
Abstract
Aberrant activity of growth factor receptors has been implicated in the pathogenesis of a wide variety of malignancies. The negative regulation of signaling by growth factor receptors is mediated in large part by the ubiquitination, internalization, and degradation of the activated receptor. Over the past few years, considerable insight into the mechanisms that control receptor downregulation has been gained. There are also data suggesting that mutations that lead to inhibition of downregulation of growth factor receptors could play a role in the pathogenesis of cancer. Therapies directed at enhancing the degradation of growth factor receptors offer a promising approach to the treatment of malignancies.
Similar content being viewed by others

Introduction
Inappropriate activity of growth factor receptors such as the epidermal growth factor receptor (EGFR) family, which comprises EGFR, ErbB-2/Her2/Neu, ErbB-3/Her3, and ErbB-4/Her4, is associated with the development of a wide variety of human cancers, including breast cancer [1]. An understanding of the mechanisms that negatively regulate growth factor signaling, and characterization of the function of such negative regulators in both normal and transformed cells can lead to identification of therapeutic targets for treatment of human cancers. Indeed, there are data to suggest that some existing therapies may act, at least in part, to enhance such negative regulatory mechanisms (although they were not developed specifically to enhance receptor degradation). In the present review, the role of ubiquitination in the negative regulation of growth factor receptor signaling is discussed. Much of the information regarding downregulation of growth factor receptors has been generated by studies of EGFR. However, the basic mechanisms described for EGFR are likely to regulate signaling by many growth factor receptors. Evidence is also presented that suggests that tumor cells may subvert the ubiquitin-based negative regulation of growth factor receptors. In addition, therapeutic options are discussed.
Ubiquitination and the regulation of cellular proteins
Ubiquitin is a highly conserved, 76-amino-acid protein that can be covalently conjugated to the lysine residues on a wide assortment of proteins and thus regulate their function (for review [2]). Ubiquitination of proteins occurs via sequential activation and conjugation of ubiquitin to target proteins by the ubiquitin-activating enzyme, a ubiquitin-conjugating enzyme, and a ubiquitin protein ligase (E3) [2]. The E3 protein confers specificity to the ubiquitination process and directs the conjugation of ubiquitin to one or more lysines of the specific target protein. Ubiquitin can form multi-ubiquitin chains, in which the first ubiquitin is attached via an isopeptide linkage to a lysine of the target protein and the subsequent ubiquitin molecules are conjugated to lysine 48 of the preceding ubiquitin molecule. Such lysine-48-linked multi-ubiquitin chains of four or more ubiquitin molecules can efficiently target proteins for degradation by the proteasome. Proteins may also be mono-ubiquitinated, or have di-ubiquitinated or multi-ubiquitinated chains that are linked via lysine 63 on the ubiquitin molecule. Rather than target proteins for proteasomal degradation, the latter ubiquitin modifications serve to regulate protein trafficking or function [2, 3]. Endocytosis and targeting of proteins to lysosomal degradation can be regulated by mono-ubiquitination or lysine-63-linked ubiquitin chains [2, 3]. Thus, ubiquitination can serve either to target proteins to proteasomes or to lysosomes for degradation [4–6].
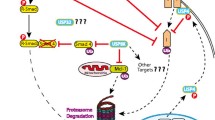
There are two ubiquitin-dependent mechanisms of growth factor receptor degradation: ligand-dependent degradation (discussed below) and ligand-independent degradation (discussed under Growth factor receptor degradation as cancer therapy) (Fig. 1). Both mechanisms have proven accessible to therapeutic interventions.

Ubiquitin (Ub)-mediated pathways of growth factor receptor (GFR) downregulation. There are two ubiquitin-mediated pathways of GFR degradation: ligand-dependent degradation (shown to the right) and ligand-independent degradation (shown to the left). Ligand-dependent degradation: upon ligand-induced activation of the mature GFR, Cbl proteins are recruited to the GFR and mediate ubiquitination, internalization, and degradation of the GFR (see text for details). Cin85, endophillins (End), and Eps 15 are involved in the internalization of the GFR. Regulatory proteins include protein kinase C (PKC) and Sprouty (spty). Ligand-independent degradation: a complex of protein chaperones (the stabilizing chaperone complex), including heat shock protein (Hsp)90, associates with both the nascent GFR in the endoplasmic reticulum and the mature GFR on the cell membrane and helps stabilize the protein. Drugs such as the benzoquinone ansamycins or tyrosine kinase inhibitor (TKIs) induce a change in the chaperone complex composition to one that favors degradation of the bound proteins (the degrading chaperone complex). The altered chaperone complex recruits an E3 ligase, which ubiquitinates the bound proteins, which in turn leads to their degradation by the proteasome (see text for details). E3 proteins are shown in green. The red color indicates current or potential agents that might enhance receptor degradation and thus be of clinical utility as treatment for cancer. E2, ubiquitin conjugation enzyme.
Ligand-dependent growth factor receptor downregulation
Upon ligand activation of many receptor tyrosine kinases such as the EGFR there is a rapid decrease in the cell surface number of the receptors and an eventual decrease in the cellular content of activated receptors – a process known as 'downregulation' [5]. This process can be divided into two distinct phases: internalization of the membrane receptor and degradation of the internalized receptor.
In nontransformed cells, inactive growth factor receptors may be concentrated in specific membrane regions know as caveolae [7]. Caveolae are small invaginations in the cell membrane that are enriched with caveolins, spin-golipid, and cholesterol. Caveolae may serve to spatially organize signaling molecules that interact with the receptors upon activation. In transformed cells that overexpress growth factor receptors, however, the receptors may not be concentrated in caveolae. This has been demonstrated for the EGFR in A431 cells [8], but no data exist regarding other members of the EGFR family. Upon ligand-mediated activation of the growth factor receptors, the receptors exit the calveolae and localize to another type of specific membrane region known as the clathrin-coated pit. Clathrin-coated pits are membrane invaginations coated by a lattice of clathrin proteins, and these pits are the major sites of endocytosis of activated membrane receptors [5, 9]. These invaginations pinch off to form clathrin-coated vesicles, thus internalizing the membrane proteins localized within these clathrin-coated pits. Specific amino acid sequences within membrane protein cytoplasmic tails target proteins to the clathrin-coated pits, where they undergo endocytosis [5, 9].
On the basis of recent publications, a mechanism for internalization of EGFR can be described (Fig. 1). Upon activation of the EGFR by ligand, Cbl proteins are recruited rapidly to the EGFR and mediate ubiquitination of the EGFR [10]. Cbl proteins are a family of proteins that are conserved throughout metazoans and have E3 activity, and which can also serve as adaptor proteins [10]. All Cbl proteins contain an amino-terminal phosphotyrosine binding (PTB) domain and a C3HC4 RING finger. The RING finger is essential for the E3 activity of Cbl proteins [10]. Cbl proteins can bind to the EGFR via two types of interactions. They can interact directly via their PTB domain with a phosphorylated tyrosine in the EGFR cytoplasmic tail (Y1045) or they can interact indirectly via binding to Grb2 [11, 12]. Upon EGFR activation, Cbl proteins are tyrosine phosphorylated by Src kinases [13]. However, phosphorylation of Cbl proteins is not required for their interaction with EGFR. Phosphorylation of Cbl proteins does enhance binding to Grb2 (which can bind via both SH2 and SH3 interactions with Cbl proteins). In addition, phosphorylation of Cbl proteins enhances interaction with another adaptor protein, namely Cbl interacting protein of 85 kDa (CIN85) [14, 15]. CIN85 contains three SH3 domains that interact with the carboxyl-terminal proline-rich domains of Cbl proteins [16–18]. CIN85 in turn is constitutively associated with endophilins [14, 15]. Endophilins have been implicated in the control of clathrin-mediated endocytosis by binding to regulatory components of endocytosis such as dynamin, amphiphysin, and synoptojanin (for references [14, 15]). Thus, binding of Cbl to the activated EGFR and recruitment of CIN85 couples EGFR to clathrin-mediated endocytosis [14].
The role of ubiquitination in the endocytosis of EGFR has not been clearly demonstrated. Cbl proteins may mediate endocytosis by serving as adaptor proteins as outlined above. However, a number of lines of evidence suggest that ubiquitination of the EGFR probably plays a role in endocytosis of the receptor. First, it has been established that ubiquitination is required for internalization of a number of membrane receptors in yeast [3]. Second, the growth hormone receptor, like EGFR, undergoes ligand-induced ubiquitination and downregulation, and inhibition of ubiquitination prevents internalization of the growth hormone receptor [19–21]. Third, Cbl association with and ubiquitination of EGFR occur at the cell membrane before internalization [22]. Finally, Eps15, an EGFR tyrosine kinase substrate that associates with the clathrin adaptor protein AP-2, has recently been shown to bind to ubiquitinated proteins via a ubiquitin-interacting motif [23, 24]. Inhibition of Eps15 function prevents EGFR internalization [24]. Eps15, therefore, could bind to the ubiquitinated EGFR via this ubiquitin-interacting motif and serve to direct EGFR to the clathrin-coated pit. Both Cbl proteins and Eps15 appear to have overlapping roles in targeting the activated, ubiquitinated EGFR for clathrin-mediated endocytosis. Whether these are part of one mechanism or represent alternative pathways remains to be determined.
Upon internalization, EGFR can be found in a vesicular structure near the cell periphery known as the early endosome [5]. From the early endosome, the growth factor receptor can either be recycled to the plasma membrane or it can be degraded. The decision to recycle or degrade appears to be regulated in part by the E3 activity of Cbl proteins. Ubiquitination of the EGFR increases its trafficking to the multivesicular body and eventually to the lysosome, where it is degraded [5, 25]. Cbl proteins have been shown to enhance ligand-induced degradation of EGFR. Recent work has further demonstrated that the Cbl proteins and other proteins within the active signaling complex, such as Grb2 and Shc, are coordinately degraded [26]. Thus Cbl proteins appear to play a second role in determining the sorting to the degradation pathway. This enhanced degradation of activated EGFR requires tyrosine kinase activity by the EGFR, interaction of Cbl proteins with phosphorylated tyrosines in the carboxyl-terminal tail of EGFR, and the E3 activity of Cbl proteins [12].
Although most data suggest that the EGFR is degraded in the lysosome, experiments using lysosome or proteasome inhibitors have demonstrated that both block degradation of the receptor as well as the complex of proteins degraded along with the receptor [26, 27]. The relationship between ubiquitination of plasma membrane proteins and their internalization, trafficking, and degradation is complex. In yeast, there are membrane proteins that are targeted for degradation in the vacuole by ubiquitination but whose degradation is independent of proteasome function [3, 28]. In mammalian cells, ligand-induced internalization and degradation of growth hormone receptor requires an intact ubiquitinating system, intact proteasome function, and intact lysosome function [19–21]. However, ubiquitination of growth hormone receptor itself is not required for degradation, but inhibition of proteasome function prevents internalization and degradation of the receptor [21]. Similar to the growth hormone receptor, degradation of EGFR is blocked by both proteasome and lysosome inhibitors [26, 27, 29]. The role of the proteasome in EGFR degradation, while implicated by inhibitor studies, is unclear. One possibility is that proteasome inhibitors may inhibit lysosomal degradation of the EGFR by depleting pools of free ubiquitin. This could inhibit Cbl-mediated ubiquitination of the EGFR and prevent trafficking of the ubiquitinated EGFR complex to the lysosomal compartment. In yeast, depletion of free ubiquitin has been shown to inhibit both proteasomal and lysosomal degradation [30]. However, although degradation of the EGFR complex is blocked by proteasome inhibitors, EGF still induces polyubiquitination of EGFR in the presence and absence of proteasome inhibitors [25]. This is consistent with a role for the proteasome in degradation of the EGFR complex. Thus, it is possible that the entire EGFR signaling complex traffics to the lysosome, where it is coordinately degraded by both lysosomal and proteasomal mechanisms. Alternatively, proteasomal degradation of regulatory proteins may be necessary for trafficking of the EGFR to the lysosome.
Many other proteins are known to play a role in downregu-lation of growth factor receptors [5]. For example, protein kinase C has been shown to phosphorylate EGFR on threonine 654, resulting in decreased ligand-induced ubiquitination, decreased degradation, and increased recycling of EGFR (Fig. 1) [31]. The precise mechanism by which protein kinase C mediated phosphorylation of EGFR changes the fate of the internalized receptor from destruction to recycling remains to be determined. Another recently described family of regulatory molecules is the human Sprouty protein family. In studies in human cells, Sprouty proteins were shown to enhance EGFR signaling by sequestering Cbl proteins, thus preventing Cbl-mediated ubiquitination and degradation (Fig. 1) [32].
Growth factor receptor downregulation and carcinogenesis
As noted above, the Cbl proteins are critical regulators of receptor downregulation. Mutant forms of c-Cbl that act as oncogenes have been identified. The v-Cbl oncogene, the first Cbl protein identified, is the transforming gene of the Cas-NS-1 murine retrovirus, which causes pre-B-cell lymphomas and myeloid leukemias in mice and which transforms NIH 3T3 cells [10]. v-Cbl is a truncated version of its cellular homologue c-Cbl. v-Cbl retains only the amino-terminal PTB domain and acts as a dominant negative inhibitor of c-Cbl-induced receptor downregulation [27]. Two other transforming forms of c-Cbl have been identified in cell lines derived from carcinogen-induced murine lymphomas, namely 70Z Cbl and p95Cbl [33, 34]. Both of those transforming Cbl proteins have deletions that encompass part of the linker region between the PTB domain and the RING finger and some or all of the RING finger. The RING finger is essential for E3 activity, and these mutant proteins cannot function as an E3. Thus, the likely mechanism of transformation by these variants of c-Cbl is inhibition of downregulation of activated growth factor receptors [10]. In human gastrointestinal tumors with microsatellite instability, one study described expansion of a trinucleotide repeat in the c-Cbl gene in 1 out of 11 tumors analyzed [35]. The functional significance of this mutation has not been characterized, and transforming forms of the other mammalian Cbl proteins (Cbl-b and Cbl-c) have not been described in either human or murine tumors.
Two recent reports have identified oncogenic mutations in growth factor receptors that eliminate the binding site for Cbl proteins. The Met receptor is the receptor for hepatocyte growth factor and is ubiquitinated and downregulated by c-Cbl [36]. The oncogenic TPR-Met receptor was generated by a carcinogen-induced chromosomal translocation in a nontumorigenic human sarcoma cell line. This oncogenic receptor lacks the tyrosine binding site for Cbl [36]. TPR-Met is constitutively active but it is not ubiquitinated and fails to bind c-Cbl. This suggests that the failure to downregulate the receptor contributes to the transforming ability of TPR-Met. Indeed, mutation of only the Cbl-binding site of the Met receptor is sufficient to promote ligand-independent oncogenic activation of the Met receptor [36].
The v-Fms oncogene is a mutated version of the feline colony stimulating factor-1 (CSF-1) receptor. c-Cbl has been shown to ubiquitinate the CSF-1 receptor upon ligand activation [37]. Interestingly, the v-Fms oncogene has deleted a carboxyl-terminal tyrosine (Y977), which is the binding site for c-Cbl [37]. As a result, c-Cbl does not ubiquitinate v-Fms or inhibit transformation by v-Fms. Reintroduction of the Cbl-binding site into v-Fms reduces the transforming potency of v-Fms, and coexpression of c-Cbl inhibited transformation by this form of the v-Fms oncogene [37]. Mutation of the equivalent tyrosine in the human CSF-1 receptor enhanced the ability of the receptor to transform fibroblasts [38]. Point mutations in the Cbl-binding site have been found in children with secondary myelodysplasia and acute myeloid leukemia [39, 40].
Mutations that interfere with downregulation of activated growth factor receptors and that result in malignant transformation demonstrate the importance of receptor down-regulation in normal homeostasis. No mutations in proteins involved in receptor downregulation have yet been described in human epithelial malignancies, including breast cancer. However, the amplification and overexpres-sion of ErbB-2 in a variety of human malignancies may, in fact, affect downregulation of EGFR. Of the receptors in the EGFR family, only EGFR undergoes ligand-induced downregulation [41]. ErbB-2 does not have a ligand that activates it. Instead, it is activated via heterodimerization with ligand-bound EGFR, ErbB-3, or ErbB-4 [42]. Homod-imerization of EGFR by epidermal growth factor results in EGFR downregulation [41]. However, heterodimerization of ErbB-2 with the activated EGFR prevents binding of Cbl proteins and inhibits downregulation of EGFR [43]. This results in more potent signaling by EGFR/ErbB-2 het-erodimers as compared with EGFR homodimers [43]. Amplification of ErbB-2, then, would be predicted to increase the fraction of EGFR in heterodimers upon activation, decrease the degradation of the activated EGFR, and increase the potency of the signal upon ligand binding. Further investigation of receptor downregulation in human breast cancer is warranted based on observations such as these.
Growth factor receptor degradation as cancer therapy
Therapies that enhance growth factor receptor downregu-lation have been tested for activity in human cancers (although none were originally developed to target down-regulation).
Monoclonal antibodies that target cell surface receptors have been developed to inhibit receptor function, either by blocking activity or by inducing an immune response to the cells bearing those receptors. In vitro studies have shown that a variety of anti-ErbB-2 antibodies can inhibit cell proliferation and block the growth of tumor xenografts of cells that overexpress ErbB-2 [44, 45]. Trastuzumab (Herceptin®, Genentech, South San Francisco, CA, USA) – the humanized version of one of these antibodies – has clinical activity alone and in combination with chemotherapy in metastatic breast cancer, and has been approved for use in patients whose tumors overexpress ErbB-2 [46, 47]. ErbB-2 overexpressing breast and ovarian cells exposed to Trastuzumab have marked downregulation of ErbB-2, resulting in a selective decrease in activation of the pro-survival kinase AKT [48]. Downregulation of the ErbB-2 receptor and AKT activity has been shown to increase sensitivity of the cells to apoptosis induced by the tumor necrosis factor family of ligands [48, 49]. Recent work has shown that the downregulation induced by these antibodies involves recruitment of Cbl proteins to and subsequent ubiquitination of ErbB-2 [45]. Mechanistically, these antibodies are weak agonists and presumably induce activation of ErbB-2 by homodimerization [50], thus exposing the Cbl-binding sites in a manner similar to homodimerization of EGFR by epidermal growth factor. This in turn results in ubiquitination and downregulation [45]. In contrast, the normal mechanism of activation of ErbB-2 by heterodimerization with one of the other family members results in masking of the Cbl-binding sites [43]. It is likely that the synergistic interaction between Trastuzumab and chemotherapy in patients is due in part to enhanced receptor downregulation.
The ligand-independent mechanism of receptor degradation was discovered in the course of studying the mechanism of action of the benzoquinone ansamycins (e.g. herbimycin A; geldanamycin; and 17-allylamino, 17-demethoxygeldanamycin [17-AAG]) (Fig. 1). This class of drugs was initially identified as inhibitors of the Src tyrosine kinase [51, 52]. Subsequent studies revealed that they had no direct effect on tyrosine kinase activity but instead enhanced degradation of a wide range of proteins, including EGFR and ErbB-2 [51, 52]. These drugs target the molecular chaperones Hsp90 and Grp94 [51, 52]. The chaperone proteins are involved in the stabilization and folding, trafficking, and degradation of proteins. The drugs bind to these chaperone proteins and force the chaperone complex to adopt a protein destabilizing conformation. This results in the ubiquitination and degradation of the proteins that bind to the chaperones [51, 52]. These drugs appear to be able to enhance degradation of both the newly synthesized receptors that have not yet reached the plasma membrane and the mature plasma membrane receptors [53–56].
The mechanism of degradation of membrane receptors by the benzoquinone ansamycins is distinct from ligand-induced degradation. In contrast to the lysosomal mechanism of ligand-induced degradation described above, ligand-independent degradation is mediated by the proteasome [51, 57]. Drug-induced degradation of membrane receptors is independent of kinase activity of the growth factor receptors but requires the presence of the kinase domain [56]. The chaperone complex associates with the membrane receptors via the kinase domain of the receptor, and mutations that inactivate the kinase appear to enhance the association of the chaperones with growth factor receptors [56, 58]. Drug-induced degradation is unaffected by deletion of amino acid sequences carboxyl-terminal to the kinase domain [56]. In contrast, ligand-dependent degradation of growth factor receptors requires activation of kinase activity (mutations that abolish kinase activity block downregulation) and requires phosphorylation of tyrosines in the carboxyl-terminal tails of the receptors (which act as a binding site for Cbl proteins) [12, 45, 59]. In addition, Cbl proteins do not appear to mediate ubiquitination of proteins induced by the benzoquinone ansamycins. A unique E3 (the U-box protein " carboxyl terminus of Hsc70-interacting protein" or CHIP) has been shown to be a chaperone-dependent E3-ligase for several proteins degraded by the drugs (reviewed in [60]). Recent work suggests that CHIP also mediates the drug-induced polyubiquitination of ErbB-2 (Y Yarden, personal communication). Phase I clinical trials are currently underway to explore the use of 17-AAG in cancer patients.
Recent observations suggest that the ligand-independent mechanism of receptor degradation may be induced by small molecule tyrosine kinase inhibitors (TKIs; Fig. 1) [58]. Geldanamycin was observed to enhance ubiquitina-tion and degradation of a kinase dead mutant of EGFR but not of wild-type EGFR. This observation led those investigators to test whether TKIs, which inactivate the kinase biochemically, would have a similar effect to gel-danamycin. Subsequent studies demonstrated that an irreversible TKI (CI-1033) enhanced endocytosis, ubiquitination, and proteasomal degradation of mature ErbB-2 molecules. In addition, the TKI enhanced degradation of nascent ErbB-2 molecules. Mechanistically, it appears that TKIs perturb the structure of the chaperone protein complex associated with ErbB-2. Like the benzoquinone ansamycins, TKIs alter the composition and structure of the chaperone complex to one that favors ubiquitination and degradation of the associated ErbB-2. Thus, the ligand-independent ubiquitination and degradation of membrane receptors may be relevant to the clinical efficacy of TKIs.
The efficacy of agents that affect growth factor receptor downregulation both in experimental models (for Trastuzumab, 17-AAG, and TKIs) and in clinical trials (for Trastuzumab) provides evidence that the development of other agents designed to target downregulation of growth factor receptors is warranted. Obvious directions would be to expand upon the examples above. The development of antibodies and/or drugs that bind to the extracellular domains of growth factor receptors and induce ligand-dependent downregulation can be envisioned for a variety of receptors that are known to be important in the pathogenesis of cancer. Another approach would be to develop small molecules that enhance the ligand-activated down-regulation pathway. One possible approach would be to identify inhibitors of protein kinase C, because this would potentially enhance sorting of the receptors to the degradation pathway. Another approach would be to inhibit the function of proteins such as Sprouty, which negatively regulate Cbl function. Similarly, additional drugs that inhibit the function of Hsp90, Grp94, and other chaperone proteins may have clinical efficacy. As more is learned about the downregulation pathway, additional targets should become apparent.
Of note is that different receptors appear more susceptible to either ligand-dependent or ligand-independent degradation. For example, the EGFR is downregulated to a greater degree than is ErbB-2, ErbB-3, or ErbB-4 in response to ligand activation, but it is relatively insensitive to downregulation induced by benzoquinone ansamycins [41, 58]. In contrast, ErbB-2 is more sensitive than the EGFR to downregulation induced by benzoquinone ansamycins or the TKIs [56, 58]. The development of effective agents to induce downregulation will depend on a solid understanding of the mechanisms relevant to each target receptor.
Another therapeutic approach that is dependent on receptor downregulation for its efficacy is the use of recombinant toxins (for review [61, 62]). In this approach, a toxin such as Pseudomonas exotoxin A lacking its cell-binding domain is targeted to specific cancer cells by conjugation to either a growth factor or an antibody directed at a cell surface receptor [61, 62]. Normally, these toxins bind to cell surface receptors and are internalized via clathrin-coated pits into the endosomal compartment, where they are processed and released into the cytosol [61, 62]. For the recombinant toxin conjugates to be active, they must be efficiently internalized by the cell surface protein to which they are targeted by the fusion partner. Recombinant toxins targeted to growth factor receptors of the EGFR family have been used in experimental models and in phase I clinical trials [63–67]. The importance of internalization was confirmed in studies of a betacellulin–Pseudomonas toxin fusion protein [65]. This recombinant toxin binds to and activates both EGFR and ErbB-4, but is efficiently internalized only upon binding to EGFR. The efficacy of the toxin is much greater in cells that express EGFR as compared with cells that express ErbB-4 [65]. The addition of agents that induce internalization of the growth factor receptor either through the ligand-dependent or the ligand-independent mechanism may enhance the efficacy of the recombinant toxins.
Conclusion
The past several years have seen a dramatic improvement in our understanding of pathways that control growth factor receptor downregulation. Concurrent with this improved understanding is an appreciation that genetic alterations in cancer cells can directly affect receptor downregulation and that this may contribute to the pathogenesis of cancer. Finally, agents that alter downregulation of growth factor receptors have shown promise as therapies for cancer. Thus, novel agents designed specifically to enhance growth factor receptor downregulation may provide a useful class of agents in the treatment of cancer.
Abbreviations
- 17-AAG:
-
17-allylamino, 17-demethoxygeldanamycin
- CHIP:
-
carboxyl terminus of Hsc70-interacting protein
- CIN85:
-
Cbl interacting protein of 85 kDa
- CSF-1:
-
colony stimulating factor-1
- E3:
-
ubiquitin protein ligase
- EGFR:
-
epidermal growth factor receptor
- Hsp:
-
heat shock protein
- PTB:
-
phosphotyrosine binding
- SH:
-
Src homology domain
- TKI:
-
tyrosine kinase inhibitor.
References
Blume-Jensen P, Hunter T: Oncogenic kinase signalling. Nature. 2001, 411: 355-365. 10.1038/35077225.
Weissman AM: Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol. 2001, 2: 169-178. 10.1038/35056563.
Hicke L: Getting' down with ubiquitin: turning off cell-surface receptors, transporters and channels. Trends Cell Biol. 1999, 9: 107-112. 10.1016/S0962-8924(98)01491-3.
Lemmon SK, Traub LM: Sorting in the endosomal system in yeast and animal cells. Curr Opin Cell Biol. 2000, 12: 457-466. 10.1016/S0955-0674(00)00117-4.
Waterman H, Yarden Y: Molecular mechanisms underlying endocytosis and sorting of ErbB receptor tyrosine kinases. FEBS Lett. 2001, 490: 142-152. 10.1016/S0014-5793(01)02117-2.
Hershko A, Ciechanover A: The ubiquitin system. Annu Rev Biochem. 1998, 67: 425-479. 10.1146/annurev.biochem.67.1.425.
Anderson RG: The caveolae membrane system. Annu Rev Biochem. 1998, 67: 199-225. 10.1146/annurev.biochem.67.1.199.
Waugh MG, Lawson D, Hsuan JJ: Epidermal growth factor receptor activation is localized within low-buoyant density, non-caveolar membrane domains. Biochem J. 1999, 337: 591-597. 10.1042/0264-6021:3370591.
Sorkin A: Endocytosis and intracellular sorting of receptor tyrosine kinases. Front Biosci. 1998, 3: d729-d738.
Thien CB, Langdon WY: Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol. 2001, 2: 294-307. 10.1038/35067100.
Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, Elson A, Jovin T, Yarden Y: A mutant EGF-receptor defective in ubiquity-lation and endocytosis unveils a role for Grb2 in negative signaling. Embo J. 2002, 21: 303-313. 10.1093/emboj/21.3.303.
Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, Lipkowitz S, Yarden Y: Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell. 1999, 4: 1029-1040.
Feshchenko EA, Langdon WY, Tsygankov AY: Fyn, Yes, and Syk phosphorylation sites in c-Cbl map to the same tyrosine residues that become phosphorylated in activated T cells. J Biol Chem. 1998, 273: 8323-8331. 10.1074/jbc.273.14.8323.
Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I: Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature. 2002, 416: 183-187. 10.1038/416183a.
Petrelli A, Gilestro GF, Lanzardo S, Comoglio PM, Migone N, Giordano S: The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-Met. Nature. 2002, 416: 187-190. 10.1038/416187a.
Watanabe S, Take H, Takeda K, Yu ZX, Iwata N, Kajigaya S: Characterization of the CIN85 adaptor protein and identification of components involved in CIN85 complexes. Biochem Biophys Res Commun. 2000, 278: 167-174. 10.1006/bbrc.2000.3760.
Take H, Watanabe S, Takeda K, Yu ZX, Iwata N, Kajigaya S: Cloning and characterization of a novel adaptor protein, CIN85, that interacts with c-Cbl. Biochem Biophys Res Commun. 2000, 268: 321-328. 10.1006/bbrc.2000.2147.
Borinstein SC, Hyatt MA, Sykes VW, Straub RE, Lipkowitz S, Boulter J, Bogler O: SETA is a multifunctional adapter protein with three SH3 domains that binds Grb2, Cbl, and the novel SB1 proteins. Cell Signal. 2000, 12: 769-779. 10.1016/S0898-6568(00)00129-7.
Govers R, ten Broeke T, van Kerkhof P, Schwartz AL, Strous GJ: Identification of a novel ubiquitin conjugation motif, required for ligand-induced internalization of the growth hormone receptor. Embo J. 1999, 18: 28-36. 10.1093/emboj/18.1.28.
Strous GJ, van Kerkhof P, Govers R, Ciechanover A, Schwartz AL: The ubiquitin conjugation system is required for ligand-induced endocytosis and degradation of the growth hormone receptor. Embo J. 1996, 15: 3806-3812.
van Kerkhof P, Govers R, Alves dos Santos CM, Strous GJ: Endocytosis and degradation of the growth hormone receptor are proteasome-dependent. J Biol Chem. 2000, 275: 1575-1580. 10.1074/jbc.275.3.1575.
de Melker AA, van der Horst G, Calafat J, Jansen H, Borst J: c-Cbl ubiquitinates the EGF receptor at the plasma membrane and remains receptor associated throughout the endocytic route. J Cell Sci. 2001, 114: 2167-2178.
Polo S, Sigismund S, Faretta M, Guidi M, Capua MR, Bossi G, Chen H, De Camilli P, Di Fiore PP: A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins. Nature. 2002, 416: 451-455. 10.1038/416451a.
Salcini AE, Chen H, Iannolo G, De Camilli P, Di Fiore PP: Epidermal growth factor pathway substrate 15, Eps15. Int J Biochem Cell Biol. 1999, 31: 805-809. 10.1016/S1357-2725(99)00042-4.
Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH: Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol. 2002, 156: 843-854. 10.1083/jcb.200106056.
Ettenberg SA, Magnifico A, Cuello M, Nau MM, Rubinstein YR, Yarden Y, Weissman AM, Lipkowitz S: Cbl-b-dependent coordinated degradation of the epidermal growth factor receptor signaling complex. J Biol Chem. 2001, 276: 27677-27684. 10.1074/jbc.M102641200.
Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, Beguinot L, Geiger B, Yarden Y: c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998, 12: 3663-3674.
Rotin D, Staub O, Haguenauer-Tsapis R: Ubiquitination and endocytosis of plasma membrane proteins: role of Nedd4/Rsp5p family of ubiquitin-protein ligases. J Membr Biol. 2000, 176: 1-17. 10.1007/s002320001079.
Carpenter G, Cohen S: 125I-labeled human epidermal growth factor. Binding, internalization, and degradation in human fibroblasts. J Cell Biol. 1976, 71: 159-171.
Swaminathan S, Amerik AY, Hochstrasser M: The Doa4 deubiquitinating enzyme is required for ubiquitin homeostasis in yeast. Mol Biol Cell. 1999, 10: 2583-2594.
Bao J, Alroy I, Waterman H, Schejter ED, Brodie C, Gruenberg J, Yarden Y: Threonine phosphoylation diverts internalized EGF-receptors from a degradative pathway to the recycling endosome. J Biol Chem. 2000, 275: 26178-26186. 10.1074/jbc.M002367200.
Egan JE, Hall AB, Yatsula BA, Bar-Sagi D: The bimodal regulation of epidermal growth factor signaling by human Sprouty proteins. Proc Natl Acad Sci USA. 2002, 99: 6041-6046. 10.1073/pnas.052090899.
Andoniou CE, Thien CBF, Langdon WY: Tumour induction by activated abl involves tyrosine phosphorylation of the product of the cbl oncogene. EMBO. 1994, 13: 4515-4523.
Bisson SA, Ujack EE, Robbins SM: Isolation and characterization of a novel, transforming allele of the c-Cbl proto-oncogene from a murine macrophage cell line. Oncogene. 2002, 21: 3677-3687. 10.1038/sj.onc.1205510.
Calin G, Herlea V, Barbanti-Brodano G, Negrini M: The coding region of the Bloom syndrome BLM gene and of the CBL proto-oncogene is mutated in genetically unstable sporadic gastrointestinal tumors. Cancer Res. 1998, 58: 3777-3781.
Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, Langdon WY, Park M: Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol Cell. 2001, 8: 995-1004. 10.1016/S1097-2765(01)00378-1.
Mancini A, Koch A, Wilms R, Tamura T: c-Cbl associates directly with the C-terminal tail of the receptor for the macrophage colony-stimulating factor, c-Fms, and down-modulates this receptor but not the viral oncogene v-Fms. J Biol Chem. 2002, 277: 14635-14640. 10.1074/jbc.M109214200.
Roussel MF, Shurtleff SA, Downing JR, Sherr CJ: A point mutation at tyrosine-809 in the human colony-stimulating factor 1 receptor impairs mitogenesis without abrogating tyrosine kinase activity, association with phosphatidylinositol 3-kinase, or induction of c-fos and junB genes. Proc Natl Acad Sci USA. 1990, 87: 6738-6742.
Ridge SA, Worwood M, Oscier D, Jacobs A, Padua RA: FMS mutations in myelodysplastic, leukemic, and normal subjects. Proc Natl Acad Sci USA. 1990, 87: 1377-1380.
Baker A, Cachia P, Ridge S, McGlynn H, Clarke R, Whittaker J, Jacobs A, Padua RA: FMS mutations in patients following cytotoxic therapy for lymphoma. Leuk Res. 1995, 19: 309-318. 10.1016/0145-2126(94)00128-W.
Baulida J, Kraus MH, Alimandi M, Di Fiore PP, Carpenter G: All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J Biol Chem. 1996, 271: 5251-5257. 10.1074/jbc.271.31.18989.
Pinkas-Kramarski R, Alroy I, Yarden Y: ErbB receptors and EGF-like ligands: Cell lineage determination and oncogenesis through combinatorial signaling. J Mammary Gland Biol Neo-plasia. 1997, 2: 97-107. 10.1023/A:1026343528967.
Muthuswamy SK, Gilman M, Brugge JS: Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol Cell Biol. 1999, 19: 6845-6857.
Sliwkowski MX, Lofgren JA, Lewis GD, Hotaling TE, Fendly BM, Fox JA: Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin Oncol. 1999, 26: 60-70.
Klapper LN, Waterman H, Sela M, Yarden Y: Tumor-inhibitory antibodies to HER-2/erbB-2 may act by recruiting c-cbl and enhancing ubiquitination of her-2. Cancer Res. 2000, 60: 3384-3388.
Baselga J, Tripathy D, Mendelsohn J, Baughman S, Benz CC, Dantis L, Sklarin NT, Seidman AD, Hudis CA, Moore J, Rosen PP, Twaddell T, Henderson IC, Norton L: Phase II study of weekly intravenous trastuzumab (Herceptin) in patients with HER2/neu-overexpressing metastatic breast cancer. Semin Oncol. 1999, 26: 78-83.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L: Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001, 344: 783-792. 10.1056/NEJM200103153441101.
Cuello M, Ettenberg SA, Clark AS, Keane MM, Posner RH, Nau MM, Dennis PA, Lipkowitz S: Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. 2001, 61: 4892-4900.
Hudziak RM, Lewis GD, Winget M, Fendly BM, Shepard HM, Ullrich A: p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol Cell Biol. 1989, 9: 1165-1172.
Kumar R, Shepard HM, Mendelsohn J: Regulation of phosphorylation of the c-erbB-2/HER2 gene product by a monoclonal antibody and serum growth factor(s) in human mammary carcinoma cells. Mol Cell Biol. 1991, 11: 979-986.
Blagosklonny MV: Hsp-90-associated oncoproteins: multiple targets of geldanamycin and its analogs. Leukemia. 2002, 16: 455-462. 10.1038/sj.leu.2402415.
Neckers L: Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002, 8 (suppl): S55-S61. 10.1016/S1471-4914(02)02316-X.
Sepp-Lorenzino L, Ma Z, Lebwohl DE, Vinitsky A, Rosen N: Herbimycin A induces the 20 S proteasome- and ubiquitin-dependent degradation of receptor tyrosine kinases. J Biol Chem. 1995, 270: 16580-16587. 10.1074/jbc.270.28.16580.
Mimnaugh EG, Chavany C, Neckers L: Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J Biol Chem. 1996, 271: 22796-22801. 10.1074/jbc.271.9.4974.
Tikhomirov O, Carpenter G: Geldanamycin induces ErbB-2 degradation by proteolytic fragmentation. J Biol Chem. 2000, 275: 26625-26631. 10.1074/jbc.M003114200.
Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, Neckers L: Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem. 2001, 276: 3702-3708. 10.1074/jbc.M006864200.
Supino-Rosin L, Yoshimura A, Yarden Y, Elazar Z, Neumann D: Intracellular retention and degradation of the epidermal growth factor receptor, two distinct processes mediated by benzoquinone ansamycins. J Biol Chem. 2000, 275: 21850-21855. 10.1074/jbc.M001834200.
Citri A, Alroy I, Lavi S, Rubin C, Xu W, Grammatikakis N, Patterson C, Neckers L, Fry DW, Yarden Y: Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. Embo J. 2002, 21: 2407-2417. 10.1093/emboj/21.10.2407.
Labow MA, Baim SB, Shenk T, Levine AJ: Conversion of the lac repressor into an allosterically regulated transcriptional activator for mammalian cells. Mol Cell Biol. 1990, 10: 3343-3356.
Cyr DM, Hohfeld J, Patterson C: Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci. 2002, 27: 368-375. 10.1016/S0968-0004(02)02125-4.
Pastan I: Targeted therapy of cancer with recombinant immunotoxins. Biochim Biophys Acta. 1997, 1333: C1-C6. 10.1016/S0304-419X(97)00021-8.
Pastan I, Chaudhary V, FitzGerald DJ: Recombinant toxins as novel therapeutic agents. Annu Rev Biochem. 1992, 61: 331-354. 10.1146/annurev.bi.61.070192.001555.
Batra JK, Kasprzyk PG, Bird RE, Pastan I, King CR: Recombinant anti-erbB2 immunotoxins containing Pseudomonas exotoxin. Proc Natl Acad Sci USA. 1992, 89: 5867-5871.
Lorimer IA, Wikstrand CJ, Batra SK, Bigner DD, Pastan I: Immunotoxins that target an oncogenic mutant epidermal growth factor receptor expressed in human tumors. Clin Cancer Res. 1995, 1: 859-864.
Mixan B, Cohen BD, Bacus SS, Fell HP, Siegall CB: Betacellulin-Pseudomonas toxin fusion proteins bind but are not cytotoxic to cells expressing HER4; correlation of EGFR for cytotoxic activity. Oncogene. 1998, 16: 1209-1215. 10.1038/sj.onc.1201632.
Dean GS, Pusztai L, Xu FJ, O'Briant K, DeSombre K, Conaway M, Boyer CM, Mendelsohn J, Bast RC: Cell surface density of p185(c-erbB-2) determines susceptibility to anti-p185(c-erbB-2)-ricin A chain (RTA) immunotoxin therapy alone and in combination with anti-p170(EGFR)-RTA in ovarian cancer cells. Clin Cancer Res. 1998, 4: 2545-2550.
Pai-Scherf LH, Villa J, Pearson D, Watson T, Liu E, Willingham MC, Pastan I: Hepatotoxicity in cancer patients receiving erb-38, a recombinant immunotoxin that targets the erbB2 receptor. Clin Cancer Res. 1999, 5: 2311-2315.
Acknowledgement
I thank Allan Weissman and the members of my group (Jing Bao, Ashley Coats, Isaac Darko, Gareth Davies, and Marion Nau) for useful discussion and critical reading of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lipkowitz, S. The role of the ubiquitination-proteasome pathway in breast cancer: Ubiquitin mediated degradation of growth factor receptors in the pathogenesis and treatment of cancer. Breast Cancer Res 5, 8 (2002). https://doi.org/10.1186/bcr541
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr541