
Abstract
Systemic lupus erythematosus (SLE) is a complex disease characterized by the appearance of autoantibodies against nuclear antigens and the involvement of multiple organ systems, including the kidneys. The precise immunological events that trigger the onset of clinical manifestations of SLE are not yet well understood. However, research using various mouse strains of spontaneous and inducible lupus in the last two decades has provided insights into the role of the immune system in the pathogenesis of this disease. According to our present understanding, the immunological defects resulting in the development of SLE can be categorized into two phases: (a) systemic autoimmunity resulting in increased serum antinuclear and antiglomerular autoantibodies and (b) immunological events that occur within the target organ and result in end organ damage. Aberrations in the innate as well as adaptive arms of the immune system both play an important role in the genesis and progression of lupus. Here, we will review the present understanding - as garnered from studying mouse models - about the roles of various immune cells in lupus pathogenesis.
Similar content being viewed by others

Introduction
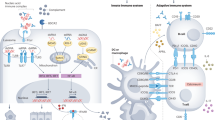
Our understanding of the pathogenesis mechanisms leading to lupus has been augmented by the analysis of several different murine models over the past four decades. Various mouse models of spontaneous lupus have been employed in an effort to understand the cellular and genetic mechanisms behind induction of systemic lupus erythematosus (SLE). The classic models of spontaneous lupus include the F1 hybrid of the New Zealand Black (NZB) and New Zealand White (NZW) strains, called NZB/W F1, and its derivatives, such as NZM2328 and NZM2410, and the MRL/lpr and BXSB/Yaa strains. The goal of this review is to discuss the systemic and local immune responses - as learned from the studies employing various mouse model systems - that lead to the development of lupus. The cellular and molecular mechanisms that contribute to the pathogenesis of lupus are discussed in the following sections and are illustrated in Figure 1.

Pathogenic mechanisms of systemic lupus. Different subpopulations of immune cells interplay to directly or indirectly regulate other subpopulations by releasing different cytokines and other mediators of inflammation, thereby contributing to the progression of the systemic disease. The hyperactivated leukocytes as well as the pathogenic autoantibodies and immune complexes resulting from the systemic disease ultimately play an important role in local autoimmunity and end organ disease within target organs like the kidneys. Ab, antibody; BAFF, B-cell activation factor; DC, dendritic cell; IC, immune complex; IFN-I, interferon-I; IL-10, interleukin-10; MΦ, macrophage; MHC, major histocompatibility complex; self-Ag, self-antigen; TCR, T-cell receptor; TGF-β, transforming growth factor-beta.
Systemic autoimmunity in systemic lupus erythematosus: handling of self-antigens
The presence of high titers of autoantibodies against nuclear antigens is the hallmark of SLE. Whereas there is little evidence to indicate whether self-antigens in SLE are aberrant in sequence or structure, there is evidence that the aberrant handling of self-antigens could facilitate lupus pathogenesis. Although apoptotic bodies are normally rapidly cleared with the help of the complement and other systems, defects in this clearance system could lead to the accumulation of apoptotic bodies and their prevalence in serum [1]. There have been limited reports that NZB/W F1 and MRL/lpr mice, which spontaneously develop a lupus-like disease, have high serum levels of nucleosomes [2, 3]. Non-autoimmune C3H, BALB/c, and C57BL/6 mouse strains, when injected with apoptotic bodies, developed serum autoantibodies similar to those seen in SLE [4]. Although these studies warrant independent confirmation, they support the notion that apoptotic cells could harbor the immunogens responsible for antinuclear antibody formation. Moreover, mice with defects in genes that play an important role in the clearance of apoptotic bodies, like DNase I, serum amyloid protein P (SAP), sIgM, or tyrosine kinase c-mer, all develop SLE-like disease with elevated levels of antinuclear antibodies [5–8]. These data support the hypothesis that the inefficient clearance of apoptotic bodies could be one factor leading to the development of SLE. The important role of the complement system and FcR in the process of clearing apoptotic materials has also been recently documented. Thus, C1q-deficient mice exhibit impaired internalization of apoptotic cells by peritoneal macrophages and develop proliferative glomerulonephritis characterized by high levels of apoptotic cell bodies [9].
Cells of the adaptive immune system as mediators of systemic autoimmunity in systemic lupus erythematosus
B lymphocytes in systemic lupus erythematosus
Autoantibodies are major contributors to end organ damage as illustrated by the glomerulonephritis associated with antinuclear and anti-glomerular antibodies, congenital heart block associated with anti-Ro antibody, and thrombosis associated with anti-cardiolipin antibodies [10]. However, B cells can contribute to SLE pathogenesis through additional pathways. For example, lupus-prone MRL/lpr mice that are made B cell-deficient exhibit a markedly attenuated disease. These mice have an absence of autoantibodies, as expected, but also display a dramatic lack in T-cell activation. This study, by Shlomchik and colleagues [11], implied an essential role for B cells, but not necessarily autoantibodies, in the pathogenesis of lupus [11]. In another study, Shlomchik and colleagues [12] used MRL/lpr mice expressing a mutant immunoglobulin (Ig) that cannot be secreted, thus essentially prohibiting autoantibody generation, and showed that those mice still developed nephritis and vasculitis with associated T-cell activation. That study convincingly showed that B cells may contribute to the development of lupus in ways that are independent of autoantibodies. The additional role of B cells as antigen-presenting cells that mediate T-cell activation in lupus comes from MRL/lpr mice studies showing that B-cell deficiency leads to reduced CD4+ and CD8+ T-cell activation [13].
Besides secreting proinflammatory cytokines like interleukin-6 (IL-6) and interferon-gamma (IFN-γ), B cells may secrete IL-10, which has an anti-inflammatory function. Therefore, B cells that secrete IL-10 (or B10 cells) have been called regulatory B cells and were first reported in B10.PL mice in which IL-10-producing B cells were found to regulate the severity of experimental auto-immune encephalitis [14]. In murine lupus, IL-10 deficiency was found to contribute to more severe disease with an increase in Th1 cytokine levels [15]. A recent study using CD19-/- NZB/W mice reported that B10 cells could induce expansion of regulatory T cells, a subset of T cells with immunosuppressive function [16]. However, data from human lupus reveal a different facet of IL-10. In the case of human SLE, studies suggest that IL-10 promotes rather than inhibits lupus [17]. A better understanding of the role of IL-10-producing B cells and the mechanism of action of IL-10 is required to explain the differences between the murine and human data.
Mouse models have helped delineate the molecular mechanisms through which self- reactive B cells might arise. Sle1 is a lupus susceptibility locus derived from the NZM2410 mouse strain. Using Sle1 congenic C57Bl6 mice, Kumar and colleagues [18] reported that the auto-immune-associated variant of the Ly108 gene, a member of the SLAM family of immune cell receptors which lies within the Sle1 locus, leads to loss of B-cell tolerance because of impaired negative selection of autoreactive B cells at the immature B-cell stage. On the other hand, increased B-cell signaling, especially in the case of mature peripheral B cells, has been implicated in the pathogenesis of humoral autoimmunity. Mice that are deficient in various inhibitory molecules that dampen B-cell receptor (BCR) signaling, such as SHP-1 [19], Lyn [20], or FcγRIIb [21], develop systemic autoimmunity. Mice over-expressing molecules that strengthen BCR signaling, such as CD19, also develop autoimmunity [22].
Recent studies have implicated a class of immune system receptors called Toll-like receptors (TLRs) in the synergistic activation of B cells by BCR and TLR signaling. Simultaneous engagement of BCRs that are specific for DNA by nuclear antigens that are abundant in apoptotic material and TLR9 on B cells causes activation of both BCR and TLR signaling pathways [23]. In the NZB/W and MRL/lpr mouse models of lupus, activation of TLR9 with a synthetic DNA ligand causes accelerated nephritis with increased anti-DNA antibody levels [24]. On the other hand, TLR9-deficient autoimmune mouse models exhibit lower levels of anti-double-stranded DNA (anti-dsDNA) and anti-chromatin antibody [25]. Similarly, B cells possessing BCRs specific for RNA could be synergistically activated by BCR and TLR7 signaling [26]. These findings were supported by research in the BXSB mouse model of lupus. Duplication of the TLR7 gene in the Yaa lupus susceptibility locus leads to increased gene dosage effects that contribute to the lupus-like phenotype seen in this model [27].
Defects that may lead to increased survival of autoreactive B cells have also been found to contribute to the development of autoimmunity. B-cell activation factor (BAFF) is a major B-cell survival factor that functions particularly at the transitional B-cell stage. BAFF transgenic mice develop a lupus-like disease with expansion of the peripheral B-cell pool and heightened production of autoantibodies [28]. Increased BAFF levels are thought to infringe the negative selection checkpoint at the T1 transitional stage, thereby allowing the survival and maturation of autoreactive B cells in the periphery and their participation in the germinal center reaction. These findings are supported by the observations that lupus-prone mice exhibit elevated serum BAFF levels and that administration of TACI-Ig, which acts as a soluble receptor for BAFF, is effective in treating murine lupus [29].
T lymphocytes in systemic lupus erythematosus
The obligatory role of T cells in lupus pathogenesis has been demonstrated by using several genetically engineered mouse models [30]. Lupus T cells are likely to contribute to disease through contact-dependent mechanisms (mediated by CD40L:CD40, OX40L:OX40, and so on) as well as released cytokines [30]. One important cytokine secreted by the Th1 cells is IFN-γ, which plays a pivotal role in the production of pathogenic isotypes of antinuclear autoantibodies in murine lupus models. Expression of IFN-γ in the skin of transgenic mice leads to the production of anti-DNA and anti-Sm auto-antibodies [31]. A deficiency of IFN-γ in MRL/lpr and NZB/W lupus-prone mice leads to reduced anti-DNA antibody levels [32, 33]. In support of these findings, blocking the IFN-γ receptor with monoclonal antibodies or soluble IFN-γ receptor reduces disease activity in lupus-prone mice [34, 35].
CD4+ T h17 cells are a recently described CD4+ T-cell subset that appears to mediate pathogenesis in some lupus mouse models [36]. However, it has become clear that the contribution of Th17 cells to autoimmunity is far more complex, depending on the disease and mouse model [37]. Studies in patients with SLE or related diseases have demonstrated that Th17 cells may have a role in disease development in some, but not all, patients [38]. Given the heterogeneity of the disease, it may be possible that Th17 cells play a role in lupus pathogenesis in a genetically defined subset of subjects or in particular end organ manifestations and that anti-IL -17 therapy may be helpful specifically in those patients. In addition, the regulatory T cell subset appears to modulate the breakdown of T-cell tolerance in lupus [39]. Clearly, we need to gain a better understanding of how pathogenic T cells (Th17 cells) and regulatory T cells, perhaps in yinyang fashion, dictate disease activity in lupus.
Innate immune cells as mediators of systemic autoimmunity in systemic lupus erythematosus
The important role of the innate immune system in SLE pathogenesis has only recently been appreciated. Described below are the possible roles played by three key cell types in the innate immune system.
Monocytes/macrophages
Monocytes/macrophages from patients with lupus have been found to have a reduced capacity to phagocytose apoptotic materials [40], and their numbers were reduced in the inflammatory exudates from patients with SLE [41]. It was postulated, on the basis of those studies, that the defective phagocytosis of monocytes/macrophages contributed to inefficient clearing of apoptotic materials, leading to autoimmunity [42], a notion that is supported by mice lacking the myeloid cell receptors, Tyro3, Axl, and Mer receptor tyrosine kinases [43, 44]. Whereas c-mer knockout mice exhibited increased anti-dsDNA autoantibody levels without any significant renal pathology [43], Tyro3/Axl/Mer triple-knockout mice exhibited high levels of autoantibodies along with multiple end organ involvement [44].
Monocytes/macrophages can contribute to disease in another important way: through the elaboration of proinflammatory cytokines and chemokines. This is illustrated by macrophages from B6.Sle3 congenic mice, in which Sle3 is a lupus susceptibility locus derived from the NZM2410 lupus-prone strain [45]. These murine studies showed that monocytes/macrophages in lupus probably have defects beyond abnormal phagocytosis and that these cells may take part in aberrant regulation of the immune system. A study by Kilmon and colleagues [46] demonstrated that macrophages regulate B-cell tolerance by secreting regulatory factors like IL-6 and CD40L and that macrophages from lupus-prone MRL/lpr mice are less capable of repressing Ig secretion coincident with reduced secretion of these repressive factors. Collectively, these findings implicate a more significant role for monocytes/macrophages in the pathogenesis of lupus, but further studies in patients with SLE are necessary to substantiate these findings.
Polymorphonuclear cells in systemic lupus erythematosus
A neutrophil-specific gene expression signature was reported to correlate positively with SLE disease activity in patients with SLE [47], suggesting that these cells may play an important role in SLE pathogenesis. The exact mechanism by which this occurs is not yet clear. However, a recent study demonstrated that neutrophil extracellular traps, which are networks of DNA, histones, and neutrophil proteins that function primarily to trap microorganisms, may be involved in SLE pathogenesis, possibly by generating autoantigens and serving as adjuvants [48]. The role of neutrophils in murine lupus, unlike that in human lupus, is poorly studied.
Dendritic cells in systemic lupus erythematosus
Studies in mouse models have been pivotal in determining the role that dendritic cells (DCs) play in the pathogenesis of lupus. One of the earliest findings implicating the putative role of DCs in the breakdown of tolerance was that transferring DCs from normal donor mice to normal recipient mice could cause loss of tolerance in the host, leading to the appearance of anti-DNA and anticardiolipin autoantibodies [49]. A study by Chen and colleagues [50] showed that mice with defective DC apoptosis accumulated large numbers of DCs and exhibited chronic lymphocyte activation and systemic autoimmunity.
Other studies using murine models have helped in identifying the genetic loci responsible for imparting the abnormal phenotypes seen in DCs from lupus-prone mice. One such locus is Sle3, which is a lupus susceptibility locus derived from the NZM2410 strain of lupus-prone mice. C57BL/6 (B6) mice congenic for Sle3 develop moderate glomerulonephritis along with modest levels of antinuclear antibodies [51]. Moreover, the DCs from B6.Sle3 congenics are less susceptible to apoptosis, have a more mature phenotype, and are more proinflammatory. They are also more efficient in stimulating T cells in vitro and eliciting autoantibodies in vivo compared with DCs from normal B6 mice [45]. These studies provided insights into genetic factors and immune mechanisms related to abnormal DC activation in murine lupus.
Studies have also shown that DCs play an important role in SLE pathogenesis by secreting potent proinflammatory cytokines, such as type I IFNs (IFN-α) [52]. Immune complexes (ICs) purified from sera of patients with SLE were found to stimulate plasmacytoid DCs (pDCs) to produce IFN-α [53]. Later studies revealed a possible role of TLRs as potent inducers of IFN-α production by activated pDCs in lupus. IFN-α secreted by activated pDCs was found to directly influence B cells to produce antibodies of the IgG subclass against soluble autoantigens in mice [54]. This cytokine also upregulated BAFF expression by monocytes and mature DCs, which promotes both the survival of autoreactive B cells and their differentiation into plasmablasts [55]. Together, these reciprocal events lead to a pathogenic cycle, in which both the innate and adaptive immune systems cooperate and feedback upon each other, eventually leading to high titers of autoantibodies.
Local autoimmunity in systemic lupus erythematosus
SLE is an autoimmune disease that affects multiple organ systems, including the kidney, skin, heart, lung, and hematopoietic and nervous systems. As reviewed below and illustrated in Figure 1, the players mediating local autoimmunity and renal disease in lupus can be classified broadly as (a) ICs and infiltrating cells of hematological origin and (b) resident cells of the kidneys.
Role of immune complexes and infiltrating cells of hematological origin in lupus nephritis
Anti-DNA and DNA ICs were the first ICs isolated from kidney elutes of patients with SLE and mouse models [56]. Intraperitoneal injection of human and murine anti-DNA antibodies was found to trigger lupuslike kidney disease [57], establishing a direct role of these antibodies in the pathogenesis of lupus nephritis. Similarly, transgenic overexpression of Ig anti-DNA antibodies in mouse models can lead to renal pathology similar to that seen in human lupus nephritis, highlighting a causal role for autoantibodies in the development of lupus nephritis [58].
Various mechanisms have been proposed to explain how anti-DNA antibodies may lead to renal disease in lupus [59]. Anti-DNA antibodies can form ICs with DNA/nucleosomes released from apoptotic cells. Eventually, ICs become deposited in the kidney, leading to the initiation of a local inflammatory response through FcγR-or complement-mediated mechanisms or both. Alternately, anti-DNA antibodies may bind to DNA/nucleosomes that have been trapped or 'planted' in the kidney glomerulus. A third mechanism suggests possible cross-reactivity between anti-DNA antibodies and local renal antigens like laminin, alpha-actinin, and heparan sulfate [60].
Other studies have shown that anti-DNA antibodies are not an absolute requirement for development of SLE. NZM2328.Lc4 mice exhibit lupus-like disease with glomerular IC deposits and eventually develop fatal lupus nephritis. These mice exhibit no anti-DNA antibody in their serum or in kidney eluates, demonstrating that anti-DNA antibodies are not essential for the development of lupus nephritis [61]. Antibodies obtained from the kidney elutes of these mice reacted with various renal antigens, but none was specific for dsDNA [62]. Another study eluted Igs from renal tissues obtained from patients with lupus at autopsy and screened the Igs against a panel of 14 different antigens, which included dsDNA, chromatin, Sm, SSA, SSB, and histones [63]. These authors found that only 0.3% to 41.3% of total kidney-eluted IgG reacted with these antigens. Therefore, a vast majority of renal antibodies bind to yet-unknown antigens. In support of this idea, a study using a glomerular proteome array showed that serum from lupus mice and patients reacts with multiple nuclear and matrix-derived autoantigens [64].
Role of lymphoid cells in lupus nephritis
Lymphoid aggregates with T cells and B cells have been observed within the kidneys afflicted by lupus nephritis, although their intrarenal roles remain to be fully appreciated. T cells also play a major role in pathogenesis of SLE. Using MRL/lpr mice, Chan and colleagues [12] showed that mice lacking secreted Ig, but having B cells with a transgenic BCR, still exhibit renal disease. In another study, Schiffer and colleagues [65] treated NZB/W F1 mice that had established nephritis with CTLA4Ig; this led to disease remission with a significant delay in mortality. Using a transgenic mouse model system, Heymann and colleagues [66] demonstrated that both cytotoxic T (Tc) cells and Th cells have direct roles in glomerulonephritis. The authors used transgenic mice expressing the model antigens ovalbumin and hen egg lysozyme in glomerular epithelial cells called podocytes. Co-injection of ovalbumin-specific transgenic CD8+ Tc and CD4+ Th cells into these mice resulted in periglomerular mononuclear infiltrates and inflammation of parietal epithelial cells, similar to lesions frequently observed in human chronic glomerulonephritis. The ovalbumin-specific CD8+ Tc cells were found to be activated and expanded in the renal lymph nodes, and the activation of T cells in turn could be prevented by depletion of DCs. Crosstalk between tubulointerstitial DCs and Th cells resulted in intrarenal cytokine and chemokine production and in recruitment of more Tc cells, monocyte-derived DCs, and macrophages. That study was the first direct evidence that glomerular antigen-specific Tc and Th cells can induce glomerulonephritis in a process regulated by DCs. Although similar studies in the context of lupus nephritis are eagerly awaited, the above studies suggest that type IV hypersensitivity-driven mechanisms are likely to be important in mediating autoimmune nephritis.
Kidneys of nephritic NZM2328 mice show high levels of expression of Th1-type cytokines like IFN-γ and IL-12 [67]. Administration of IFN-γ, a Th1 cytokine, to NZB/W F1 mice exacerbates disease, whereas the deficiency of IFN-γ in MRL/lpr mice ameliorates nephritis [32, 34]. Similarly, inhibition of IL-18, a cytokine that induces IFN-γ and other Th1 cytokines, was found to cause a significant reduction in lymphoproliferation and IFN-γ production and resulted in a decrease in lupus nephritis [68]. These studies reinforce the concept that type IV hypersensitivity reactions may also contribute to lupus nephritis.
Other studies have shown a possible role for IL-4, a Th2-type cytokine, in glomerulosclerosis in which transgenic overexpression of IL-4 was found to facilitate glomerulosclerosis [69]. In various studies, IL-4 was found to have a direct effect on fibroblast proliferation, collagen gene expression, and collagen synthesis [70]. Moreover, inhibition of IL-4 signaling in various lupus-prone mouse models was found to protect mice from developing advanced nephritis in the presence of anti-DNA antibody and infiltrating inflammatory cells [32]. Therefore, IL-4 appears to play a pivotal role in the pathogenesis of glomerulosclerosis and chronic renal fibrosis in lupus by acting directly upon resident renal cells.
Role of myeloid cells in lupus nephritis
Infiltration of DCs in the glomeruli and tubulointerstitial spaces of the kidneys has been reported in various murine models of nephritis, including NZB/W F1 [67], anti-glomerular basement membrane (anti-GBM) [71], and mice engineered to express a foreign antigen on podocytes [66]. DCs may promote lupus nephritis by several mechanisms. One mechanism may be through migration of DCs to local lymph nodes and presentation of renal antigens to autoreactive lymphocytes in the local lymph nodes, thereby initiating an autoimmune response against renal autoantigens [72]. DCs may also secrete a variety of different proinflammatory and profibrotic cytokines like IL-6, IL-1, IL-18, IFN-α, tumor necrosis factor-alpha (TNF-α), and transforming growth factor-beta to promote renal inflammation and fibrosis [73]. Despite these possibilities, we do not currently understand the degree to which intrarenal DCs are important for lupus pathogenesis or the exact mechanisms through which they operate.
Similar to DCs, macrophages are recruited to the nephritic kidneys of patients with SLE and lupus-prone mice [74, 75] and influence the progression of lupus nephritis. In a study using poly(I:C)-induced lupus nephritis in NZB/W mice, proliferative glomerulonephritis was found to be associated with renalinfiltrating macrophages and renal expression of IFN-inducible genes, matrix metalloproteinases (MMPs), and growth factors [76]. Glomerular crescent formation and renal MMP and growth factor expression were dependent on renal macrophages that expressed IL-10, MMPs, osteopontin, and various growth factors, including platelet-derived growth factor-C (PDGF-C) and heparin-binding epidermal growth factor-like growth factor (HB-EGF) [76]. These findings accord a potentially important role for macrophages as mediators of aggressive nephritis in lupus by driving the expression of various cytokines and growth factors in the kidney. Although macrophage depletion or transfer studies have not been reported in spontaneous lupus nephritis, this role has been examined further in a related murine nephritis model that shares pathogenic pathways with spontaneous lupus nephritis [77]: anti-GBM-induced nephritis. In the latter model, depletion and adoptive transfer studies have demonstrated a critical role for macrophages in the pathogenesis of nephritis [78, 79].
By implanting renal tubular epithelial cells genetically modified to produce colony-stimulating factor-1 (CSF-1) under the renal capsule of MRL/lpr mice, Kelley and colleagues [80] showed that CSF-1 and TNF-α released by tubular epithelial cells were responsible for promoting renal pathology by fostering macrophage accumulation. In a study using NZB/W lupus-prone mice, Schiffer and colleagues [75] identified an activated type II macrophage population as a key marker for proteinuria onset and disease remission and suggested that therapy for lupus nephritis include strategies that prevent both activation of monocytes and their migration to the kidney.
Neutrophils, another myeloid cell subtype, have also been shown to infiltrate nephritic kidneys and contribute to the pathogenesis of experimental nephritis [81], although their role in lupus nephritis is less well defined. Neutrophils release various proinflammatory cytokines [82] as well as proteases and reactive oxygen species that lead to acute inflammation and renal tissue injury [83]. Further studies in murine models are clearly warranted.
Role of resident renal cells in lupus nephritis
It is clear that many intrinsic renal cells, including podocytes and tubular epithelial cells, have the capacity to costimulate intrarenal T cells via ICAM-I, MHC-II, B-7, and/or CD40-mediated mechanisms [84–86] or by releasing pathogenic cytokines [87]. However, the extent to which these intrarenal costimulatory mechanisms and cytokines are operative in spontaneous lupus nephritis remains unknown. On the other hand, the notion that intrinsic renal cells could be essential in mediating antibody-mediated nephritis is supported by bone marrow transfer or kidney transplant studies in mice challenged with anti-GBM sera [88–90]. These studies have defined the roles of MHC-II, INF-α, and Fn14 expression on intrinsic renal cells in mediating nephritis. Overall, we have a very limited understanding of the role that intrinsic renal cells play in spontaneous lupus nephritis.
Conclusions
As discussed in this review, mouse models have proven to be indispensable for understanding the immunological and molecular basis of systemic autoimmunity in lupus. However, it is becoming increasingly clear that local autoimmunity in target organs and the responses that target organs play are equally important in disease pathogenesis. Murine models are also proving to be instrumental in helping us unravel the intricate mechanisms that underlie the end organ responses in lupus, although the specific roles of various cell types warrant systematic investigation. Mouse models have also enhanced our understanding of the respective roles of various cells and molecules in the innate immune system and the interplay between the innate and adaptive immune systems in lupus pathogenesis. The challenge ahead is to better define the cellular and molecular players orchestrating lupus and to translate our improved understanding of lupus pathogenesis into better rationalized therapeutics targeting selected cells or molecules (or both) that facilitate lupus.
Note
Autoimmune Basis of Rheumatic Diseases
This article is part of a series on Systemic lupus erythematosus, edited by David Pisetsky, which can be found online at http://arthritis-research.com/series/lupus
This series forms part of a special collection of reviews covering major autoimmune rheumatic diseases, available at: http://arthritis-research.com/series/abrd
Abbreviations
- anti-dsDNA:
-
anti-double-stranded DNA
- BAFF:
-
B-cell activation factor
- BCR:
-
B-cell receptor
- CSF-1:
-
colony-stimulating factor-1
- DC:
-
dendritic cell
- GBM:
-
glomerular basement membrane
- IC:
-
immune complex
- IFN:
-
interferon
- Ig:
-
immunoglobulin
- IL:
-
interleukin
- MMP:
-
matrix metalloproteinase
- NZB:
-
New Zealand Black
- NZW:
-
New Zealand White
- pDC:
-
plasmacytoid dendritic cell
- SLE:
-
systemic lupus erythematosus
- Tc:
-
cytotoxic T
- TLR:
-
Toll-like receptor
- TNF-α:
-
tumor necrosis factor-alpha.
References
Manderson AP, Botto M, Walport MJ: The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004, 22: 431-456. 10.1146/annurev.immunol.22.012703.104549.
Lambert PH, Dixon FJ: Pathogenesis of the glomerulonephritis of NZB/W mice. J Exp Med. 1968, 127: 507-522. 10.1084/jem.127.3.507.
Licht R, van Bruggen MC, Oppers-Walgreen B, Rijke TP, Berden JH: Plasma levels of nucleosomes and nucleosome-autoantibody complexes in murine lupus: effects of disease progression and lipopolyssacharide administration. Arthritis Rheum. 2001, 44: 1320-1330. 10.1002/1529-0131(200106)44:6<1320::AID-ART224>3.0.CO;2-X.
Mevorach D, Zhou JL, Song X, Elkon KB: Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med. 1998, 188: 387-392. 10.1084/jem.188.2.387.
Walport MJ: Lupus, DNase and defective disposal of cellular debris. Nat Genet. 2000, 25: 135-136. 10.1038/75963.
Cohen PL, Caricchio R, Abraham V, Camenisch TD, Jennette JC, Roubey RA, Earp HS, Matsushima G, Reap EA: Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002, 196: 135-140. 10.1084/jem.20012094.
Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J: Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci USA. 2000, 97: 1184-1189. 10.1073/pnas.97.3.1184.
Bickerstaff MC, Botto M, Hutchinson WL, Herbert J, Tennent GA, Bybee A, Mitchell DA, Cook HT, Butler PJ, Walport MJ, Pepys MB: Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nat Med. 1999, 5: 694-697. 10.1038/9544.
Botto M, Dell'Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ: Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998, 19: 56-59. 10.1038/ng0598-56.
Grammer AC, Dorner T, Lipsky PE: Abnormalities in B cell activity and the immunoglobulin repertoire in human systemic lupus erythematosus. Mol Pathol Autoimmune Dis. 2001, 2: 282-318.
Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D: The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994, 180: 1295-1306. 10.1084/jem.180.4.1295.
Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ: A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999, 189: 1639-1648. 10.1084/jem.189.10.1639.
Chan OT, Sh lomchik MJ: Cutting edge: B cells promote CD8+ T cell activation in MRL-Fas(lpr) mice independently of MHC class I antigen presentation. J Immunol. 2000, 164: 1658-1662.
Wolf SD, Dittel BN, Hardardottir F, Janeway CA: Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J Exp Med. 1996, 184: 2271-2278. 10.1084/jem.184.6.2271.
Yin Z, Bahtiyar G, Zhang N, Liu L, Zhu P, Robert ME, McNiff J, Madaio MP, Craft J: IL-10 regulates murine lupus. J Immunol. 2002, 169: 2148-2155.
Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, Sato S, Tedder TF, Fujimoto M: Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol. 2010, 184: 4801-4809. 10.4049/jimmunol.0902385.
Llorente L, Richaud-Patin Y, García-Padilla C, Claret E, Jakez-Ocampo J, Cardiel MH, Alcocer-Varela J, Grangeot-Keros L, Alarcón-Segovia D, Wijdenes J, Galanaud P, Emilie D: Clinical and biologic effects of anti-interleukin-10 monoclonal antibody administration in systemic lupus erythematosus. Arthritis Rheum. 2000, 43: 1790-1800. 10.1002/1529-0131(200008)43:8<1790::AID-ANR15>3.0.CO;2-2.
Kumar KR, Li L, Yan M, Bhaskarabhatla M, Mobley AB, Nguyen C, Mooney JM, Schatzle JD, Wakeland EK, Mohan C: Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. 2006, 312: 1665-1669. 10.1126/science.1125893.
Shultz LD, Rajan TV, Greiner DL: Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotechnol. 1997, 15: 302-307. 10.1016/S0167-7799(97)01060-3.
Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, Stacker SA, Dunn AR: Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell. 1995, 83: 301-311. 10.1016/0092-8674(95)90171-X.
Bolland S, Ravetch JV: Spontaneous autoimmune disease in Fc(gamma) RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000, 13: 277-285. 10.1016/S1074-7613(00)00027-3.
Taylor DK, Ito E, Thorn M, Sundar K, Tedder T, Spatz LA: Loss of tolerance of anti-dsDNA B cells in mice overexpressing CD19. Mol Immunol. 2006, 43: 1776-1790. 10.1016/j.molimm.2005.11.003.
Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A: Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003, 19: 837-847. 10.1016/S1074-7613(03)00323-6.
Hasegawa K, Hayashi T: Synthetic CpG oligodeoxynucleotides accelerate the development of lupus nephritis during preactive phase in NZB × NZWF1 mice. Lupus. 2003, 12: 838-845. 10.1191/0961203303lu483oa.
Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ: Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005, 202: 321-331. 10.1084/jem.20050338.
Krieg AM, Vollmer J: Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev. 2007, 220: 251-269. 10.1111/j.1600-065X.2007.00572.x.
Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S: Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006, 312: 1669-1672. 10.1126/science.1124978.
Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL: Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999, 190: 1697-1710. 10.1084/jem.190.11.1697.
Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, Xu W, Parrish-Novak J, Foster D, Lofton-Day C, Moore M, Littau A, Grossman A, Haugen H, Foley K, Blumberg H, Harrison K, Kindsvogel W, Clegg CH: TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000, 404: 995-999. 10.1038/35010115.
Peng SL, Craft J: T cells in murine lupus: propagation and regulation of disease. Mol Biol Rep. 1996, 23: 247-251. 10.1007/BF00351176.
Seery JP, Carroll JM, Cattell V, Watt FM: Antinuclear autoantibodies and lupus nephritis in transgenic mice expressing interferon gamma in the epidermis. J Exp Med. 1997, 186: 1451-1459. 10.1084/jem.186.9.1451.
Peng SL, Moslehi J, Craft J: Roles of interferon-gamma and interleukin-4 in murine lupus. J Clin Invest. 1997, 99: 1936-1946. 10.1172/JCI119361.
Haas C, Ryffel B, Le Hir M: IFN-gamma is essential for the development of autoimmune glomerulonephritis in MRL/Ipr mice. J Immunol. 1997, 158: 5484-5491.
Jacob CO, van der Meide PH, McDevitt HO: In vivo treatment of (NZB × NZW)F1 lupus-like nephritis with monoclonal antibody to gamma interferon. J Exp Med. 1987, 166: 798-803. 10.1084/jem.166.3.798.
Ozmen L, Roman D, Fountoulakis M, Schmid G, Ryffel B, Garotta G: Soluble interferon-gamma receptor: a therapeutically useful drug for systemic lupus erythematosus. J Interferon Res. 1994, 14: 283-284. 10.1089/jir.1994.14.283.
Kang HK, Liu M, Datta SK: Low-dose peptide tolerance therapy of lupus generates plasmacytoid dendritic cells that cause expansion of autoantigen-specific regulatory T cells and contraction of inflammatory Th17 cells. J Immunol. 2007, 178: 7849-7858.
Li X: Act1 modulates autoimmunity through its dual functions in CD40L/BAFF and IL-17 signaling. Cytokine. 2008, 41: 105-113. 10.1016/j.cyto.2007.09.015.
Garrett-Sinha LA, John S, Gaffen SL: IL-17 and the Th17 lineage in systemic lupus erythematosus. Curr Opin Rheumatol. 2008, 20: 519-525. 10.1097/BOR.0b013e328304b6b5.
Hoffman RW: T cells in the pathogenesis of systemic lupus erythematosus. Clin Immunol. 2004, 113: 4-13. 10.1016/j.clim.2004.05.001.
Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR: Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998, 41: 1241-1250. 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H.
Passero FC, Myers AR: Decreased numbers of monocytes in inflamatory exudates in systemic lupus erythematosus. J Rheumatol. 1981, 8: 62-68.
Jin O, Sun L, Kavikondala S, Lau C: Innate immunity and systemic lupus erythematosus. APLAR J Rheumatol. 2006, 9: 359-364. 10.1111/j.1479-8077.2006.00229.x.
Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK: Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001, 411: 207-211. 10.1038/35075603.
Lu Q, Lemke G: Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science. 2001, 293: 306-311. 10.1126/science.1061663.
Zhu J, Liu X, Xie C, Yan M, Yu Y, Sobel ES, Wakeland EK, Mohan C: T cell hyperactivity in lupus as a consequence of hyperstimulatory antigenpresenting cells. J Clin Invest. 2005, 115: 1869-1878. 10.1172/JCI23049.
Kilmon MA, Wagner NJ, Garland AL, Lin L, Aviszus K, Wysocki LJ, Vilen BJ: Macrophages prevent the differentiation of autoreactive B cells by secreting CD40 ligand and interleukin-6. Blood. 2007, 110: 1595-1602. 10.1182/blood-2006-12-061648.
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V: Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003, 197: 711-723. 10.1084/jem.20021553.
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A: Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010, 107: 9813-9818. 10.1073/pnas.0909927107.
Georgiev M, Agle LM, Chu JL, Elkon KB, Ashany D: Mature dendritic cells readily break tolerance in normal mice but do not lead to disease expression. Arthritis Rheum. 2005, 52: 225-238. 10.1002/art.20759.
Chen M, Wang YH, Wang Y, Huang L, Sandoval H, Liu YJ, Wang J: Dendritic cell apoptosis in the maintenance of immune tolerance. Science. 2006, 311: 1160-1164. 10.1126/science.1122545.
Fairhurst AM, Wandstrat AE, Wakeland EK: Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Adv Immunol. 2006, 92: 1-69.
Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL: Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005, 202: 1131-1139. 10.1084/jem.20050914.
Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD: Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005, 115: 407-417.
Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, Tough DF: Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol. 2006, 176: 2074-2078.
Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, Cerutti A: DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002, 3: 822-829.
Ceppellini R, Polli E, Celada F: A DNA-reacting f actor in serum of a patient with lupus erythematosus diffusus. Proc Soc Exp Biol Med. 1957, 96: 572-574.
Liang Z, Xie C, Chen C, Kreska D, Hsu K, Li L, Zhou XJ, Mohan C: Pathogenic profiles and molecular signatures of antinuclear autoantibodies rescued from NZM2410 lupus mice. J Exp Med. 2004, 199: 381-398. 10.1084/jem.20030132.
Tsao BP, Ohnishi K, Cheroutre H, Mitchell B, Teitell M, Mixter P, Kronenberg M, Hahn BH: Failed self-tolerance and autoimmunity in IgG anti-DNA transgenic mice. J Immunol. 1992, 149: 350-358.
Yung S, Chan TM: Anti-DNA antibodies in the pathogenesis of lupus nephritis--the emerging mechanisms. Autoimmun Rev. 2008, 7: 317-321. 10.1016/j.autrev.2007.12.001.
Sekine H, Watanabe H, Gilkeson GS: Enrichment of anti-glomerular antigen antibody-producing cells in the kidneys of MRL/MpJ-Fas(lpr) mice. J Immunol. 2004, 172: 3913-3921.
Waters ST, McDuffie M, Bagavant H, Deshmukh US, Gaskin F, Jiang C, Tung KS, Fu SM: Breaking tolerance to double stranded DNA, nucleosome, and other nuclear antigens is not required for the pathogenesis of lupus glomerulonephritis. J Exp Med. 2004, 199: 255-264. 10.1084/jem.20031519.
Waters ST, Fu SM, Gaskin F, Deshmukh US, Sung SS, Kannapell CC, Tung KS, McEwen SB, McDuffie M: NZM2328: a new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci. Clin Immunol. 2001, 100: 372-383. 10.1006/clim.2001.5079.
Mannik M, Merrill CE, Stamps LD, Wener MH: Multiple autoantibodies form the glomerular immune deposits in patients with systemic lupus erythematosus. J Rheumatol. 2003, 30: 1495-1504.
Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, Mohan C: Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005, 115: 3428-3439. 10.1172/JCI23587.
Schiffer L, Sinha J, Wang X, Huang W, von Gersdorff G, Schiffer M, Madaio MP, Davidson A: Short term administration of costimulatory blockade and cyclophosphamide induces remission of systemic lupus erythematosus nephritis in NZB/W F1 mice by a mechanism downstream of renal immune complex deposition. J Immunol. 2003, 171: 489-497.
Heymann F, Meyer-Schwesinger C, Hamilton-Williams EE, Hammerich L, Panzer U, Kaden S, Quaggin SE, Floege J, Grone HJ, Kurts C: Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J Clin Invest. 2009, 119: 1286-1297. 10.1172/JCI38399.
Bagavant H, Deshmukh US, Wang H, Ly T, Fu SM: Role for nephritogenic T cells in lupus glomerulonephritis: progression to renal failure is accompanied by T cell activation and expansion in regional lymph nodes. J Immunol. 2006, 177: 8258-8265.
Bossù P, Neumann D, Del Giudice E, Ciaramella A, Gloaguen I, Fantuzzi G, Dinarello CA, Di Carlo E, Musiani P, Meroni PL, Caselli G, Ruggiero P, Boraschi D: IL-18 cDNA vaccination protects mice from spontaneous lupus-like autoimmune disease. Proc Natl Acad Sci USA. 2003, 100: 14181-14186. 10.1073/pnas.2336094100.
Ruger BM, Erb KJ, He Y, Lane JM, Davis PF, Hasan Q: Interleukin-4 transgenic mice develop glomerulosclerosis independent of immunoglobulin deposition. Eur J Immunol. 2000, 30: 2698-2703. 10.1002/1521-4141(200009)30:9<2698::AID-IMMU2698>3.0.CO;2-1.
Singh RR: IL-4 and many roads to lupuslike autoimmunity. Clin Immunol. 2003, 108: 73-79. 10.1016/S1521-6616(03)00145-1.
Krüger T, Benke D, Eitner F, Lang A, Wirtz M, Hamilton-Williams EE, Engel D, Giese B, Müller-Newen G, Floege J, Kurts C: Identification and functional characterization of dendritic cells in the healthy murine kidney and in experimental glomerulonephritis. J Am Soc Nephrol. 2004, 15: 613-621. 10.1097/01.ASN.0000114553.36258.91.
Fiore N, Castellano G, Blasi A, Capobianco C, Loverre A, Montinaro V, Netti S, Torres D, Manno C, Grandaliano G, Ranieri E, Schena FP, Gesualdo L: Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Mol Immunol. 2008, 45: 259-265. 10.1016/j.molimm.2007.04.029.
Monrad S, Kaplan MJ: Dendritic cells and the immunopa thogenesis of systemic lupus erythematosus. Immunol Res. 2007, 37: 135-145. 10.1007/BF02685895.
Hooke DH, Gee DC, Atkins RC: Leukocyte analysis using monoclonal antibodies in human glomerulonephritis. Kidney Int. 1987, 31: 964-972. 10.1038/ki.1987.93.
Schiffer L, Bethunaickan R, Ramanujam M, Huang W, Schiffer M, Tao H, Madaio MP, Bottinger EP, Davidson A: Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. J Immunol. 2008, 180: 1938-1947.
Triantafyllopoulou A, Franzke CW, Seshan SV, Perino G, Kalliolias GD, Ramanujam M, van Rooijen N, Davidson A, Ivashkiv LB: Proliferative lesions and metalloproteinase activity in murine lupus nephritis mediated by type I interferons and macrophages. Proc Natl Acad Sci USA. 2010, 107: 3012-3017. 10.1073/pnas.0914902107.
Fu Y, Du Y, Mohan C: Experimental anti-GBM disease as a tool for studying spontaneous lupus nephritis. Clin Immunol. 2007, 124: 109-118. 10.1016/j.clim.2007.05.007.
Holdsworth SR, Neale TJ, Wilson CB: Abrogation of macrophage-dependent injury in experimental glomerulonephritis in the rabbit. Use of an antimacrophage serum. J Clin Invest. 1981, 68: 686-698. 10.1172/JCI110304.
Ikezumi Y, Hurst LA, Masaki T, Atkins RC, Nikolic-Paterson DJ: Adoptive transfer studies demonstrate that macrophages can induce proteinuria and mesangial cell proliferation. Kidney Int. 2003, 63: 83-95.
Moore KJ, Yeh K, Naito T, Kelley VR: TNF-alpha enhances colony-stimulating factor-1-induced macrophage accumulation in autoimmune renal disease. J Immunol. 1996, 157: 427-432.
Henson PM: Pathologic mechanisms in neutrophil-mediated injury. Am J Pathol. 1972, 68: 593-612.
Scapini P, Carletto A, Nardelli B, Calzetti F, Roschke V, Merigo F, Tamassia N, Pieropan S, Biasi D, Sbarbati A, Sozzani S, Bambara L, Cassatella MA: Proinflammatory mediators elicit secretion of the intracellular B-lymphocyte stimulator pool (BLyS) that is stored in activated neutrophils: implications for inflammatory diseases. Blood. 2005, 105: 830-837. 10.1182/blood-2004-02-0564.
Johnson RJ, Lovett D, Lehrer RI, Couser WG, Klebanoff SJ: Role of oxidants and proteases in glomerular injury. Kidney Int. 1994, 45: 352-359. 10.1038/ki.1994.45.
Baudeau C, Delarue F, He CJ, Nguyen G, Adida C, Peraldi MN, Sraer JD, Rondeau E: Induction of MHC class II molecules HLA-DR, -DP and -DQ and ICAM 1 in human podocytes by gamma-interferon. Exp Nephrol. 1994, 2: 306-312.
Coers W, Brouwer E, Vos JT, Chand A, Huitema S, Heeringa P, Kallenberg CG, Weening JJ: Podocyte expression of MHC class I and II and intercellular adhesion molecule-1 (ICAM-1) in experimental pauci-immune crescentic glomerulonephritis. Clin Exp Immunol. 1994, 98: 279-286.
Yokoyama H, Zheng X, Strom TB, Kelley VR: B7(+)-transfectant tubular epithelial cells induce T cell anergy, ignorance or proliferation. Kidney Int. 1994, 45: 1105-1112. 10.1038/ki.1994.147.
Tipping PG, Holdsworth SR: Cytokines in glomeruloneph ritis. Semin Nephrol. 2007, 27: 275-285. 10.1016/j.semnephrol.2007.02.002.
Li S, Kurts C, Kontgen F, Holdsworth SR, Tipping PG: Major histocompatibility complex class II expression by intrinsic renal cells is required for crescentic glomerulonephritis. J Exp Med. 1998, 188: 597-602. 10.1084/jem.188.3.597.
Molano A, Lakhani P, Aran A, Burkly LC, Michaelson JS, Putterman C: TWEAK stimulation of kidney resident cells in the pathogenesis of graft versus host induced lupus nephritis. Immunol Lett. 2009, 125: 119-128. 10.1016/j.imlet.2009.06.010.
Timoshanko JR, Sedgwick JD, Holdsworth SR, Tipping PG: Intrinsic renal cells are the major source of tumor necrosis factor contributing to renal injury in murine crescentic glomerulonephritis. J Am Soc Nephrol. 2003, 14: 1785-1793. 10.1097/01.ASN.0000073902.38428.33.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Pathak, S., Mohan, C. Cellular and molecular pathogenesis of systemic lupus erythematosus: lessons from animal models . Arthritis Res Ther 13, 241 (2011). https://doi.org/10.1186/ar3465
Published:
DOI: https://doi.org/10.1186/ar3465