
Chapter Summary
The expansion of the synovial lining of joints in rheumatoid arthritis (RA) and the subsequent invasion by the pannus of underlying cartilage and bone necessitate an increase in the vascular supply to the synovium, to cope with the increased requirement for oxygen and nutrients. The formation of new blood vessels – termed 'angiogenesis' – is now recognised as a key event in the formation and maintenance of the pannus in RA. This pannus is highly vascularised, suggesting that targeting blood vessels in RA may be an effective future therapeutic strategy. Disruption of the formation of new blood vessels would not only prevent delivery of nutrients to the inflammatory site, but could also lead to vessel regression and possibly reversal of disease. Although many proangiogenic factors are expressed in the synovium in RA, the potent proangiogenic cytokine vascular endothelial growth factor (VEGF) has been shown to a have a central involvement in the angiogenic process in RA. The additional activity of VEGF as a vascular permeability factor may also increase oedema and hence joint swelling in RA. Several studies have shown that targeting angiogenesis in animal models of arthritis ameliorates disease. Our own study showed that inhibition of VEGF activity in murine collagen-induced arthritis, using a soluble VEGF receptor, reduced disease severity, paw swelling, and joint destruction. Although no clinical trials of anti-angiogenic therapy in RA have been reported to date, the blockade of angiogenesis – and especially of VEGF – appears to be a promising avenue for the future treatment of RA.
Similar content being viewed by others

Introduction
Inflammatory joint diseases such as rheumatoid arthritis (RA) are not only a major cause of disability, but are also frequently associated with increased morbidity and mortality. For example, patients with RA have been found to have a higher prevalence of angina pectoris and stroke than nonpatients. Research into the mechanisms underlying musculoskeletal disorders and into the development of newer and more effective therapeutic drugs is thus highly desirable.
RA is a chronic and destructive disease, which typically affects the peripheral joints but may affect any synovial joint in the body. The synovium in RA becomes inflamed and increases greatly in mass, because of hyperplasia of the lining cells. The volume of synovial fluid increases, resulting in joint swelling and pain. Blood-derived cells, including T cells, B cells, macrophages, and plasma cells, infiltrate the sublining of the synovium. Although RA shares these histological features (namely, infiltration and hyperplasia) with other inflammatory arthritides, a particularly characteristic feature of RA is the predilection for the synovium to become locally invasive at the synovial interface with cartilage and bone. This invasive and destructive front (termed 'pannus') causes the erosions observed in RA. Progressive destruction of the articular cartilage, subchondral bone, and periarticular soft tissues eventually combine to produce the deformities characteristic of longstanding RA. These deformities result in functional deterioration and profound disability in the long term.
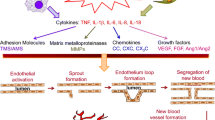
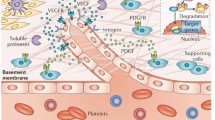
An attribute of RA that has long been recognised but has only recently risen to prominence, because of an increased understanding of the underlying mechanisms, is the role of the vasculature in these invasive and destructive processes. There are abundant blood vessels in RA synovium (Fig. 1) and, given the features of RA outlined above – infiltration by blood-derived cells, hyperplasia, oedema, invasiveness – it is perhaps obvious that these vessels are likely to be involved in the development of RA. Indeed, the endothelial cells lining the blood vessels appear to be an active target for the action of cytokines and mitogens, permeability factors, and matrix-degrading enzymes, and the cells' response to these factors both maintains and promotes RA. In particular, the formation of new blood vessels – 'angiogenesis' – has been suggested to be of importance in the pathogenesis of RA, in that the expansion of synovial tissue necessitates a compensatory increase in the number and density of synovial blood vessels. The arthritic synovium is in fact a very hypoxic environment, which is a potent signal for the generation of new blood vessels.

Expression of CD31 and von Willebrand factor in RA synovium. Frozen or paraffin-embedded sections were stained using antibodies against human CD31 or von Willebrand factor (vWf). Samples were then incubated with biotinylated anti-mouse or anti-goat immunoglobulin, followed by streptavidin–horseradish peroxidase. Immune complexes were detected using 3,3'-diaminobenzidine.
It is now generally accepted that angiogenesis is central to maintaining and promoting RA. It is also possible that a potential method of attenuating development of the pannus is to interfere with its blood supply. This possibility is supported by several recent studies in animal models of arthritis which have suggested that blocking angiogenesis during the course of RA might actually be of therapeutic benefit.
This chapter focuses on the regulation of angiogenesis in RA, on the application of angiogenesis inhibitors in animal models of arthritis, and on the potential for development of new vascular-targeted therapies for treatment of RA.
Historical background
A review in 1982 suggested that in RA, "microcirculatory compromise, concomitant with an increase in metabolic needs of synovial tissue, may initiate tissue injury via anoxia and acidosis, resulting in hydrolytic enzyme release, increased vascular permeability and acceleration of inflammatory processes" [1]. Since that paper by Rothschild and Masi, the number of publications on the PubMed database at the National Library of Medicine citing 'angiogenesis' (or 'angiogenic') and 'arthritis' has risen exponentially, from just 5 in the years 1980–1984, to 130 in 1995–1999. In 2000 and 2001 alone, there were 99 such references (at the time of writing, November 2001).
Changes in the density of blood vessels in the synovium and alterations in endothelial proliferative responses in RA have been shown in a range of studies. For example, the number of synovial blood vessels has been found to correlate with hyperplasia of synovial cells, infiltration of mononuclear cells, and indices of joint tenderness [2]. A morphometric study has suggested that capillaries are distributed more deeply in RA synovium than in normal tissue, although the blood-volume fraction was greater in normal knees than in RA [3]. Another group noted that although perivascular mononuclearcell infiltration and increased thickness of the synovial lining layer were observed in tissue from both inflamed and noninflamed joints of RA patients, vascular proliferation was seen only in tissues from inflamed joints [4]. Endothelial cells lining blood vessels within RA synovium have been shown to express cell-cycle-associated antigens such as PCNA and Ki67, and integrin αvβ3, which is associated with vascular proliferation [5, 6]. Indices of endothelial proliferation and cell death were shown to be higher in synovia from patients with RA than in controls or individuals with osteoarthritis (OA) [6].
The above observations suggest that in RA synovium, there is active endothelial proliferation. This is not surprising, since a consequence of the synovial hyperplasia associated with RA is an increase in the distance between the proliferating cells and the nearest blood vessels. This situation results in local hypoxia and hypoperfusion. Interestingly, it was first reported more than three decades ago that oxygen tension is lowin synovial fluid samples taken from knee joints of people with RA. Lund-Olesen reported that synovial fluid PO2 was 27 mmHg in patients with RA, versus 43 mmHg in patients with OA and 63 mmHg in controls [7]. The augmented proliferation of the synovial cells imposes an additional demand on the vasculature, further promoting the hypoxic state. Although an increase in local blood flow has been reported, this is unlikely to be sufficient to compensate for the increased requirement for oxygen and nutrients. The increase in synovial fluid volume is also likely to compound the hypoxic state in RA, by reducing synovial capillary flow. Resting intra-articular pressure in chronically inflamed joints has been found to be higher than in normal joints, and this effect would be compounded during movement of joints, inducing acute ischemia in the synovial environment [8].
Such a combination of increased metabolic demand and hypoxia is a potent signal for angiogenesis (Fig. 2). Under normal circumstances, the adult vasculature is mostly quiescent, and angiogenesis does not take place except during wound healing and the female reproductive cycle. Disregulated angiogenesis contributes to the pathology of a number of disease states, during which tissue proliferation outstrips the supply of nutrients and oxygen. These include tumour formation, and indeed parallels have been drawn between tumours and the arthritic synovium, with its attendant features of hyperplasia, oedema, angiogenesis, and invasiveness. In one of the earliest reports concerning angiogenesis and arthritis, Brown and colleagues reported that synovial fluids from patients with RA contained a low-molecular-weight angiogenesis factor apparently identical with that derived from tumours [9]. Subsequently, it was shown that synovial fluids from patients with either RA or OA induced morphological changes in endothelial cells in culture, including the formation of tubular networks morphologically resembling capillaries [10].

Why does angiogenesis occur in RA? A consequence of the synovial hyperplasia associated with RA is an increase in the distance between the proliferating cells and the nearest blood vessels. This results in hypoxia and hypoperfusion. The augmented proliferation of the synovial cells imposes an additional demand on the vasculature, further promoting hypoxia. This drives angiogenesis, and hence infiltration and hyperplasia.
Angiogenesis thus contributes to the development and maintenance of RA. Identification of the angiogenic factors has progressed over the intervening years (reviewed [11–14]), and some of the better-characterised proangiogenic stimuli in RA are reviewed in the next section.
Expression of proangiogenic factors in arthritis
A range of growth factors, cytokines, and chemokines are capable of influencing angiogenesis in RA synovium. Many of these substances are thought to act indirectly, by upregulating the expression of more potent and specific angiogenic stimuli (Table 1).
Several growth factors, which are capable of promoting angiogenesis, are in fact broad-range mitogens. Typical examples are the fibroblast growth factors (FGFs), namely FGF-1 (acidic FGF) and FGF-2 (basic FGF). These polypeptide mitogens elicit a variety of responses depending on the target cell type, including proliferation, migration, and differentiation. Both FGF-1 and FGF-2 are expressed in RA: in macrophages, lining cells, and endothelial cells. Similarly, platelet-derived growth factor (PDGF), which is also a potent mitogen for many cell types, including fibroblasts and smooth muscle cells, is expressed in RA synovium. The heparin-binding cytokine hepatocyte growth factor (HGF; scatter factor) has been reported to be expressed in RA. Hepatocyte growth factor promotes directed and random migration of many epithelial cell types and of vascular endothelial cells, and has been found at significant levels in RA synovial fluids (reviewed [15, 16])
In contrast, vascular endothelial growth factor (VEGF) is a relatively endothelial-cell-specific angiogenic factor. The ever-increasing VEGF family is now known to contain at least six related cytokines, although the original member, VEGF, remains the most extensively studied. Alternative mRNA splicing of a single gene yields distinct isoforms of VEGF, with differing properties (Fig. 3). Expression of VEGF is elevated in a range of angiogenesis-associated disease states, such as malignancies, retinal neovascularisation, and psoriasis. VEGF exerts its effects through tyrosine kinase receptors Flt-1 (fms-like tyrosine kinase receptor; also known as VEGF-R1) and Flk-1/KDR (fetal liver kinase receptor/kinase-insert-domain-containing receptor; also known as VEGF-R2) [17, 18]. Additional receptors appear to act as co-receptors. For example, neuropilin-1 acts as a co-receptor for VEGF-R2, enhancing the binding and biological activity of the VEGF-165 isoform. A key feature of VEGF is the upregulation of this growth factor by hypoxia [19]. Several distinct molecular mechanisms are thought to be involved in hypoxia-induced upregulation of VEGF expression, including transcriptional control, through transcription factors such as hypoxia-inducible factor-1 (HIF-1), and post-transcriptional stabilisation of VEGF mRNA [20].

The VEGF family. The binding of VEGF ligands and their splice variants to cell-surface receptors.
The dual activities of VEGF as an endothelial-cell mitogen and a modulator of changes in vascular permeability are of relevance in the pathogenesis of RA. VEGF levels are markedly higher in the serum and synovial fluids of patients with RA than in either patients with OA or normal controls [21–25]. Serum VEGF concentrations in RA patients correlate with levels of C-reactive protein, a marker of inflammation and disease activity [23]. Expression of VEGF mRNA by cells of the lining layer in RA has been reported, and immunohistochemical analyses of synovial biopsies in RA revealed expression of VEGF by synovial lining layers and endothelial cells lining small blood vessels within the pannus [21, 26, 27]. Synovial fluid neutrophils express VEGF at higher levels than are found in fluids from patients with OA [28]. Moreover, microvascular endothelial cells in the vicinity of VEGF-positive cells express mRNA for VEGF receptors [26, 29].
Perhaps the most relevant property of VEGF in the context of angiogenesis and RA is the upregulation of this growth factor by hypoxia. The hypoxic state in the RA joint suggests that the formation of new blood vessels in the pannus may be driven by hypoxia-induced expression of VEGF. Expression of hypoxia-inducible factor-1α by macrophages in RA synovium, predominantly close to the intimal layer but also in the subintimal area, has been described [30]. We have reported that dissociated cells of the synovial membrane in RA respond to hypoxia by upregulating VEGF production. Cells of the synovial membrane in RA were isolated by enzymatic digestion, and after overnight adherence were placed in either normoxic (mean PO2 140 mmHg) or hypoxic (mean PO2 60 mmHg) conditions. After 24 hours in hypoxia, release of VEGF was selectively upregulated, whereas production of IL-1β and IL-8 was unaffected. These observations suggest that a component of the formation of new blood vessels observed in RA may result from hypoxia-driven induction of VEGF [23]. To investigate the relation between tissue oxygen levels and synovial VEGF production in inflammatory arthritis in humans, we examined patients undergoing knee arthroscopy. Synovial PO2 levels were significantly lower in patients with active RA than in patients without RA, and release of VEGF from synovial cells prepared from tissue biopsies was likewise greater for patients with RA. It would appear, therefore, that reduced intra-articular PO2 is likely to be a stimulus for local VEGF production [31].
We have also recently shown that VEGF is important in the development of joint destruction in RA. We observed a significant correlation between serum VEGF at presentation with early RA and the magnitude of radiological deterioration within the first year, calculated using radiographs of hands and feet, taken at initial presentation and at follow-up after 1 year. Radiographs were scored according to the van der Heijde modification of Sharp's method. Patients with radiological deterioration less than the median rate (change after 1 year = 1.5) had lower circulating VEGF concentrations (358 pg/ml) than those with greater than the median rate of radiological deterioration (change after 1 year = 7.5; serum VEGF= 638 pg/ml; P <0.001) [32]. These results suggest that high serum VEGF levels at an early stage of disease are associated with the increased subsequent damage to joints observed by radiography.
More recent studies have addressed the role in arthritis of another important family of molecules involved in angiogenesis, namely the angiopoietins. These molecules, together with their cell-surface receptors Tie-1 and Tie-2, play a key role in development of the vasculature and have been implicated in the control of vessel stabilisation and regression. The patterns of expression of the best-characterised molecules, angiopoietin (Ang)-1 and Ang-2, during embryonic development and during pathological angiogenesis suggest that Ang-1 may act to stabilise new vessels formed in response to VEGF. In contrast, Ang-2 may destabilise blood vessels, which would lead to new vessel sprouts in the presence of VEGF or to regression of vessels in the absence of VEGF. Expression of Tie-1 and Tie-2 in RA synovium has been reported [33]. Detectable levels of mRNA for Ang-1 and its receptors have been shown in specimens of synovial tissue from patients with juvenile RA, in which expression was significantly higher than in tissues from patients with OA or other noninflammatory controls [34]. These observations are perhaps surprising, given that administration of Ang-1 was shown to protect adult mouse vasculature from leaking, countering the permeability activity of VEGF [35].
The levels of an angiogenesis inhibitor, endostatin, were recently reported for patients with RA. VEGF levels in the serum and joint fluid from patients with RA were higher than in patients without RA, whereas endostatin levels were comparable between the groups [36]. My coworkers and I have found that serum levels of the soluble form of the VEGF Flt-1 receptor are raised in RA, as well as in self-limiting arthritis [32]. An inverse relation between the cytokine and its soluble receptor might be predicted. However, raised levels of sFlt-1 observed in RA are presumably insufficient to inhibit VEGF activity. These observations suggest that there may be an imbalance in RA favouring proangiogenic stimuli, whereas inhibitors of angiogenesis such as endostatin are not elevated, or, as in the case of the soluble VEGF Flt-1 receptor, are not increased enough to block the effects of stimuli such as VEGF.
In summary, the invasive pannus in RA is highly vascularised, and numerous growth factors are expressed, which might promote the formation of new blood vessels. Subsequent sections examine the signalling mechanisms involved in the induction of VEGF expression in the context of RA, and the development of new therapies targeting blood vessels in RA.
Angiogenesis blockade in animal models of arthritis
Angiogenesis is clearly a feature of arthritis, with VEGF playing a particularly central role in this process. It seems likely that suppression of the formation of blood vessels should retard the progression of arthritis. There is certainly considerable literature describing the ability of broadly acting angiogenesis inhibitors to modulate disease in animal models. Taxol, TNP-470, and thalidomide – compounds that exert nonspecific anti-angiogenic, as well as other, effects – have all been shown to inhibit pannus formation and neovascularisation [37–39]. For example, in a rat model of arthritis, in which disease is induced by injection of heterologous collagen, leading to synovitis, joint erosion, and associated neovascularisation, TNP-470 was found to suppress established disease. In parallel, there was a marked inhibition of pannus formation and of neovascularisation [37]. TNP-470 has recently been shown to delay onset of arthritis and greatly reduce bone and cartilage destruction if given very early in a transgenic mouse model of arthritis [40].
A hypothesis could also be made that inhibition of VEGF activity should be an effective therapy in RA. We have addressed this hypothesis using the model of collagen-induced arthritis in genetically susceptible mice. To study the association between VEGF and disease severity in murine arthritis, we measured release of this angiogenic cytokine by enzymatically dissociated murine synovial cells. Synovial cells isolated from the knee joints of naive or sham-immunised mice, or from mice immunised with collagen but without arthritis, released little or no detectable VEGF. Onset of arthritis was associated with expression of VEGF, and the levels of VEGF secreted by synovial cells isolated from joints of mice with severe arthritis were significantly higher than from mice with mild disease [41]. We additionally showed that a soluble form of the Flt-1 VEGF receptor (sFlt) significantly reduced disease severity and joint destruction in murine collagen-induced arthritis. Mice treated with a soluble form of this receptor after the onset of arthritis exhibited significantly lower clinical scores and paw swelling than untreated or control-treated animals. These sFlt-treated animals also showed significantly reduced joint inflammation and less destruction of bone and cartilage, as assessed by histology [41].
Later studies, using anti-VEGF polyclonal antibodies, showed the effectiveness of VEGF blockade in collagen-induced arthritis [42, 43]. It therefore appears that VEGF plays a unique role in mediating angiogenesis in RA. Our results using sFlt, and more recent, unpublished data using adenovirus-mediated transfer of VEGF antagonists, suggest that blockade of VEGF activity might be of therapeutic benefit in RA.
Anti-TNF-α antibody in RA: effects on angiogenesis
The findings of elevated expression of angiogenic factors in RA suggest that reducing synovial vascularity may be a desirable component of anti-RA therapies. Certain disease-modifying antirheumatic drugs (DMARDs) have been shown to inhibit angiogenesis in experimental systems. These include drugs such as methotrexate (MTX) [44], sulphasalazine, and penicillamine. Combinations of such drugs also affect production of VEGF by synovial cells in vitro. For example, bucillamine and gold sodium thiomalate inhibited VEGF production, as did a combination of bucillamine, gold sodium thiomalate, and MTX with dexamethasone [45].
Further insights into the importance of reduced angiogenesis in RA were gained from clinical trials of anti-tumour necrosis factor(TNF)-α antibody infliximab – a chimeric mouse Fv, human IgG1, κ antibody of high affinity. From the earliest trials in 1992, infliximab has shown remarkable therapeutic efficacy, reducing both clinical and laboratory indices of disease activity (reviewed [46, 47]). The effects of TNF-α on the angiogenic process are both stimulatory and inhibitory, depending on the system. For example, exposure of endothelial cells to TNF-α has been reported to induce release of VEGF and FGF-2 [48]. Production by synovial-joint cells of angiogenic cytokines such as VEGF is at least in part induced by TNF-α, as was demonstrated in a study showing reduced synovial-cell VEGF release in the presence of anti-TNF-α antibody: my colleagues and I reported that in the presence of anti-TNF-α antibody, spontaneous release of VEGF by RA synovial-membrane cells was decreased. An even greater reduction was observed in the presence of a combination of IL-1-receptor antagonist and anti-TNF-α antibody (inhibition 45%, P <0.05, versus release from untreated cells) [23]. We therefore postulated that part of the benefit of anti-TNF-α antibody in RA was gained through a reduction in synovial vascularity.
To examine this hypothesis, we measured serum VEGF levels in patients with RA who were treated with anti-TNF-α antibody, and observed significant reductions in circulating concentrations of this angiogenic cytokine. In patients receiving 10 mg infliximab per kilogram of body weight, a reduction in serum VEGF levels of more than 40% was achieved, and even 4 weeks after the treatment with the anti-TNF-α serum, VEGF concentrations were significantly below pre-infusion values. Treatment of RA patients with a combination of multiple infusions of infliximab and MTX resulted in a more prolonged decrease in serum VEGF levels than in patients who received infliximab without MTX. We found that infusion of 10 mg infliximab per kilogram of body weight without MTX reduced the levels of circulating VEGF, although these returned to pre-infusion concentrations after the final infusion. In contrast, in patients who received infliximab as well as MTX, this reduction was maintained up to the end of the trial period [23]. These observations suggest that TNF-α regulates production of VEGF in vivo, and that part of the beneficial effect of anti-TNF-α in RA may be a down-modulation in the formation of blood vessels.
In a more recent study, the effects of infliximab on synovial angiogenesis, vascularity, and VEGF expression were investigated [49]. Patients with active RA received a single dose, 10 mg per kilogram of body weight, of anti-TNF-α antibody. Synovial biopsies were taken during arthroscopic examination of the knee joint 1 day before and 2 weeks after treatment, and synovial vascularity was assessed by immunohistochemistry followed by quantitative image analysis. Anti-TNF-α therapy was found to reduce synovial vascularity as assessed by immunostaining for the presence of CD31 and von Willebrand factor. Additionally, a significant reduction in the number of αvβ3-integrin-positive vessels was found. The reduced expression of CD31, von Willebrand factor, and αvβ3 integrin after TNF-α blockade is in agreement with the concept that the balance of new vessel growth and regression is altered such that a net loss of microvessels occurs. Since the endothelial surface plays a key role in mediating cell traffic and delivery of nutrients, such alterations in vascular density may also contribute to therapeutic efficacy. My co-workers and I are currently in the process of using power colour Doppler to examine the effects of anti-TNF-α antibody treatment on synovial vascularity.
Angiogenesis: a realistic target for new therapies in RA?
Therapeutic agents and strategies are being devised to either interrupt or inhibit one or more of the pathogenic steps involved in angiogenesis, and blockade of neovascularisation has been effective in many tumour models. Clearly, angiogenesis can be targeted at several different stages, including inhibition of production of stimuli such as VEGF, binding of proangiogenic factors (using antibodies or soluble receptors), interruption of downstream signalling, blockade of matrix degradation, or even the use of anti-angiogenic stimuli such as endostatin. Many of these approaches have been used with varying degrees of success for human cancers (Table 2; for an updated list of angiogenesis inhibitors in clinical trials see [50]).
In terms of inhibiting the action of VEGF, phase I and phase 1b clinical trial data for pharmacological, safety, and pharmacokinetic studies have been reported for anti-VEGF antibody in patients with solid tumours [51]. Another approach is to use inhibitors of receptor tyrosine kinases, such as SU5416 and SU6668, designed by SUGEN, a company of the Pharmacia Corporation based in South San Francisco. SU5416 has been shown to potently inhibit VEGF-dependent tyrosine phosphorylation, ATP-dependent Flk-1 autophosphorylation, and the proliferation of human endothelial cells. Phase I clinical trials in AIDS-related Kaposi's sarcoma and various solid tumours showed SU5416 to be well tolerated. Most recently, a phase II/III research study of SU5416 in metastatic colorectal cancer completed enrolment [52]. SU6668 is less selective for Flk-1, inhibiting also signalling downstream of the PDGF and the FGF-1 receptors. Currently, SU6668 is in phase I trials for the treatment of advanced solid tumours. None of these compounds is as yet in clinical trials for RA, although our own unpublished data collected using a synthetic inhibitor with relatively greater inhibitory activity for the Flk-1/KDR VEGF receptor showed a significant reduction in clinical score and paw swelling, without any apparent side effects.
The use of anti-angiogenic molecules is less common. In a phase I trial of endostatin at the University of Texas M D Anderson Cancer Center, 25 study patients tolerated the drug well, with few toxic side effects, and two patients showed evidence of some tumour shrinkage [53]. My coworkers and I have recently begun a study in mouse collagen-induced arthritis of K1–5 (protease-activated kringles 1–5), which is related to the potent angiogenesis inhibitor angiostatin. Like several other endogenous anti-angiogenic molecules, angiostatin is a cryptic fragment of a larger molecule lacking in anti-angiogenic activity and is generated as a result of proteolytic cleavage of plasminogen. Angiostatin comprises the first four triple-loop disulfide-linked structures of plasminogen, termed kringle (K) domains. Urokinase-activated plasmin can also convert plasminogen into a molecule containing the intact K1–4 and most of the K5 domains, termed K1–5. This angiogenesis inhibitor K1–5 inhibited the proliferation of endothelial cells more effectively than angiostatin, and suppressed tumour growth and neovascularisation [54]. The effectiveness of treatment with K1–5 treatment in the mouse tumour model prompted us to examine the effects of this inhibitor in the murine model of CIA, and preliminary data are encouraging.
It is not unreasonable to suggest that targeting the newly formed vasculature of the RA pannus, in combination with other therapies such as anti-TNF-α, may lead to a more persistent reduction in pannus volume and hence modify disease progression, but confirmation of this hypothesis requires appropriate clinical trials. Although anti-TNF-α antibody has been shown to reduce serum levels of VEGF by up to 40% in patients with RA, circulating VEGF levels nonetheless remained significantly higher than in healthy individuals [23]. For example, median concentrations of serum VEGF in nonarthritic individuals were equivalent to 160 pg/ml, versus 503 pg/ml in patients with active RA. In patients who received a single infusion of 10 mg/kg infliximab, the maximal change in serum VEGF concentrations was achieved at week 3 (decrease 42%), but the median VEGF concentration was still nearly double that observed in individuals without RA (319 pg/ml). Moreover, not all patients respond to TNF-α blockade. Targeting the inflammatory and vascular components of RA, by combining TNF-α inhibition with angiogenesis blockade, could therefore increase benefit to patients with RA, without augmenting the infection risk.
Concluding remarks
Angiogenesis is, clearly, an important process in the development and perpetuation of RA. Clinical trials in cancer patients of VEGF antibody and small-molecule inhibitors of receptor tyrosine kinases, including those for VEGF, are well under way. It may well be that in the not too distant future, clinical trials of VEGF-targeted therapies may also commence for RA, either alone or in combination with established therapies such as anti-TNF-α antibody.
Naturally, there are undoubted potential drawbacks of anti-angiogenic therapy, such as reduced fertility, impaired healing of fractures, or maybe reduced formation of collateral vessels after an episode of ischaemia. Since patients with RA develop cardiovascular problems at an earlier age than their nonarthritic peers, anti-VEGF therapy might not, therefore, be desirable, in spite of the proven role for VEGF in RA and data showing promising effects of VEGF blockade in animal models. On the other hand, recombinant human VEGF increased the rate and degree of formation of atherosclerotic plaques in the thoracic aorta in a model in cholesterol-fed rabbits, and plasma levels are elevated in atherosclerotic patients [52, 53]. It is thus difficult to predict what the results of angiogenesis inhibition in RA might be, and probably only carefully designed clinical trials will answer this question. In theory, at least, anti-angiogenic treatment should not potentially increase the risk of infection, and a combination of anti-VEGF and infliximab in RA may be beneficial without augmenting potential adverse effects.
Glossary of terms
Ang = angiopoietin; FGF-1 = fibroblast growth factor-1 (acidic FGF); FGF-2 = fibroblast growth factor-2 (basic FGF); Flk-1/KDR = fetal liver kinase receptor/kinase-insert-domain-containing receptor (VEGF-R2); Flt-1 = fms-like tyrosine kinase receptor (VEGF-R1); HGF = hepatocyte growth factor; HIF-1 = hypoxia-inducible factor-1; K = kringle; sFlt-1 = soluble VEGF Flt-1 receptor; Tie = tyrosine kinase with immunoglobulin and epidermal growth factor homology domains.
References
Rothschild BM, Masi AT: Pathogenesis of rheumatoid arthritis: a vascular hypothesis. Semin Arthritis Rheum. 1982, 12: 11-31. 10.1016/0049-0172(82)90020-8.
Rooney M, Condell D, Quinlan W, Daly L, Whelan A, Feighery C, Bresnihan B: Analysis of the histologic variation of synovitis in rheumatoid arthritis. Arthritis Rheum. 1988, 31: 956-963.
Stevens CR, Blake DR, Merry P, Revell PA, Levick JR: A comparative study by morphometry of the microvasculature in normal and rheumatoid synovium. Arthritis Rheum. 1991, 34: 1508-1513.
FitzGerald O, Soden M, Yanni G, Robinson R, Bresnihan B: Morphometric analysis of blood vessels in synovial membranes obtained from clinically affected and unaffected knee joints of patients with rheumatoid arthritis. Ann Rheum Dis. 1991, 50: 792-796.
Ceponis A, Konttinen YT, Imai S, Tamulaitiene M, Li TF, Xu JW, Hietanen J, Santavirta S, Fassbender HG: Synovial lining, endothelial and inflammatory mononuclear cell proliferation in synovial membranes in psoriatic and reactive arthritis: a comparative quantitative morphometric study. Br J Rheumatol. 1998, 37: 170-178. 10.1093/rheumatology/37.2.170.
Walsh DA, Wade M, Mapp PI, Blake DR: Focally regulated endothelial proliferation and cell death in human synovium. Am J Pathol. 1998, 152: 691-702.
Lund-Olesen K: Oxygen tension in synovial fluids. Arthritis Rheum. 1970, 13: 769-776.
Jawed S, Gaffney K, Blake DR: Intra-articular pressure profile of the knee joint in a spectrum of inflammatory arthropathies. Ann Rheum Dis. 1997, 56: 686-689.
Brown RA, Weiss JB, Tomlinson IW, Phillips P, Kumar S: Angiogenic factor from synovial fluid resembling that from tumours. Lancet. 1980, 1 (8170): 682-685. 10.1016/S0140-6736(80)91173-3.
Semble EL, Turner RA, McCrickard EL: Rheumatoid arthritis and osteoarthritis synovial fluid effects on primary human endothelial cell cultures. J Rheumatol. 1985, 12: 237-241.
Paleolog EM, Fava RA: Angiogenesis in rheumatoid arthritis: implications for future therapeutic strategies. Springer Semin Immunopathol. 1998, 20: 73-94.
Paleolog EM, Miotla JM: Angiogenesis in arthritis: role in disease pathogenesis and as a potential therapeutic target. Angiogenesis. 1998, 2: 295-307. 10.1023/A:1009229508096.
Ballara SC, Miotla JM, Paleolog EM: New vessels, new approaches: angiogenesis as a therapeutic target in musculoskeletal disorders. Int J Exp Pathol. 1999, 80: 235-250. 10.1046/j.1365-2613.1999.00129.x.
Paleolog EM, Miotla JM: Rheumatoid arthritis: a target for anti-angiogenic therapy?. In The New Angiotherapy. Edited by: Edited by Fan TP, Kohn EC. Totowa, NJ. 2001, USA: Humana Press Inc;, 129-149.
Koch AE: The role of angiogenesis in rheumatoid arthritis: recent developments. Ann Rheum Dis. 2000, 59 Suppl 1: I65-71. 10.1136/ard.59.suppl_1.i65.
Walsh DA, Pearson CI: Angiogenesis in the pathogenesis of inflammatory joint and lung diseases. Arthritis Res. 2001, 3: 147-153. 10.1186/ar292.
Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z: Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13: 9-22.
Ferrara N: Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol. 2001, 280: C1358-1366.
Shweiki D, Itin A, Soffer D, Keshet E: Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992, 359: 843-845. 10.1038/359843a0.
Levy NS, Chung S, Furneaux H, Levy AP: Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem. 1998, 273: 6417-6423. 10.1074/jbc.273.11.6417.
Koch AE, Harlow LA, Haines GK, Amento EP, Unemori EN, Wong WL, Pope RM, Ferrara N: Vascular endothelial growth factor. A cytokine modulating endothelial function in rheumatoid arthritis. J Immunol. 1994, 152: 4149-4156.
Harada M, Mitsuyama K, Yoshida H, Sakisaka S, Taniguchi E, Kawaguchi T, Ariyoshi M, Saiki T, Sakamoto M, Nagata K, Sata M, Matsuo K, Tanikawa K: Vascular endothelial growth factor in patients with rheumatoid arthritis. Scand J Rheumatol. 1998, 27: 377-380. 10.1080/03009749850154429.
Paleolog EM, Young S, Stark AC, McCloskey RV, Feldmann M, Maini RN: Modulation of angiogenic vascular endothelial growth factor (VEGF) by TNFα and IL-1 in rheumatoid arthritis. Arthritis Rheum. 1998, 41: 1258-1265. 10.1002/1529-0131(199807)41:7<1258::AID-ART17>3.0.CO;2-1.
Nagashima M, Yoshino S, Ishiwata T, Asano G: Role of vascular endothelial growth factor in angiogenesis of rheumatoid arthritis. J Rheumatol. 1995, 22: 1624-1630.
Lee SS, Joo YS, Kim WU, Min DJ, Min JK, Park SH, Cho CS, Kim HY: Vascular endothelial growth factor levels in the serum and synovial fluid of patients with rheumatoid arthritis. Clin Exp Rheumatol. 2001, 19: 321-324.
Fava RA, Olsen NJ, Spencer-Green G, Yeo KT, Yeo TK, Berse B, Jackman RW, Senger DR, Dvorak HF, Brown LF: Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med. 1994, 180: 341-346.
Pufe T, Petersen W, Tillmann B, Mentlein R: Splice variants VEGF121 and VEGF165 of the angiogenic peptide vascular endothelial cell growth factor are expressed in the synovial tissue of patients with rheumatoid arthritis. J Rheumatol. 2001, 28: 1482-1485.
Kasama T, Kobayashi K, Yajima N, Shiozawa F, Yoda Y, Takeuchi HT, Mori Y, Negishi M, Ide H, Adachi M: Expression of vascular endothelial growth factor by synovial fluid neutrophils in rheumatoid arthritis (RA). Clin Exp Immunol. 2000, 121: 533-538. 10.1046/j.1365-2249.2000.01272.x.
Ikeda M, Hosoda Y, Hirose S, Okada Y, Ikeda E: Expression of vascular endothelial growth factor isoforms and their receptors Flt-1, KDR, and neuropilin-1 in synovial tissues of rheumatoid arthritis. J Pathol. 2000, 191: 426-433. 10.1002/1096-9896(2000)9999:9999<::AID-PATH649>3.0.CO;2-E.
Hollander AP, Corke KP, Freemont AJ, Lewis CE: Expression of hypoxia-inducible factor 1 alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. 2001, 44: 1540-1544. 10.1002/1529-0131(200107)44:7<1540::AID-ART277>3.0.CO;2-7.
Taylor P, Miotla JM, Etherington P, Winlove P, Young Y, Paleolog E, Maini RN: VEGF release is associated with hypoxia in inflammatory arthritis [abstract]. Arthritis Rheum. 2000, 43 Suppl 9: S296-
Ballara SC, Taylor PC, Reusch P, Marmé D, Feldmann M, Maini RN, Paleolog EM: Raised serum vascular endothelial growth factor levels are associated with destructive change in inflammatory arthritis. Arthritis Rheum. 2001, 44: 2055-2064. 10.1002/1529-0131(200109)44:9<2055::AID-ART355>3.0.CO;2-2.
Uchida T, Nakashima M, Hirota Y, Miyazaki Y, Tsukazaki T, Shindo H: Immunohistochemical localisation of protein tyrosine kinase receptors Tie-1 and Tie-2 in synovial tissue of rheumatoid arthritis: correlation with angiogenesis and synovial proliferation. Ann Rheum Dis. 2000, 59: 607-614. 10.1136/ard.59.8.607.
Scola MP, Imagawa T, Boivin GP, Giannini EH, Glass DN, Hirsch R, Grom AA: Expression of angiogenic factors in juvenile rheumatoid arthritis: correlation with revascularization of human synovium engrafted into SCID mice. Arthritis Rheum. 2001, 44: 794-801. 10.1002/1529-0131(200104)44:4<794::AID-ANR135>3.0.CO;2-7.
Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, Glazer N, Holash J, McDonald DM, Yancopoulos GD: Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000, 6: 460-463. 10.1038/74725.
Nagashima M, Asano G, Yoshino S: Imbalance in production between vascular endothelial growth factor and endostatin in patients with rheumatoid arthritis. J Rheumatol. 2000, 27: 2339-2342.
Oliver SJ, Cheng TP, Banquerigo ML, Brahn E: Suppression of collagen-induced arthritis by an angiogenesis inhibitor, AGM-1470, in combination with cyclosporin: reduction of vascular endothelial growth factor (VEGF). Cell Immunol. 1995, 166: 196-206. 10.1006/cimm.1995.9978.
Arsenault AL, Lhotak S, Hunter WL, Banquerigo ML, Brahn E: Taxol involution of collagen-induced arthritis: ultrastructural correlation with the inhibition of synovitis and neovascularization. Clin Immunol Immunopathol. 1998, 86: 280-289. 10.1006/clin.1997.4479.
Oliver SJ, Cheng TP, Banquerigo ML, Brahn E: The effect of thalidomide and 2 analogs on collagen induced arthritis. J Rheumatol. 1998, 25: 964-969.
de Bandt M, Grossin M, Weber AJ, Chopin M, Elbim C, Pla M, Gougerot-Pocidalo MA, Gaudry M: Suppression of arthritis and protection from bone destruction by treatment with TNP-470/AGM-1470 in a transgenic mouse model of rheumatoid arthritis. Arthritis Rheum. 2000, 43: 2056-2063. 10.1002/1529-0131(200009)43:9<2056::AID-ANR17>3.0.CO;2-2.
Miotla J, Maciewicz R, Kendrew J, Feldmann M, Paleolog E: Treatment with soluble VEGF receptor reduces disease severity in murine collagen-induced arthritis. Lab Invest. 2000, 80: 1195-1205.
Lu J, Kasama T, Kobayashi K, Yoda Y, Shiozawa F, Hanyuda M, Negishi M, Ide H, Adachi M: Vascular endothelial growth factor expression and regulation of murine collagen-induced arthritis. J Immunol. 2000, 164: 5922-5927.
Sone H, Kawakami Y, Sakauchi M, Nakamura Y, Takahashi A, Shimano H, Okuda Y, Segawa T, Suzuki H, Yamada N: Neutralization of vascular endothelial growth factor prevents collagen-induced arthritis and ameliorates established disease in mice. Biochem Biophys Res Commun. 2001, 281: 562-568. 10.1006/bbrc.2001.4395.
Hirata S, Matsubara T, Saura R, Tateishi H, Hirohata K: Inhibition of in vitro vascular endothelial cell proliferation and in vivo neovascularization by low dose methotrexate. Arthritis Rheum. 1989, 32: 1065-1073.
Nagashima M, Wauke K, Hirano D, Ishigami S, Aono H, Takai M, Sasano M, Yoshino S: Effects of combinations of anti-rheumatic drugs on the production of vascular endothelial growth factor and basic fibroblast growth factor in cultured synoviocytes and patients with rheumatoid arthritis. Rheumatology (Oxford). 2000, 39: 1255-1262. 10.1093/rheumatology/39.11.1255.
Maini RN, Taylor PC, Paleolog E, Charles P, Ballara S, Brennan FM, Feldmann M: Anti-tumour necrosis factor specific antibody (infliximab) treatment provides insights into the pathophysiology of rheumatoid arthritis. Ann Rheum Dis. 1999, 58 Suppl 1: I56-60.
Feldmann M, Maini RN: Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned?. Annu Rev Immunol. 2001, 19: 163-196. 10.1146/annurev.immunol.19.1.163.
Yoshida S, Ono M, Shono T, Izumi H, Ishibashi T, Suzuki H, Kuwano M: Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol Cell Biol. 1997, 17: 4015-4023.
Taylor P, Patel S, Paleolog E, McCloskey RV, Feldmann M, Maini RN: Reduced synovial vascularity following TNFα blockade in rheumatoid arthritis [abstract]. Arthritis Rheum. 1998, 41 Suppl 9: S295-
The Angiogenesis Foundation: [http://www.cancer.gov/clinical_trials/doc.aspx?viewid=B0959CBB-3004-4160-A679-6DD204BEE68C]
Margolin K, Gordon MS, Ho mgren E, Gaudreault J, Novotny W, Fyfe G, Adelman D, Stalter S, Breed J: Phase Ib trial of intravenous recombinant humanized monoclonal antibody to vascular endothelial growth factor in combination with chemotherapy in patients with advanced cancer: pharmacologic and long-term safety data. J Clin Oncol. 2001, 19: 851-856.
National Cancer Institute: [http://www.sugen.com/webpage_templates/sec.php3?page_name=trials]
SUGEN Inc.: [http://www.mdanderson.org/Featured_Sites/Endostatin/]
Cao R, Wu HL, Veitonmaki N, Linden P, Farnebo J, Shi GY, Cao Y: Suppression of angiogenesis and tumor growth by the inhibitor K1–5 generated by plasmin-mediated proteolysis. Proc Natl Acad Sci U S A. 1999, 96: 5728-5733. 10.1073/pnas.96.10.5728.
Blann AD, Belgore FM, Constans J, Conri C, Lip GY: Plasma vascular endothelial growth factor and its receptor Flt-1 in patients with hyperlipidemia and atherosclerosis and the effects of fluvastatin or fenofibrate. Am J Cardiol. 2001, 87: 1160-1163. 10.1016/S0002-9149(01)01486-2.
Celletti FL, Hilfiker PR, Ghafouri P, Dake MD: Effect of human recombinant vascular endothelial growth factor165 on progression of atherosclerotic plaque. J Am Coll Cardiol. 2001, 37: 2126-2130. 10.1016/S0735-1097(01)01301-8.
Acknowledgements
The contributions of Dr Sundeept Ballara, Dr Jadwiga Miotla, Dr Claudia Monaco, Dr Peter Taylor, and Ms Sylvia Young and the support of Professor Marc Feldmann and Professor Ravinder N Maini are gratefully acknowledged. The Kennedy Institute of Rheumatology is a Division of the Faculty of Medicine, Imperial College of Science, Technology and Medicine, and receives a Core Grant from the Arthritis Research Campaign of Great Britain.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Paleolog, E.M. Angiogenesis in rheumatoid arthritis. Arthritis Res Ther 4 (Suppl 3), S81 (2002). https://doi.org/10.1186/ar575
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar575