
Abstract
Interleukin-17 (IL-17) has been characterized as a proinflammatory cytokine produced by CD4+ CD45RO+ memory T cells. Overproduction of IL-17 was detected in the synovium of patients with rheumatoid arthritis (RA) compared with patients with osteoarthritis. This study examines differentially expressed genes after the stimulation of fibroblast-like synoviocytes of RA patients by IL-17. Among these genes we identified the following: tumor necrosis factor-stimulated gene-6 (TSG-6), IL-6, IL-8, GRO-β, and bone morphogenetic protein-6 with an expression 3.6–10.6-fold that in the unstimulated control. IL-17 augmented the expression of TSG-6, a hyaluronan-binding protein, in a time- and dose-dependent manner. IL-17 showed additive effects with IL-1β and tumour necrosis factor-α on the expression of TSG-6, IL-6 and IL-8. The mitogen-activated protein kinase p38 seems to be necessary for the regulation of TSG-6 expression by IL-17, as shown by inhibition with SB203580. Our results support the hypothesis that IL-17 is important in the pathogenesis of RA, contributing to an unbalanced production of cytokines as well as participating in connective tissue remodeling.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by a relapsing and remitting course of joint inflammation. The chronic inflammation process leads to an excessive hyperplasia of the synovium with proliferation of the synovial lining cells, the generation of new blood vessels, and diffusely scattered or nodular mononuclear cell infiltrates. The proliferation and invasive growth of fibroblast-like cells of the synovium (fibroblast-like synoviocytes; SFCs) results ultimately in the destruction of the joint [1, 2]. Cytokines such as interleukin (IL)-1β or tumour necrosis factor-α (TNF-α) are known to be involved in the perpetuation of the chronic inflammation in RA [3–6]. Overproduction of the proinflammatory cytokine IL-17 was detected in the RA synovium compared with patients with osteoarthritis [7, 8]. IL-17 is a 20–30 kDa glycosylated, homodimeric polypeptide secreted by CD4+ activated memory (CD45RO+) T cells [9, 10]. In the context of arthritis, the effects of IL-17 were associated with joint inflammation and destruction because of the IL-17-stimulated production of MMP-1 and MMP-9 and degradation of proteoglycan, and the IL-17-increased expression of IL-6 and leukemia inhibitory factor in SFCs [11–14]. Recently we showed the increased expression of CXC chemokines such as IL-8, GRO-α and GRO-β after stimulation of SFCs with IL-17 [15]. A blockade of IL-17 in vivo by treatment with a fusion protein of IL-17 receptor with human IgG1 Fc in adjuvant-induced arthritis decreased joint inflammation and bone erosion [16].
To gain knowledge about the effects of IL-17 in SFCs of patients with RA, we studied the expression and modulation of selected genes differentially expressed after stimulation with IL-17. Our results show that IL-17 is an important member of the cytokine network involved in RA.
Materials and methods
Cell culture
SFCs were obtained from nine patients with classical or definite RA (range of ages 27–71 years, with a mean age (± SEM) of 51.9 ± 16.4 years) undergoing surgical synovectomy, by dissociating the minced tissue enzymatically with HBSS (Hank's buffered saline solution) containing 0.5 mg/ml collagenase type II (Sigma, Deisenhofen, Germany), 0.15 mg/ml DNase I (Boehringer Mannheim, Germany) and 5 mM Ca2+. The cells were cultured in RPMI 1640 medium containing 10% fetal calf serum, antibiotics and glutamine, as described previously [17]. Cells were used at confluence at the third to fifth passage. The inhibitors calphostin C (20 μg/ml; Sigma, Deisenhofen, Germany), PD98059 (10 μM; Alexis, Grünberg, Germany), SB203580 (1 μM; Calbiochem, Schwalbach, Germany) and genistein (10 μg/ml; Gibco BRL, Karlsruhe, Germany) were added to the cultures 30 min before incubation with IL-17. Rabbit neutralizing polyclonal anti-hIL-17 antibody (2 μg/ml; Cell Concepts) was co-incubated for 30 min with recombinant IL-17 and added to the cultures.
cDNA synthesis and cDNA array
Isolation of total cellular RNA was described previously [18]. RNA was treated with DNase I (Qiagen) and resuspended in water. The first strand of DNA was synthesized (after a 10 min incubation at 20°C) at 42°C for 50 min by using 500 ng of total RNA in 5.5 μl of diethyl pyrocarbonate water (0.1% diethyl pyrocarbonate-treated water), 2 μl of 5 × first-strand buffer (250 mM Tris/HCl pH 8.3, 375 mM KCl, 15 mM MgCl2), 0.5 μl of dNTP mix (10 mM of each of dATP, dCTP, dGTP, and dTTP), 1 μl of 0.1 M dithiothreitol, 0.5 μl (50 pmol) of random primer (Roche, Mannheim, Germany), and 0.5 μl of Superscript™ II-RT (200 U/μl; Invitrogen, Karlsruhe, Germany). For quantitative RT–PCR, standard RNA and total RNA were converted into cDNA in separate tubes in triplicate.
Total RNA (4-μg, pooled from cultured SFCs of a female RA patient 41 years old) served as starting material for the preparation of a [α-32P]dCTP-labeled cDNA with the cDNA Synthesis Primer Mix (Clontech). For investigating differential gene expression, cDNA was hybridized to the Atlas™ Human 1.2 Array (Clontech) in accordance with the user manual. This array includes 1176 human cDNAs, housekeeping genes and negative controls immobilized on a nylon membrane. After hybridization and washing, the array membrane was exposed to a phosphorimaging screen. Data analysis was performed with AtlasImage Software 1.0. Expression values of transcripts were normalized to the total signal intensity on the membrane. In agreement with the indications of the manufacturers, transcripts with a ratio of normalized expression levels of more than 2 or less than 0.5 were regarded as modulated.
Construction of RNA standards2
The standards were constructed by previously described procedures [15]. In brief, for the construction of standard RNA, a composite primer was synthesized (see Table 1 for primer sequences). Primer 1 contained a sequence for the SP6 RNA polymerase and also one of the specific sequences of the appropriate gene. The product of the PCR amplification with primers 1 and 2 was gel-purified (QIA quick Gel Extraction Kit; Qiagen, Hilden, Germany) followed by transcription in vitro by the SP6 promoter with the Roche transcription system. The recombinant RNA was quantified by the measurement of A260 and used as a standard (after cDNA synthesis) in the quantitative RT–PCR reaction.
Quantitative PCR analysis
For quantification, 1 μl of the reverse transcriptase reaction mixture was added to 25 μl of reaction mixture consisting of 1 × reaction buffer, 1.5 U of Taq polymerase (Qiagen), 1.8 mM MgCl2, 0.1 × SYBR Green (Biozym, Hess. Oldendorf, Germany), dNTPs (each at 200 μM), and primers 3 and 2 (each at 0.5 μM) (Table 1). A negative control without template was included. Samples of six dilutions of the standard cDNA and of the target cDNA were run in triplicate in a Rotor-Gene 2000 (LTF, Wasserburg, Germany). Initial denaturation at 95°C for 300 s was followed by 40 cycles denaturation at 95°C for 15 s, annealing at 60°C for 30 s, and elongation at 72°C for 20 s. The fluorescence intensity of the double-strand-specific SYBR Green, reflecting the amount of PCR product formed, was read after each elongation step at 82°C. RNA amounts were determined with the software Rotor-Gene version 4.04 in quantitation mode.
Western blotting
Supernatants of IL-17-treated SFCs (20 ng/ml) were collected and, after being washed twice with ice-cold PBS, cells were harvested by scraping into ice-cold RIPA buffer (1 × PBS, 1% Nonidet P40, 0.5% sodium deoxycholate). Inhibitors were added in the following concentrations: 1 mM PMSF, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM Na3VO4, and 1 mM NaF (Sigma). The cell lysate was transferred to microcentrifuge tubes and incubated on ice for 60 min; centrifugation was for 20 min at 14,000 r.p.m. and 4°C. Protein concentration in the supernatant was quantified with the BCA (bicinchoninic acid) Protein Assay Reagent Kit (KMF, Leipzig, Germany), and 40 μg of cell lysate protein or cell culture supernatant was used for Western blot analysis. Proteins were electroblotted from NuPAGE gels (NOVEX, Frankfurt-Hoechst, Germany) onto Hybond ECL (enhanced chemiluminescence) membrane (Amersham, Freiburg, Germany). The membrane was blocked for 1 hour with 5% milk in Tris-buffered saline containing Tween 20 (TBST; pH 7.5, 0.1% Tween 20) at 23 ± 2°C. Blots were incubated with the primary antibody (against TNF-stimulated gene-6 [anti-TSG-6], 1:1000 dilution; kindly provided by Dr MT Bayliss, Oxford, UK) in TBST with 5% milk at 23 ± 2°C for 2 hours. Blots were washed three times and then incubated for 1 hour with the secondary antibody (1:1000 dilution; Dianova, Hamburg, Germany) coupled with horseradish peroxidase. Immunodetection was accomplished with ECL Western blotting detection reagents (Amersham) for chemiluminescent detection. Immunoreactivity was quantified by scanning densitometry with the software Scan Pack version 3.0 (Biometra).
Measurement of IL-6 and IL-8
SFCs were cultured for 72 hours in the presence of 20 ng/ml IL-17. Supernatants were collected and assayed for IL-6 and IL-8 with the Chemiluminescence-Enzyme Immunoassay System, Immulite (DPC Biermann, Bad Nauheim, Germany).
Statistical analysis
Data are expressed as means ± SEM. The Wilcoxon rank sum test was used to determine whether two experimental values were significantly different.
Results
Transcriptional activation by IL-17
Cultured SFCs were stimulated for 24 hours with 20 ng/ml IL-17 and differentially expressed genes were analyzed with cDNA array technology. As expected, a set of stimulated transcripts was represented by cytokines and growth factors, including IL-6, IL-8, GRO-β, and bone morphogenetic protein-6 (BMP-6). For the first time we identified the gene of hyaluronan-binding protein TSG-6 as an IL-17-target gene. The expression of the oncogene c-myc was downregulated. The differential expression of the genes was verified by real-time RT–PCR. As shown in Figure 1, we confirmed the upregulation of transcripts of IL-6 (4.2-fold), IL-8 (7.06-fold), GRO-β (10.6-fold), and BMP-6 (3.67-fold) in SFCs obtained from six different RA patients. The mRNA expression of TSG-6 was 4.08-fold higher after stimulation with IL-17 in SFCs of nine different patients with RA. Furthermore, we confirmed the inhibition of c-myc expression.

IL-17-induced gene expression. Fibroblast-like synoviocytes were cultured for 24 hours in the presence of IL-17 (20 ng/ml). Results of quantitative RT–PCR. Six different patients were measured each in duplicate, apart from nine different patients for TSG-6 measurements. *P < 0.05 in comparison with the unstimulated control.
IL-17 shows additive effects with IL-1β and TNF-α on the expression of IL-6 and IL-8
To confirm the array data and the results of real-time RT–PCR on the protein level, we measured the secretion of IL-6 and IL-8 in SFCs after stimulation with IL-17 (20 ng/ml). After 72 hours, the secreted IL-6 and IL-8 amounts were 40.4-fold and 27.7-fold, respectively, those of the untreated control SFCs. We detected an increase in IL-6 level after stimulation with IL-1β (10 ng/ml) and TNF-α (10 ng/ml) to 372-fold and 109-fold, respectively, and in combination with IL-17 to 434-fold and 432-fold, respectively. We also found an augmentation of IL-8 protein secretion after treatment with IL-1β or TNF-α to 1185-fold and 295-fold, respectively. Combinations of IL-1β and IL-17, or TNF-α and IL-17, showed additive effects on IL-8 secretion of 1654-fold and 1593-fold, respectively (Fig. 2).

Combined effects of IL-17 (20 ng/ml) plus IL-1β (10 ng/ml) or IL-17 plus TNF-α (10 ng/ml) on the expression of IL-6 and IL-8. Fibroblast-like synoviocytes were cultured for 72 hours in the presence of cytokines, and the concentrations of IL-6 or IL-8 in supernatants were determined. Measurements were made on synoviocytes from four different patients. *P < 0.05 in comparison with the results of IL-17 stimulation.
Upregulation of hyaluronan-binding protein TSG-6 by IL-17
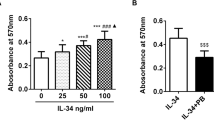
To learn more about the IL-17-stimulated expression of TSG-6, we studied the effect of different IL-17 concentrations (1–100 ng/ml) and of different incubation times (3–48 hours). After 24 hours of IL-17 incubation, significantly higher TSG-6 mRNA levels were observed at IL-17 concentrations between 20 and 100 ng/ml (Fig. 3). We also quantified the transcript level of TSG-6 after IL-17 stimulation with combinations of the cytokines IL-1β and TNF-α. As shown in Figure 4, we detected an upregulation of TSG-6 expression with IL-1β (11.1-fold) as well as with TNF-α (12.9-fold), whereas with IL-17 (20 ng/ml) alone the increase in TSG-6 amount was 4.08-fold. Combinations of IL-1β and IL-17, or TNF-α and IL-17, showed additive effects on TSG-6 expression of 13.6-fold and 16.6-fold, respectively. An IL-17 concentration of 50 ng/ml also synergized with IL-1β and TNF-α to induce the TSG-6 transcript levels. Co-incubation with an anti-IL-17 antibody markedly decreased the IL-17-induced expression of TSG-6 mRNA.

IL-17 stimulates the expression of TSG-6 mRNA. (a) Time course of TSG-6 mRNA expression after stimulation with 20 ng/ml IL-17. (b) Dose-dependent stimulation of TSG-6 mRNA expression after stimulation with IL-17 for 24 hours. Results of quantitative RT–PCR are given as percentages of the basal control (culture without IL-17 set at 100%). Results are from four different patients, each measured in duplicate. *P < 0.05 in comparison with the unstimulated control.

Combined effects of IL-17 (20 or 50 ng/ml) plus IL-1β (10 ng/ml) or IL-17 (20 or 50 ng/ml) plus TNF-α (10 ng/ml) on the expression of TSG-6 mRNA. Results of quantitative RT–PCR are given as percentages of the basal control (culture without IL-17 set at 100%). Results are from six different patients, each measured in duplicate. *P < 0.05 in comparison with the results of IL-1 stimulation.
Having observed IL-17-mediated TSG-6 transcript stimulation, it was important to assess whether protein production was, in fact, stimulated. As shown in Figure 5, and in agreement with mRNA data, exposure of SFCs to 20 ng/ml IL-17 for 48 hours was a potent inducer of TSG-6 protein secreted in the cell culture supernatant (shown in Fig. 5) as well as in the cell extract (data not shown). TSG-6 was detected in both its 35 and 120 kDa forms; the latter was identified as a complex of TSG-6 with the serum protein inter-α-inhibitor. The concentrations of both forms were increased in response to IL-17. Additive effects of IL-17 and IL-1β were observed for all patients tested, whereas with TNF-α we detected in four of six patients additive effects in TSG-6 protein expression (Fig. 5).

Combined effects of IL-17 (20 ng/ml) plus IL-1β (10 ng/ml) or IL-17 (20 ng/ml) plus TNF-α (10 ng/ml) on the expression of TSG-6 protein. Fibroblast-like synoviocytes (SFCs) were cultured for 48 hours with the cytokines indicated below. Cells were lysed, separated, blotted, and probed with a TSG-6 antibody as described in Materials and methods. Lanes 1–6, SFCs of patient 1; lanes 7–12, SFCs of patient 2. Lanes 1 and 7, control; lane 2, IL-17; lanes 3 and 8, IL-1β; lanes 4 and 9, IL-17 plus IL-1β; lanes 5 and 10, TNF-α; lanes 6 and 11, IL-17 plus TNF-α; lane 12, IL-17 plus anti-IL-17 antibody. The intensity was analyzed densitometrically and normalized to the intensity of the appropriate control.
To investigate further the intracellular signaling pathways activated by IL-17 (20 ng/ml) and responsible for inducing TSG-6 expression, SFCs were preincubated separately with cell-permeable inhibitors of MAP kinase/ERK kinase-1/2 (PD98059), p38 (SB203580), protein kinase C (calphostin C), and tyrosine kinase (genistein), followed by the addition of IL-17 and then an analysis of TSG-6 mRNA concentration. Only the inhibitor SB203580 significantly decreased the mRNA expression of TSG-6 stimulated by IL-17 in SFCs; with genistein, calphostin C, and PD98059 we measured no significant decrease in the amount of TSG-6 mRNA (Fig. 6).

Effects of protein kinase inhibitors on the expression of TSG-6 mRNA. Fibroblast-like synoviocytes were cultured for 24 hours with or without IL-17 (20 ng/ml) and the appropriate inhibitor. Total RNA (0.5 μg) was used for cDNA synthesis in a volume of 10 μl; 1.5 μl of the synthesized cDNA was used for real-time PCR as described. Results are given as a percentage of the basal control (culture without cytokine set at 100%). Results are from four different patients, each measured in duplicate. *P < 0.05 in comparison with the results of IL-17 stimulation.
Discussion
IL-17 was found at high levels in the RA synovium, and the concentration of this cytokine in synovial fluid of RA patients is elevated [7, 8]. For the first time we identified an increase in TSG-6 after stimulation of SFCs with IL-17. TSG-6 is a hyaluronan-binding protein found in the synovial fluids of arthritis patients. TSG-6 has a significant homology to the hyaluronan-binding regions present in cartilage link protein, aggrecan, and the adhesion receptor CD44 [19]. The 35 kDa glycoprotein has a role in extracellular matrix remodeling, leucocyte migration, and cell proliferation [20–22]. TSG-6 forms a covalent complex with the serine protease inhibitor inter-α-inhibitor, and thereby increases its anti-plasmin activity. This suggests a role for TSG-6 in the regulation of the plasmin/plasminogen activator system and therefore the control of growth factor and matrix metalloproteinase activation [23, 24]. In TSG-6 transgenic mice with an antigen-induced arthritis, TSG-6 shows a cartilage-specific constitutive expression and provides chondroprotective, but not anti-inflammatory, effects [25]. Similar results were obtained in TSG-6 transgenic mice with collagen type II-induced arthritis. TSG-6 was locally expressed at sites of inflammation and joint destruction, and resulted in potent inhibition of joint destruction [26]. These findings support the hypothesis that endogenously produced TSG-6 can be part of a negative feedback loop in the inflammatory response [21]. IL-17 most probably has a dual role at sites of inflammation, supporting the local inflammatory response but simultaneously delivering the anti-inflammatory TSG-6 protein.
Among the set of upregulated genes after treatment with IL-17, as expected we found IL-6 and the CXC chemokines IL-8 and GRO-β. IL-6, a pleiotropic cytokine in rheumatoid joints produced predominantly by SFCs and synovial macrophages, has a major role in cellular activation [27, 28]. CXC chemokines are potent neutrophil chemoattractants and have been detected in synovial fluids, synovial tissues, and sera of RA patients (reviewed in [29]). With the use of cDNA microarray technology in RA tissue, high chemokine expression has been verified, including that of the CXC chemokines IL-8 and GRO-α [30]. IL-8 induces synovial inflammation [31], might be involved in the regulation of collagen turnover in SFCs [32], and can stimulate angiogenesis [29].
Furthermore, we identified the BMP-6 gene as a gene upregulated by IL-17. BMP-6 is a member of the transforming growth factor-β superfamily with a role in chondrogenesis and the osteoblastic diffentiation process and also serves as a mediator of estrogen's osteogenic action [33–35]. The expression of the proto-oncogene c-myc is not disease-specific in synovial cells because equal levels in samples of patients with RA or osteoarthritis have been reported [36]. Interestingly, antisense oligonucleotides of c-myc can induce apoptosis and downregulation of Fas expression in synoviocytes [37].
The present study demonstrates the potency of IL-17, IL-1β, and TNF-α alone and also in combination to induce the synthesis of IL-6, IL-8, and TSG-6 by SFCs. The three cytokines activate the common transcription factor NF-κB in SFCs and in a variety of other cell types [15]. Indeed, combination of IL-17 with IL-1β often leads to synergistic or additive effects [9, 11, 38, 39]. In contrast, Lubberts et al. reported an IL-1-independent role of IL-17 in synovial inflammation and joint destruction in the autoimmune collagen-induced arthritis model. Local overexpression of IL-17 in the knee joint of mice immunized with collagen type II resulted in elevated levels of IL-1β in the synovium. Blocking IL-1 with neutralizing antibodies had no effect on the IL-17-induced inflammation and joint damage, implying a pathway independent of IL-1 [40]. The interaction of the cytokines IL-1β, IL-17, and TNF-α sustained inflammatory processes within the joint and amplified the involvement of T cells in the pathogenesis of RA.
We and others have found that IL-17 is capable of stimulating the mitogen-activated protein kinase (MAPK) signaling pathways ERK1/2 and p38 as well as the NF-κB pathway [41–43]. However, with the inhibitors genistein, calphostin C, or PD98059 we observed no significant decrease in the amounts of TSG-6 mRNA. MAPK p38 seems necessary for the expression of TSG-6, shown by inhibition with SB203580, and is involved in the IL-17-enhanced production of inducible nitric oxide synthase and secretion of chemokines [15, 42], and has a role in the MMP-9 expression induced by IL-17 [14].
Conclusion
Our results support the hypothesis that IL-17 might have a significant role in the pathogenesis of RA and might contribute to an unbalanced production of cytokines as well as participating in connective tissue remodeling. However, a deeper understanding of the effects of the IL-17 seems necessary for the understanding of its functions as well as for a development of therapeutic approaches including IL-17 and IL-17 receptor as a target.
Abbreviations
- BMP-6:
-
bone morphogenetic protein-6
- IL-17:
-
interleukin-17
- MAPK:
-
mitogen-activated protein kinase
- RT–PCR:
-
reverse transcriptase–polymerase chain reaction
- RA:
-
rheumatoid arthritis
- SFC:
-
fibroblast-like synoviocytes
- TBST:
-
tris-buffered saline containing Tween 20
- TNF-α:
-
tumour necrosis factor-α
- TSG-6:
-
product of TNF-stimulated gene-6.
References
Firestein GS: Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors?. Arthritis Rheum. 1996, 39: 1781-1790.
Weyand CM, Goronzy JJ: The molecular basis of rheumatoid arthritis. J Mol Med. 1997, 75: 772-785. 10.1007/s001090050167.
Breedveld FC: New insights in the pathogenesis of rheumatoid arthritis. J Rheumatol. 1998, 53 (suppl): S3-S7.
Feldmann M, Brennan FM, Maini RN: Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996, 14: 397-440. 10.1146/annurev.immunol.14.1.397.
Feldmann M, Brennan FM, Maini RN: Rheumatoid arthritis. Cell. 1996, 85: 307-310. 10.1016/S0092-8674(00)81109-5.
Koch AE, Kunkel SL, Strieter RM: Cytokines in rheumatoid arthritis. J Invest Med. 1995, 43: 28-38.
Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P: Human interleukin-17: a T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999, 42: 963-970. 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E.
Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W: High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000, 164: 2832-2838.
Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das MB, Rouvier E, Golstein P, Banchereau J, Lebecque S: T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996, 183: 2593-2603. 10.1084/jem.183.6.2593.
Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, Armitage RJ: Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995, 155: 5483-5486.
Chabaud M, Fossiez F, Taupin JL, Miossec P: Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998, 161: 409-414.
Chabaud M, Garnero P, Dayer JM, Guerne PA, Fossiez F, Miossec P: Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine. 2000, 12: 1092-1099. 10.1006/cyto.2000.0681.
Dudler J, Renggli-Zulliger N, Busso N, Lotz M, So A: Effect of interleukin 17 on proteoglycan degradation in murine knee joints. Ann Rheum Dis. 2000, 59: 529-532. 10.1136/ard.59.7.529.
Jovanovic DV, Martel-Pelletier J, Di Battista JA, Mineau F, Jolicoeur FC, Benderdour M, Pelletier JP: Stimulation of 92-kd gelatinase (matrix metalloproteinase 9) production by interleukin-17 in human monocyte/macrophages: a possible role in rheumatoid arthritis. Arthritis Rheum. 2000, 43: 1134-1144. 10.1002/1529-0131(200005)43:5<1134::AID-ANR24>3.0.CO;2-#.
Kehlen A, Thiele K, Riemann D, Langner J: Expression, modulation and signalling of IL-17 receptor in fibroblast- like synoviocytes of patients with rheumatoid arthritis. Clin Exp Immunol. 2002, 127: 539-546. 10.1046/j.1365-2249.2002.01782.x.
Bush KA, Farmer KM, Walker JS, Kirkham BW: Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis Rheum. 2002, 46: 802-805. 10.1002/art.10173.
Schwachula A, Riemann D, Kehlen A, Langner J: Characterization of the immunophenotype and functional properties of fibroblast-like synoviocytes in comparison to skin fibroblasts and umbilical vein endothelial cells. Immunobiology. 1994, 190: 67-92.
Chomczynski P, Sacchi N: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987, 162: 156-159. 10.1006/abio.1987.9999.
Bajorath J: Molecular organization, structural features, and ligand binding characteristics of CD44, a highly variable cell surface glycoprotein with multiple functions. Proteins. 2000, 39: 103-111. 10.1002/(SICI)1097-0134(20000501)39:2<103::AID-PROT1>3.3.CO;2-7.
Lee TH, Wisniewski HG, Vilcek J: A novel secretory tumor necrosis factor-inducible protein (TSG-6) is a member of the family of hyaluronate binding proteins, closely related to the adhesion receptor CD44. J Cell Biol. 1992, 116: 545-557. 10.1083/jcb.116.2.545.
Wisniewski HG, Hua JC, Poppers DM, Naime D, Vilcek J, Cronstein BN: TNF/IL-1-inducible protein TSG-6 potentiates plasmin inhibition by inter-alpha-inhibitor and exerts a strong anti-inflammatory effect in vivo. J Immunol. 1996, 156: 1609-1615.
Maier R, Wisniewski HG, Vilcek J, Lotz M: TSG-6 expression in human articular chondrocytes. Possible implications in joint inflammation and cartilage degradation. Arthritis Rheum. 1996, 39: 552-559.
Wisniewski HG, Vilcek J: TSG-6: an IL-1/TNF-inducible protein with anti-inflammatory activity. Cytokine Growth Factor Rev. 1997, 8: 143-156. 10.1016/S1359-6101(97)00008-7.
Wisniewski HG, Burgess WH, Oppenheim JD, Vilcek J: TSG-6, an arthritis-associated hyaluronan binding protein, forms a stable complex with the serum protein inter-alpha-inhibitor. Biochemistry. 1994, 33: 7423-7429. 10.1021/bi00189a049.
Glant TT, Kamath RV, Bardos T, Gal I, Szanto S, Murad YM, Sandy JD, Mort JS, Roughley PJ, Mikecz K: Cartilage-specific constitutive expression of TSG-6 protein (product of tumor necrosis factor alpha-stimulated gene 6) provides a chondroprotective, but not antiinflammatory, effect in antigen-induced arthritis. Arthritis Rheum. 2002, 46: 2207-2218. 10.1002/art.10555.
Mindrescu C, Dias AA, Olszewski RJ, Klein MJ, Reis LF, Wisniewski HG: Reduced susceptibility to collagen-induced arthritis in DBA/1J mice expressing the TSG-6 transgene. Arthritis Rheum. 2002, 46: 2453-2464. 10.1002/art.10503.
Hirth A, Skapenko A, Kinne RW, Emmrich F, Schulze-Koops H, Sack U: Cytokine mRNA and protein expression in primary-culture and repeated-passage synovial fibroblasts from patients with rheumatoid arthritis. Arthritis Res. 2002, 4: 117-125. 10.1186/ar391.
Firestein GS, Alvaro-Gracia JM, Maki R, Alvaro-Garcia JM: Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J Immunol. 1990, 144: 3347-3353.
Szekanecz Z, Strieter RM, Kunkel SL, Koch AE: Chemokines in rheumatoid arthritis. Springer Semin Immunopathol. 1998, 20: 115-132. 10.1007/BF00832002.
Heller RA, Schena M, Chai A, Shalon D, Bedilion T, Gilmore J, Woolley DE, Davis RW: Discovery and analysis of inflammatory disease-related genes using cDNA microarrays. Proc Natl Acad Sci USA. 1997, 94: 2150-2155. 10.1073/pnas.94.6.2150.
Endo H, Akahoshi T, Takagishi K, Kashiwazaki S, Matsushima K: Elevation of interleukin-8 (IL-8) levels in joint fluids of patients with rheumatoid arthritis and the induction by IL-8 of leukocyte infiltration and synovitis in rabbit joints. Lymphokine Cytokine Res. 1991, 10: 245-252.
Unemori EN, Amento EP, Bauer EA, Horuk R: Melanoma growth-stimulatory activity/GRO decreases collagen expression by human fibroblasts. Regulation by C-X-C but not C-C cytokines. J Biol Chem. 1993, 268: 1338-1342.
Sekiya I, Colter DC, Prockop DJ: BMP-6 enhances chondrogen-esis in a subpopulation of human marrow stromal cells. Biochem Biophys Res Commun. 2001, 284: 411-418. 10.1006/bbrc.2001.4898.
Yamamoto N, Furuya K, Hanada K: Progressive development of the osteoblast phenotype during differentiation of osteoprogenitor cells derived from fetal rat calvaria: model for in vitro bone formation. Biol Pharm Bull. 2002, 25: 509-515. 10.1248/bpb.25.509.
Plant A, Tobias JH: Increased bone morphogenetic protein-6 expression in mouse long bones after estrogen administration. J Bone Miner Res. 2002, 17: 782-790.
Roivainen A, Pirila L, Yli-Jama T, Laaksonen H, Toivanen P: Expression of the myc-family proto-oncogenes and related genes max and mad in synovial tissue. Scand J Rheumatol. 1999, 28: 314-318. 10.1080/03009749950155517.
Hashiramoto A, Sano H, Maekawa T, Kawahito Y, Kimura S, Kusaka Y, Wilder RL, Kato H, Kondo M, Nakajima H: C-myc antisense oligodeoxynucleotides can induce apoptosis and down-regulate Fas expression in rheumatoid synoviocytes. Arthritis Rheum. 1999, 42: 954-962. 10.1002/1529-0131(199905)42:5<954::AID-ANR14>3.0.CO;2-J.
Kehlen A, Thiele K, Riemann D, Rainov N, Langner J: Interleukin-17 stimulates the expression of IκB α mRNA and the secretion of IL-6 and IL-8 in glioblastoma cell lines. J Neuroimmunol. 1999, 101: 1-6. 10.1016/S0165-5728(99)00111-3.
Chabaud M, Page G, Miossec P: Enhancing effect of IL-1, IL-17, and TNF-α on macrophage inflammatory protein-3α production in rheumatoid arthritis: regulation by soluble receptors and Th2 cytokines. J Immunol. 2001, 167: 6015-6020.
Lubberts E, Joosten LA, Oppers B, van Den BL, Coenen-De Roo CJ, Kolls JK, Schwarzenberger P, van De Loo FA, van Den Berg WB: IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001, 167: 1004-1013.
Attur MG, Patel RN, Abramson SB, Amin AR: Interleukin-17 upregulation of nitric oxide production in human osteoarthritis cartilage. Arthritis Rheum. 1997, 40: 1050-1053.
Martel-Pelletier J, Mineau F, Jovanovic D, Di Battista JA, Pelletier JP: Mitogen-activated protein kinase and nuclear factor κB together regulate interleukin-17-induced nitric oxide production in human osteoarthritic chondrocytes: possible role of transactivating factor mitogen-activated protein kinase-activated protein kinase (MAPKAPK). Arthritis Rheum. 1999, 42: 2399-2409. 10.1002/1529-0131(199911)42:11<2399::AID-ANR19>3.0.CO;2-Y.
Shalom-Barak T, Quach J, Lotz M: Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-κB. J Biol Chem. 1998, 273: 27467-27473. 10.1074/jbc.273.42.27467.
Acknowledgements
We thank Sandra Fuhrmann for technical assistance, Kersten Fischer (Department of Urology, Halle) for quantification of IL-6 and IL-8 protein, Mike Schlicker (Department of Human Genetics, Halle) for support in the array procedure, and Markus Benicke (Department of Pathology, Leipzig) for help with the AtlasImage software.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Rights and permissions
About this article
Cite this article
Kehlen, A., Pachnio, A., Thiele, K. et al. Gene expression induced by interleukin-17 in fibroblast-like synoviocytes of patients with rheumatoid arthritis: upregulation of hyaluronan-binding protein TSG-6. Arthritis Res Ther 5, R186 (2003). https://doi.org/10.1186/ar762
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar762