencing can be maintained for generations after the RNA trigger has been eliminated from the plants, and MET1 function is required for silencing to be heritable in the absence of the RNA trigger [17, 20–22]. Also, mutations affecting H3 K9 methylation and RNA-dependent DNA methylation have little effect on CG methylation at most loci [6–8, 11, 23]. CG methylation can thus be maintained passively, without the need for an active signal.
Non-CG methylation is generally found on CNG motifs or more rarely on asymmetrical motifs (CNN), where N is any nucleotide. Most CNG methylation in Arabidopsis is maintained by CHROMOMETHYLASE3 (CMT3) [13, 14], so named because of the presence of a chromodomain within the catalytic domain [24]. CMT3 mutants lack virtually all CNG methylation in pericentric heterochromatin, and a number of transposable elements that reside there are reactivated [18]. But at some silent regions that span only a few nucleosomes, loss of CMT3 function leads to only partial loss of methylation on CNG and asymmetric motifs [8, 25]. At these sequences, the remainder of non-CG methylation is catalyzed by the DRM family of methyltransferases. This effect is most pronounced at loci consisting of tandem direct repeats, such as FWA and MEA-ISR, where mutations in CMT3 have only a minor effect on non-CG methylation whereas DRM loss-of-function mutations eliminate this methylation completely [25].
The first clues about how CMT3 might be recruited came from the discovery of its partial dependence on the KYP/SUVH4 H3 K9 methyltransferase [6, 7]. Mutations in kyp/suvh4 mimic the cmt3 phenotype with respect to DNA methylation and transposon reactivation, although the effect is weaker than that of cmt3 mutants. A potential mechanism for how histone methylation may target CMT3 is suggested by the fact that CMT3 contains a chromodomain. CMT3 could therefore have the ability to directly interact with methylated histone H3.
Further insight into the mechanism of CMT3 regulation was provided by the recovery of an allele of AGO4 from the screen for suppressors of silencing of the SUP locus [8]. The ago4 mutant plants exhibited substantial loss of CNG methylation, dramatic loss of asymmetric methylation and decreased H3 K9 methylation at several silent regions that span only a few nucleosomes. Experiments with PAI silencing also provide evidence that siRNA-mediated silencing plays a role in targeting CMT3 [26]. An upstream promoter is responsible for making a transcript that reads through the inverted repeat responsible for silencing PAI, thus creating a long double-stranded RNA. Silencing of this promoter leads to loss of both non-CG methylation and silencing at PAI. Crosses that remove the inverted repeat produce similar results [20]. Additionally, highly transcribed inverted-repeat loci designed to trigger RNA-dependent DNA methylation induce high levels of non-CG methylation, which is largely lost when the inverted repeat is removed by crossing. These observations suggest that CNG and asymmetric methylation, unlike CG methylation, need to be actively maintained.
Establishment of methylation versus active maintenance
So far we have described two general modes of maintenance DNA methylation: passive and active. Passive maintenance is self-perpetuating, and clearly distinct from de novo methylation on a naive template. Active maintenance, on the other hand, may be nothing more than recurring rounds of de novo methylation. Alternatively, the requirements for active maintenance methylation and de novo methylation may be different, despite the fact that they both require an active signal. At least two lines of evidence indicate that the latter is indeed the case. First, the Arabidopsis de novo methyltransferases of the DRM family are absolutely required for the establishment of the RNA-directed DNA methylation that is triggered by a number of loci involving inverted repeats, but DRM proteins have only a partial role in the active maintenance of asymmetric and CNG methylation at these loci [25, 27, 28]. The rest of the non-CG methylation is maintained by CMT3, which is not required for establishment of methylation. CMT3 is therefore capable, at least in some cases, of responding to an RNA signal in order to actively maintain, but not to establish, DNA methylation.
A second line of evidence comes from the effects of the ddm1 mutation on DNA methylation. After erasure of methylation by passage through a ddm1 mutant background, restoration of DDM1 activity by crossing into a wild-type background does not restore methylation [16]. Restoration of DDM1 function is therefore not sufficient to regain methylation and silencing, despite the observation that many of the transposons in question are associated with siRNAs. Thus, in the same cell, the same silencing signal is sufficient to maintain DNA methylation and silencing of transposons on one set of chromosomes but is not sufficient to efficiently initiate DNA methylation and silencing of the same sequences on a different set of chromosomes.
Maintaining a silent chromatin state
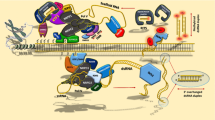
How do these various processes fit together in order to maintain silent chromatin? Like Dnmt1, MET1 is thought to maintain CG methylation following DNA replication [18]. Old histones are evenly distributed between the two products of replication, so each chromatid has a memory not only of the original DNA-methylation state but also of the original histone-modification state. New nucleosomes are deposited after replication by the Chromatin Assembly Factor 1 (CAF1) chaperone complex. The H3 K9 methylation state of old nucleosomes would provide cues for CAF1 to deposit methylated nucleosomes [29] (see Figure 1a). The modified regions recruit CMT3 in order to maintain CNG methylation. In support of a role for replication-coupled nucleosome assembly in helping to maintain silent chromatin, mutations in CAF1 components have recently been shown to destabilize heterochromatic silencing in Arabidopsis [30]. Mutation of the DDM1 chromatin remodeler could thus simultaneously disrupt the maintenance of both DNA and histone methylation, leading to a profound loss of silencing.

A model for the maintenance of chromatin silencing. (a) Replication-coupled maintenance of a silent region of chromatin. Solid lines indicate DNA; cylinders represent nucleosomes (light, old; dark, newly added); circles represent other proteins or protein complexes; flags indicate histone methylation; M indicates DNA methylation (with new methylation in bold); the large oval represents DNA polymerase. Before replication (1), a silenced region is marked by histone methylation and DNA methylation on CG and CNG motifs. As the polymerase moves along the leading strand from left to right (2), methylation on CG dinucleotides is passively maintained behind the replication fork by MET1 (3). Old nucleosomes are randomly distributed between the two chromatids and new nucleosomes are added by the CAF1 chaperone complex (4). In the top chromatid in the diagram (A), there are two adjacent nucleosomes that are methylated on H3 K9, thus providing cues for CAF1 to deposit a new nucleosome that is methylated on H3 K9 by KYP (5). On the bottom chromatid (B), however, the nucleosome distribution leads to loss of epigenetic information at the edge of the silent domain, so new nucleosomes are deposited by CAF1 without H3 K9 methylation (6). CMT3 is therefore able to use the cues provided by H3 K9 methylation to properly maintain CNG methylation on the top chromatid (7), but not the bottom (8). Chromatin remodeling by DDM1 enables both DNA and histone methylation, perhaps by allowing access of other proteins to the DNA. (b) RNA-based reinforcement of silencing. The bottom (B) chromatid from (a) is shown after the replication fork has passed completely. Now, siRNAs homologous to the silent region guide H3 K9 methylation by KYP and DNA methylation by DRM. H3 K9 methylation also allows the maintenance of CNG methylation by CMT3. The problem shown in (a) is thus solved: the silent domain is fully maintained, despite random nucleosome distribution during replication.
Such replication-coupled maintenance may be all that is necessary to maintain silencing of large regions of chromatin. Regions that are only a few nucleosomes in length might be difficult to maintain, however, because the 'unit' of chromatin memory is a nucleosome and the distribution of old nucleosomes to daughter chromatids is random [31]. For example, if two adjacent nucleosomes are distributed to one daughter chromatid, then all histone-associated information is lost from the corresponding region of the other chromatid (Figure 1a). Therefore, to maintain stable silencing, DNA and histone modifications that are limited to small regions may need to be occasionally reinforced by active targeting of siRNAs to the homologous DNA (Figure 1b). In accordance with this idea, mutations in genes affecting siRNA-mediated silencing and de novo methylation have the strongest effects on short stretches of silent chromatin interspersed in otherwise active regions [8, 11, 25, 32]. One of these genes encodes a putative ATP-dependent chromatin remodeling protein, thus providing a DDM1 counterpart where silent regions are of limited extent [32].
Thus, in this model, siRNAs would have a dual role in silencing: they would provide triggers for establishing silent regions, but when these regions are too small to maintain themselves siRNAs would provide active reinforcement for maintenance of silencing. This latter process would be especially important for the silencing of newly integrated transposable elements, where chromatin-based silencing alone may be unstable. Stable silencing of such elements requires reinforcement by siRNAs; it amounts to 'kicking them when they're down'.
References
Bird A: DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16: 6-21. 10.1101/gad.947102.
Yoder JA, Walsh CP, Bestor TH: Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13: 335-340. 10.1016/S0168-9525(97)01181-5.
Lachner M, Jenuwein T: The many faces of histone lysine methylation. Curr Opin Cell Biol. 2002, 14: 286-298. 10.1016/S0955-0674(02)00335-6.
Richards EJ, Elgin SC: Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell. 2002, 108: 489-500. 10.1016/S0092-8674(02)00644-X.
Tamaru H, Selker EU: A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature. 2001, 414: 277-283. 10.1038/35104508.
Jackson JP, Lindroth AM, Cao X, Jacobsen SE: Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002, 416: 556-560. 10.1038/nature731.
Malagnac F, Bartee L, Bender J: An Arabidopsis SET domain protein required for maintenance but not establishment of DNA methylation. EMBO J. 2002, 21: 6842-6852. 10.1093/emboj/cdf687.
Zilberman D, Cao X, Jacobsen SE: ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science. 2003, 299: 716-719. 10.1126/science.1079695.
Volpe TA, Kidner C, Hall IM, Teng G, Grewal SIS, Martienssen RA: Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002, 297: 1833-1837. 10.1126/science.1074973.
Pal-Bhadra M, Bhadra U, Birchler JA: RNAi related mechanisms affect both transcriptional and posttranscriptional transgene silencing in Drosophila. Mol Cell. 2002, 9: 315-327. 10.1016/S1097-2765(02)00440-9.
Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, Jacobsen SE, Carrington JC: Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004, 2: E104-10.1371/journal.pbio.0020104.
Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D: RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004, 303: 672-676. 10.1126/science.1093686.
Lindroth AM, Cao X, Jackson JP, Zilberman D, McCallum CM, Henikoff S, Jacobsen SE: Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science. 2001, 292: 2077-2080. 10.1126/science.1059745.
Bartee L, Malagnac F, Bender J: Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev. 2001, 15: 1753-1758. 10.1101/gad.905701.
Tompa R, McCallum CM, Delrow J, Henikoff JG, van Steensel B, Henikoff S: Genome-wide profiling of DNA methylation reveals transposon targets of CHROMOMETHYLASE3. Curr Biol. 2002, 12: 65-68. 10.1016/S0960-9822(01)00622-4.
Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, et al: Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004, 430: 471-476. 10.1038/nature02651.
Zilberman D, Cao X, Johansen LK, Xie Z, Carrington JC, Jacobsen SE: Role of Arabidopsis ARGONAUTE4 in RNA-directed DNA methylation triggered by inverted repeats. Curr Biol. 2004, 14: 1214-1220. 10.1016/j.cub.2004.06.055.
Bender J: DNA methylation and epigenetics. Annu Rev Plant Physiol Plant Mol Biol. 2004, 55: 41-68.
Wassenegger M: RNA-directed DNA methylation. Plant Mol Biol. 2000, 43: 203-220. 10.1023/A:1006479327881.
Luff B, Pawlowski L, Bender J: An inverted repeat triggers cytosine methylation of identical sequences in Arabidopsis. Mol Cell. 1999, 3: 505-511. 10.1016/S1097-2765(00)80478-5.
Jones L, Ratcliff F, Baulcombe DC: RNA-directed transcriptional gene silencing in plants can be inherited independently of the RNA trigger and requires Met1 for maintenance. Curr Biol. 2001, 11: 747-757. 10.1016/S0960-9822(01)00226-3.
Aufsatz W, Mette MF, van der Winden J, Matzke AJ, Matzke M: RNA-directed DNA methylation in Arabidopsis. Proc Natl Acad Sci USA. 2002, 99 (Suppl 4): 16499-16506. 10.1073/pnas.162371499.
Chan SW, Zilberman D, Xie Z, Johansen LK, Carrington JC, Jacobsen SE: RNA silencing genes control de novo DNA methylation. Science. 2004, 303: 1336-10.1126/science.1095989.
Henikoff S, Comai L: A DNA methyltransferase homolog with a chromodomain exists in multiple polymorphic forms in Arabidopsis. Genetics. 1998, 149: 307-318.
Cao XF, Jacobsen SE: Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci USA. 2002, 99: 16491-16498. 10.1073/pnas.162371599.
Melquist S, Bender J: Transcription from an upstream promoter controls methylation signaling from an inverted repeat of endogenous genes in Arabidopsis. Genes Dev. 2003, 17: 2036-2047. 10.1101/gad.1081603.
Cao X, Jacobsen SE: Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol. 2002, 12: 1138-1144. 10.1016/S0960-9822(02)00925-9.
Cao X, Aufsatz W, Zilberman D, Mette MF, Huang MS, Matzke M, Jacobsen SE: Role of the DRM and CMT3 methyltransferases in RNA-directed DNA methylation. Curr Biol. 2003, 13: 2212-2217. 10.1016/j.cub.2003.11.052.
Sarraf SA, Stancheva I: Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell. 2004, 15: 595-605. 10.1016/j.molcel.2004.06.043.
Takeda S, Tadele Z, Hofmann I, Probst AV, Angelis KJ, Kaya H, Araki T, Mengiste T, Scheid OM, Shibahara K, et al: BRU1, a novel link between responses to DNA damage and epigenetic gene silencing in Arabidopsis. Genes Dev. 2004, 18: 782-793. 10.1101/gad.295404.
Henikoff S, Furuyama T, Ahmad A: Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet. 2004, 20: 320-326. 10.1016/j.tig.2004.05.004.
Kanno T, Mette MF, Kreil DP, Aufsatz W, Matzke M, Matzke AJ: Involvement of putative SNF2 chromatin remodeling protein DRD1 in RNA-directed DNA methylation. Curr Biol. 2004, 14: 801-805. 10.1016/j.cub.2004.04.037.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Zilberman, D., Henikoff, S. Silencing of transposons in plant genomes: kick them when they're down. Genome Biol 5, 249 (2004). https://doi.org/10.1186/gb-2004-5-12-249
Published:
DOI: https://doi.org/10.1186/gb-2004-5-12-249