
Abstract
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease that is known to be associated with polyclonal B-cell hyperreactivity. The underlying causes of the diffuse B-cell over-reactivity are unclear, but potential candidates include (a) intrinsic hyper-reactivity leading to polyclonal B-cell activation with disturbed activation thresholds and ineffective negative selection; (b) lack of immunoregulatory functions; (c) secondary effects of an overactive inflammatory environment, such as overactive germinal center and ectopic follicular activity; and/or (d) disturbed cytokine production by non-B immune cells. These mechanisms are not mutually exclusive and may operate to varying extents and at varying times in SLE. Phenotypic and molecular studies as well as the results of recent clinical trials have begun to provide new insights to address these possibilities. Of importance, new information has made it possible to distinguish between the contribution played by abnormalities in central checkpoints that could lead to a pre-immune repertoire enriched in autoreactive B cells, on the one hand, and the possibility that autoimmunity arises in the periphery from somatic hypermutation and abnormal selection during T cell-dependent B-cell responses on the other. There is an intriguing possibility that apoptotic material bound to the surface of follicular dendritic cells positively selects autoreactive B cells that arise from non-autoreactive B-cell precursors as a result of somatic hypermutation and thereby promotes the peripheral emergence of autoimmunity.
Similar content being viewed by others


Introduction
Systemic lupus erythematosus (SLE) is considered a prototypic autoimmune disease, although it cannot be ruled out that SLE is actually a syndrome representing the common final pathway of a number of discrete genetic and molecular aberrations. The large numbers of genetic abnormalities that can induce a lupus-like syndrome in mice [1] are consistent with the possibility that SLE may be a syndrome reflective of a number of different molecular abnormalities. Most of these murine models of lupus, however, are associated with diffuse B-cell hyperactivity or defects in the clearance of apoptotic material or both. The available data from these models, therefore, suggest that abnormalities of B-cell activation or clearance of apoptotic material of a variety of sorts (or both) may be the defining molecular pathways in human SLE.
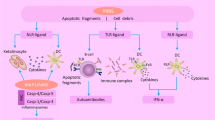
Both mice and human subjects with SLE produce a number of autoantibodies against nuclear complexes, although the profile of these antibodies can vary widely between individual subjects and murine models. Central to both human lupus and the various animal models is the production of antibodies to DNA and other components of the nucleosome. Traditionally, serologic evaluation has been used to delineate immunologic abnormalities in SLE, but it has also been known for many years that both mice and humans with SLE typically show evidence of polyclonal B-cell hyper-reactivity. More recently, analyses largely employing flow cytometry have documented the specific perturbations of B-cell maturation and differentiation in lupus and have generated new information on whether alterations in B-cell differentiation are involved primarily in immunopathology or, alternatively, may be secondary to the inflammatory environment in SLE (Figure 1). Additional insights have been derived from an analysis of the impact of certain therapies on clinical disease activity as well as their effects on abnormalities of B-cell subsets in SLE. All of these new data on human lupus, informed by advances in the study of murine lupus, have enhanced our understanding of the pathogenic role of B cells in SLE and also provided new information on the identification of potential therapeutic targets in this autoimmune disease. Moreover, an emerging understanding of the central role of B cells in lupus pathogenesis has provided the opportunity to monitor specific B-cell subsets as biomarkers of disease activity and clinical response to therapy.

Scheme of potential aberrations of T cell-dependent activation of B cells under the conditions of systemic lupus erythematosus. Intrinsic as well as extrinsic factors may lead to known B-cell hyperactivity as a result of enhanced germinal center reactions with defects in selection. As a net result, enhanced memory B cells and plasmacytosis could be explained and are consistent with abnormalities detectable in the blood of patients with active systemic lupus erythematosus.
Rationale for B cells as therapeutic targets in autoimmune diseases
Increased knowledge of the roles of B cells in normal immune responses, in particular their capacity to differentiate into antibody-secreting plasmablasts and plasma cells as well as their ability to function as antigen-presenting cells (APCs), secrete cytokines, and influence lymphoid architecture, has focused attention on them as a therapeutic target in autoimmune disease. In addition, genome-wide association studies have identified a number of candidate genes, such as BANK1, BLK, IL-21R, CD40, Lyn, PTPN22, TNFAIP3, FcγRs, and Blimp-1, that are associated with SLE and other autoimmune diseases and could predispose to increased B-cell responsiveness [2–4]. Finally, studies in genetically manipulated mice have indicated that a variety of interventions that serve to increase B-cell responsiveness can lead to the production of autoantibodies and, in the appropriate genetic strain, a lupus-like syndrome [1]. Recently, it has also been shown that disruption of genes with wide expression can result in B-cell hyper-responsiveness. In this regard, mice lacking the A20 molecule that restricts nuclear factor-kappa-B (NF-κB) activation by tumor necrosis factor (TNF) receptor family members and is encoded by the tnfaip3 gene, polymorphisms of which have been associated with SLE, develop exaggerated B-cell activity, increased germinal center (GC) activity, autoantibodies, and glomerular deposits of immunoglobulin (Ig) [5]. Together, these finding have caused the targeting of B cells to be considered potential interventions in various autoimmune diseases.
Initially, B cell-depleting therapy was employed in rheumatoid arthritis because of the belief that it would diminish rheumatoid factor production and thereby ameliorate disease activity [6] and even induce remission. Although the actual mechanism whereby B cell-depleting therapy is effective in rheumatoid arthritis remains incompletely delineated, rituximab has been approved and adopted for treatment of this autoimmune/inflammatory disease. Subsequently, trials have been carried out in SLE, ANCA+ (anti-neutrophil cytoplasmic antibody-positive) vasculitis, Sjögren's syndrome, immune thrombocytopenia, multiple sclerosis, autoimmune myositis, and other autoimmune conditions [7]. Although trials of rituximab in SLE [8] did not achieve their primary clinical endpoints, lymphocyte monitoring after B cell-targeted therapy [9] was instrumental in documenting the biologic impact of the intervention and also characteristics of the B-cell repopulation which are associated with clinical response. More recently, a monoclonal antibody to the B-cell survival factor BAFF/BLyS (B cell-activating factor/B lymphocyte stimulator) (TNFSF13b) was shown to be effective in clinical trials of non-renal lupus and was approved in the US and Europe in 2011.
B-cell subsets in human systemic lupus erythematosus
The abnormalities of certain peripheral B-cell subsets in SLE compared with healthy subjects were reviewed recently [10]. Differences from healthy subjects suggest that mechanisms that control many aspects of B-cell biology, including pre-immune B-cell maturation, negative selection at defined maturational checkpoints, and receptor editing, as well as many aspects of antigen responsiveness, including somatic hypermutation and effector B-cell generation, are diffusely abnormal in subjects with SLE. Whether this reflects an intrinsic B-cell defect or is secondary to inflammation is unclear. Reflecting abnormalities in B-cell maturation, a number of alterations in pre-immune B-cell populations in the periphery have been noted in patients with SLE. In the peripheral blood, abnormal frequencies of pre-immune B cells, including transitional B cells, pre-naïve B cells, and naïve B cells, can be detected [11]. These populations denote major stages of B-cell maturation which occur outside the bone marrow. In SLE, both transitional B cells and pre-naïve B cells are greatly expanded. These populations can be defined by a variety of markers, including CD38, CD5, CD9, CD10, CD24, and the ABCB1 transporter [11]. These results indicate a population shift within the pre-immune B-cell compartment in SLE toward the more immature B cells, and this shift is independent of disease activity.
With regard to early defects of selection against autoreactive B cells, a comprehensive study [12] showed that even patients with inactive SLE fail to remove B cells expressing self-reactive B-cell receptors (BCRs) expressed by naïve B cells. Consistent with this, earlier studies demonstrated that self-reactivity or loss of proper selection during early B-cell development from immature (CD10+CD27-IgM+ B cells) to mature (CD10-CD27-IgM+ B cells) naïve B cells is a feature in SLE [13]. Although it is tempting to conclude that SLE has defects in 'checkpoints against autoimmunity' that explain the development of pathogenic IgG autoantibodies, the possibility cannot be excluded that these 'checkpoints' are active in SLE to prevent serious autoimmunity but are simply overwhelmed by chronic polyclonal B-cell activation. Alternatively, it is possible that, since some predominantly IgM autoantibodies may be protective, defects in selection at pre-immune checkpoints represent an effort to enhance the production of non-pathogenic protective autoantibodies.
Disturbances of memory B cells in systemic lupus erythematosus
A central finding in phenotyping peripheral blood B cells in adult SLE is the substantial increase of antigen-experienced CD27+IgD- post-switched memory B cells [14], which are less susceptible to immunosuppressive therapy [10, 14]. Importantly, the peripheral memory BCR repertoire in SLE is shaped by abnormal selection, exaggerated somatic hypermutation, and increased receptor editing [15, 16]. In contrast, the pre-immune BCR initially generated by VH(D)JH recombination appears similar to that of healthy subjects [17]. Moreover, autologous stem cell transplantation in SLE provides proof of concept that the disease can undergo successful remission along with the emergence of predominating naïve T and B cells after transplantation [18]. This is consistent with the conclusion that abnormalities in the shaping of the Ig repertoire after antigenic stimulation and differentiation of memory B cells, rather than molecular disturbances in generating the initial BCR repertoire of pre-immune B cells, may be more important in lupus pathogenesis.
It is currently unclear whether the predominance of IgD-CD27+ post-switched memory B cells reflects a loss of peripheral tolerance or abnormal selection processes or whether their appearance is simply the result of enhanced activation and differentiation of naïve B cells or the lack of susceptibility of memory B cells to immunosuppressive drugs. Independently of these considerations, an enlarged pool of memory B cells poses a risk for autoimmunity since these cells have lower activation thresholds, regulation by FcγRIIb receptors may be reduced [19], and these cells can be rapidly activated in a non-antigen-specific manner by the combination of Toll-like receptor (TLR) agonists and a proliferation-inducing ligand (APRIL) (TNFSF13A) or BAFF (TNFSF13B) as well as by the combination of cytokines, such as IL-21 and BAFF [20].
Another memory B-cell subset particularly enhanced in patients with SLE, besides the large subset of IgD-CD27+ post-switched cells, has also been identified. This is a population of memory B cells that does not express CD27 [21–23]. CD27- B cells were previously thought to be exclusively naïve B cells and to express IgD [23], but in SLE an expanded population of IgD-CD27- B cells with characteristics of memory B cells could be detected. While in normal donors IgG+CD27- B cells expressing mutated BCRs have been identified both in tonsils and in very small numbers in peripheral blood [24, 25], CD27-IgD- memory B cells in the peripheral blood were increased in patients with SLE and that increase has been reported to be associated with increased disease activity and active renal disease [22]. The frequency of these IgD-CD27- memory B cells was also reported to correlate with the presence of certain autoantibodies - anti-dsDNA (anti-double-stranded DNA), anti-Smith, and anti-ribonucleoprotein - and the presence of B cells expressing the 9G4 idiotype expressed by the VH4-34 heavy chain [22] and frequently encoding anti-DNA autoantibodies [26]. Similar to transitional B cells and pre-naïve B cells and also typical of memory B cells, these CD27-IgD- B cells lack expression of the ABCB1 transporter and are, therefore, unable to extrude the fluorescent dye, rhodamine123.
Subsequently, a more homogeneous subset of IgD-CD27-CD95+ memory B cells was found among the IgD-CD27- B cells [21]. Detailed phenotypic and molecular studies revealed that this memory B-cell subset was associated with active disease. In contrast, the entire subset of CD27-IgD- B cells was found to be heterogeneous and did not correlate with lupus activity. The IgD-CD27- B cells, but not IgD-CD27-CD95+ memory B cells, were also increased in patients with bacterial infections, suggesting that the larger population is responsive to non-specific immune activation and not changes that are unique to SLE. The IgD-CD27-CD95+ B-cell subset was a more homogeneous subset within the larger IgD-CD27- pool that expressed various activation markers (CD86, HLA-DR, and CD38), chemokine receptors (CXCR3, CXCR4, and CXCR5), CD21 (CR2), and CD44. Importantly, IgD-CD27-CD95+ B cells displayed higher levels of CD86, CXCR3, HLA-DR, and CD71 expression and lower levels of CXCR4. These data are consistent with the conclusion that the IgD-CD27-CD95+ memory B cells constitute a unique subpopulation that appears in increased numbers in SLE and represents recently activated memory B cells resulting from intensive GC activity. The expression of CD95 by this memory B-cell subset appears to be an important finding since it represents a unique characteristic of this population of SLE B cells and implies that censoring of GC B cells by Fas/Fas-L interaction may be defective in this disease.
Regulatory B cells
Though not definitely proven to play an important immunoregulatory role in humans, regulatory B cells appear to be induced after antigen-dependent activation in mice and, therefore, are also considered to be part of the memory subset. In mice, a regulatory CD1dhiCD5+CD19hi B-cell subset producing IL-10 was shown to control T cell-dependent immune responses [27]. Moreover, a specific effect of anti-CD20-mediated B-cell depletion on CD4+, but not on CD8+, T-cell responses against foreign and self-antigen was demonstrated [28], implying that IL-10-producing murine B cells may play important regulatory roles on pathologic T-cell responses. Their precise role in human lupus, however, remains to be confirmed. A recent report in subjects with SLE found a population of regulatory CD19+/CD24high/CD38high B cells [29], a phenotype reminiscent of pre-immune B cells. This subset has been reported to secrete IL-10 and thereby is able to suppress Th1 and Th2 functions after CD80/CD86 as well as CD40/CD154 ligation. Interestingly, these cells, though present in numbers comparable to those of healthy controls, lack full functionality in patients with SLE. A more recent analysis [30] demonstrated that two functionally different subsets are present within the CD24highCD27+ B-cell population: B10 and B10pro, with the former ranging between 0.6% and 5% in human peripheral blood and able to produce IL-10 within 5 hours and thereby regulate monocyte function. Notably, this study was not able to identify substantial differences of B10-cell frequencies between autoimmune patients (including those with SLE) and controls. Moreover, the relationship between the aforementioned human regulatory B-cell and B10 or B10pro is not clear. Despite this, the results suggest that some B cells apparently play a key role in controlling T-cell and monocyte responses and that an analogous activity may be defective in SLE.
Disturbances of plasmablasts/plasma cells in systemic lupus erythematosus
One hallmark in adult and juvenile SLE is the significant increase of CD27++ expressing plasmablasts/plasma cells in active disease. These cells are CD19lowCD20-/CD38+++, surface Iglow, and express intracellular Ig [14, 31, 32]. Recent studies [33, 34] demonstrated that IgA+/β7 integrin+ plasmablasts/plasma cells represent steady-state Ig-secreting cells circulating in the peripheral blood of healthy subjects and contrast with antigen- and adjuvant-stimulated CD62L+/IgG+ plasmablasts/plasma cells appearing in the blood after purposeful vaccination. The steady-state plasmablasts/plasma cells also differ from the CD62L-/β7- plasma cells in the bone marrow. The bulk of the currently available data indicate that active SLE is characterized by increased numbers of circulating plasma cells in children as well as adults, indicating dysregulation of the homeostasis of these cells [14, 31, 35]. The circulating plasmacytosis of active SLE reflects the ongoing generation of plasma cells/plasmablasts within GCs since their appearance was markedly decreased when patients with SLE were treated with a monoclonal antibody to CD154 that inhibited CD40-CD154 interactions in vivo [36] and that blocked IL-6 as an important factor for B-cell differentiation [37] and, thereby, GC reactions.
Consistent with this, circulating plasma cells in patients with active SLE expressed highly mutated IgVH genes with mutations consistent with a GC pattern [38]. Notably, the plasma cells were oligoclonal and included cells that were actively secreting anti-dsDNA antibodies. An expansion of plasma cells in the kidneys of patients with lupus nephritis was also observed [39], with these cells localizing in close proximity to extrafollicular structures. This supports the conclusion that these structures, often found in autoimmunity, may contribute to the local production of autoantibodies that may be pathogenic in this disease.
Models to explain B-cell abnormalities observed in the context of systemic lupus erythematosus
It is clear that human lupus is associated with intense polyclonal B-cell activation. Abnormalities can be observed in pre-immune B-cell populations as well as post-GC memory B and plasma cells. The questions of how these abnormalities may contribute to lupus pathogenesis and how this relates to the induction of autoantibodies are unresolved, but a number of possibilities have been suggested.
1. Systemic lupus erythematosus results from an abnormal pre-immune repertoire owing to defective negative selection
One mechanism relates to the action of central B-cell tolerance checkpoints that ensure that emerging autoreactive B cells are counter-selected [40, 41]. In this regard, transgenic mice carrying pre-rearranged selfreactive Ig genes were instrumental in uncovering the mechanisms that regulate developing self-reactive B cells. However, these models did not allow estimates of the frequency at which self-reactive BCRs are generated in humans by VH(D)JH recombination with a nearly unlimited Ig gene repertoire. In humans, an understanding of the potential role of these checkpoints for selftolerance is based on the analysis of Ig genes cloned from single purified B cells at different stages during their development [12, 13, 42–44]. The results of these analyses indicate that autoantigen-binding B cells occur fairly frequently early in B-cell ontogeny but that these autoreactive B cells are culled at various stages of subsequent B-cell ontogeny. In lupus, these checkpoints appear to be defective, resulting in the emergence of higher numbers of B cells expressing autoreactive BCRs than normal. It then follows that there is a greater likelihood that either autoantigen-specific activation or polyclonal stimulation would have a greater likelihood of leading to the production of autoantibodies. This likelihood is increased by the fact that peripheral checkpoints are also defective in SLE as well as in other autoimmune diseases. However, whether these defects in peripheral checkpoints contribute to the emergence of pathogenic autoimmunity and whether these checkpoint abnormalities are primary or secondary to the profound abnormalities in B-cell function or cytokine production have not been fully delineated.
A number of observations challenge this hypothesis. The isolation of certain pre-defined B-cell subsets, followed by re-expression of their BCRs, may not reflect the natural development of these cells that normally undergo several rounds of selection before they enter the memory B-cell and plasma cell pools. Moreover, it is uncertain whether pathogenic autoantibodies emerge as a result of the central or peripheral checkpoint abnormalities. Pathogenic autoantibodies are encoded largely by highly mutated Ig genes and in part by use of certain VH genes. Importantly, these pathogenic autoantibodies usually lose autoantigen binding when reverted back to the germline configuration [44–47], implying that they did not arise from stimulation of naïve autoreactive B cells. It is notable not only that pathogenic autoantibodies are highly mutated but that studies of the BCR repertoire in human SLE demonstrated a diffusely enhanced frequency of somatic hypermutation compared with healthy subjects by using single-cell analysis [14–16, 44].
It is not clear whether germline-encoded polyreactive autoantibodies are the precursors of pathogenic autoantibodies. These polyreactive antibodies are considered to be produced by B1 B cells and/or MZ B cells, which are under less stringent T-cell control and tend to have long CDR3s as exemplified by antibodies during HIV-1 infection [48], whereas pathogenic autoantibodies have a far shorter CDR3. Moreover, the Ig genes of polyreactive autoantibodies are usually in germline configuration, whereas studies of highly mutated pathogenic autoantibodies indicate that reversion to the germline configuration abolished all autoreactivity, strongly arguing that pathogenic autoantibodies did not arise from B cells expressing polyreactive autoantibodies. All of these considerations make it unlikely that pathogenic autoantibodies routinely emerge as a result of defects in checkpoints shaping the pre-immune B-cell repertoire.
The association of certain HLA alleles with certain autoantibody specificities (that is, anti-Ro/La with HLA-DR2/3 [49] and a less robust association of HLA-DQw2 with anti-Smith [50]) suggests that T-cell activation by APCs may be essential for these responses and speaks against T cell-independent activation of B1 B cells or MZ B cells expressing a biased autoreactive BCR repertoire as an important mechanism in the generation of these autoantibodies. At least, this appears to be the case for autoantibodies to some autoantigens that may be the products of long-lived plasma cells, as defined by their resistance to anti-proliferative agents [51]. On the other hand, an HLA association with anti-DNA antibodies has not been established, suggesting that the short-lived plasmablasts producing these autoantibodies may be less dependent on T-cell antigen recognition or alternatively that the T-cell epitopes that lead to their production may be so promiscuous that an HLA association is not apparent. It should be recalled that the HLA region in general is the strongest genetic association with lupus in genome-wide association studies [52].
2. Systemic lupus erythematosus may develop as a result of enhanced germinal center activity and preferential selection by autoantigen
An alternative hypothesis postulates that pathogenic autoimmunity in SLE arises as a result of increased T cell-dependent GC-like activity that generates autoantibodies from B cells whose BCRs had no intrinsic reactivity with autoantigens in germline configuration. Intrinsic to this hypothesis are the concepts of B-cell hyper-responsiveness or exaggerated T-cell responses or both, coupled with enhanced somatic hypermutation and the accumulation of apoptotic material within GCs with the capacity to positively select autoreactive B cells. This hypothesis is supported by the findings that, in murine models and human lupus, autoimmunity can arise by somatic hypermutation from B-cell precursors with no intrinsic autoreactivity.
The importance of T cell-dependent GC reaction in SLE is emphasized by the finding of an expansion of post-GC B-cell subsets in the peripheral blood of patients with lupus [14, 22, 35, 44, 53]. Although post-GC cells are expanded in SLE, little information concerning whether this results from the activity of classic or ectopic GCs and where these GCs might reside is available. In both classic and ectopic GCs, clonal expansion of B cells along with somatic hypermutation of Ig gene rearrangements and Ig heavy-chain class-switch recombination are induced. Resulting memory B cells can be positively selected by immune complexes residing on the surface of follicular dendritic cells (FDCs) and negatively selected by the presence of soluble autoantigen. Although there are minimal data, it is possible that the stringency of negative selection might be less in ectopic GCs. The three different stages of GC formation and activity, including (a) establishment of GC structures, (b) the activity of these sites of generation of B-cell effectors, and the (c) selection of high-avidity effector cells by FDCs, evolve over time after antigen stimulation.
2a. Generation of germinal centers
Given the enhanced GC activity and increased formation of ectopic GCs known to result in the generation of antigen-experienced memory B and plasma cells, there is a possibility that factors involved in establishing GC structures, including T follicular helper (TFH) cells, IL-21, IL-6, CXCL13, TNFβ, and inducible T-cell co-stimulator (ICOS), play a critical role in SLE. In this regard, the sanroque mouse that harbors a mutation that regulates ICOS expression on T cells develops a very aggressive form of lupus. In this context, CD154/CD40 and ICOS-L/ICOS [54] as well as CD80/CD86 [55] ligation have been suggested to be necessary for the activation of B cells but also indispensable for the activation of autoreactive T cells [56] and the induction of regulatory B cells [29]. With regard to the important involvement of co-stimulatory ligands expressed on plasma cells and memory B cells in SLE, it has been shown that T and B cells from SLE blood spontaneously express CD154 [36], suggesting a potential abnormal release of activated lymphocytes from the GCs or insufficient selection permitting abnormal egress of memory B cells into the blood. Similarly, CD4+ and CD8+ T cells from patients with lupus were identified to have enhanced ICOS expression, whereas memory B cells substantially down-regulate ICOS-L presumably as a result of recent T-cell instruction [39].
The type I cytokine, IL-21, essential for co-stimulated B cells to differentiate into plasma cells [20], has also been suggested to be involved in SLE pathogenesis. This hypothesis is supported by data from IL-21R-/- mice with diminished IgG1 and IgG2b in response to immunization and those from IL-21 transgenic mice that develop hypergammaglobulinemia [57]. Moreover, significantly enhanced IL-21 was found in the BXSB-Yaa+ and the sanroque murine models of SLE [57]. In addition, blocking of IL-21 activity was employed successfully in the MRL lupus mouse consistently with observations in BXSB-Yaa+ mice crossed with the IL-21R-/- mouse in which all manifestations of SLE disappeared [20, 58]. IL-21 is constitutively produced by CXCR5+ TFH cells, a subpopulation of T cells that reside in B-cell follicles and are specialized to provide initial help to antigen-activated B cells [59]. In addition, TFH cells require ICOS stimulation for their generation, and their differentiation is regulated by the transcriptional repressor, bcl-6 [60]. Notably, the sanroque mouse that constitutively over-expresses ICOS contains an expanded population of TFH cells that produce large amounts of IL-21 inducing the lupus phenotype. These findings of the linkage between ICOS and IL-21 required for B-cell stimulation in secondary lymphoid organs represent further critical evidence that factors involved in GC induction or maintenance or both are of critical importance in human SLE. The role for GCs in SLE is further supported by data showing that polymorphisms of both IL-21 and the IL-21R are associated with SLE [61]. In this context, a direct effect of interferon-alpha on B cells is less likely, but type I interferon [62] can also induce BAFF/BLyS by myeloid cells, subsequently enhancing autoreactive B-cell survival [63]. Overall, a complex set of ligands and cytokines appear to be important to initiate GC structure functionally and define their anatomic site.
Enhanced availability of GC factors involved in the establishment of GCs in SLE may explain the formation of so-called ectopic GCs considered to be involved in the differentiation of B cells and plasma cells under autoimmune conditions. Although conclusive evidence concerning whether ectopic GCs are specific for autoimmunity is still lacking, evidence about their occurrence during infectious diseases such as viral rhinitis suggests that they are not unique to autoimmune conditions but still may contribute. Moreover, it is not clear that lupus ectopic GCs are uniquely defective in negative selection since even classic GCs in tonsils from patients with SLE were found to be defective in selection against 9G4-expressing B cells.
Ectopic GC structures may be involved in T cell-independent activation of memory B cells by co-engagement of the BCR with activating receptors for BAFF and APRIL (TACI, TNFRSF13B) or ligation of TLR9 by bacterial DNA or TLR-7 or TLR-8 by single-stranded RNA, resulting in activation of B cells by an NF-κB-dependent mechanism. Signaling via TLRs, in particular TLR9 induced by dsDNA and TLR7 by single-stranded DNA/RNA, uniquely stimulates MyD88-dependent memory B cells [64] and may contribute to plasmacytosis in SLE.
It needs to be emphasized that other cells may also be involved in the initiation of B-cell activation in autoimmune disease. In this context, a recent study [65] using conditional knockout mice for DCs on an MRL.Faslpr background found that induction of plasmablasts and autoantibodies were dependent on DCs possibly related to a role in T cell-independent extrafollicular responses. Total Ig levels remained stable in these animals. Interestingly, this study highlights that there may be differences in the contributions of interactions between DCs and T cells versus DCs and B cells in certain autoimmune diseases.
2b. Antigen-presenting cell function
B cells can act as professional APCs. This conclusion was suggested by data from animal models [66] in which a lupus-like disease, albeit mild, developed when autoimmune-prone mice were reconstituted with B cells that lacked the ability to secrete Ig, whereas no disease was seen in the complete absence of B cells. Thus, an important function of memory B cells may be antigen presentation, which is facilitated by the expression of high-avidity BCR and also major histocompatibility complex (MHC) class II and co-stimulatory molecules, and, as a result, memory B cells are likely to be able to activate and expand memory T cells.
2c. T cell-independent activation of memory B cells
In addition to having a potential role as APCs, abundant memory B-cell subsets in SLE with their characteristic hyper-responsiveness to various stimuli, such as TLR ligands, IL-21, BAFF, IL-10, BCR activation, and co-stimulatory ligand-ligand interactions, can result in an increase of Ig-producing plasma cells without strong selection criteria as required for naïve B cells. This increase in the reactivity of memory B cells could also reflect the increased production of cytokines characteristic of SLE, such as IL-6, lymphotoxin-beta, IL-4, IL-10, and IL-21. In a substantial number of patients with SLE, interferon-alpha produced by tissue-resident plasmacytoid DCs that are reduced in the blood of patients with SLE may ultimately induce the increased B-cell hyperactivity by inducing the production of BAFF/BLyS (TNFSF13b) [67].
2d. Somatic hypermutation and autoimmunity
A number of studies provided evidence that somatic hypermutation was required for the generation of disease-associated anti-nuclear antibodies (ANAs). A recent instructive study [47] using TdT-/- mice and therefore avoiding CDR3 autoreactive binding introduced by junctional diversity showed that spontaneous autoimmunity was closely related to high-avidity autoimmune B cells created from non-autoreactive precursors by somatic hypermutation. The mutational process introduced encoding codons as a result of targeted mutations toward mutational hot spots (AGC and AGT/serine codons). Consistent with earlier studies suggesting the importance of arginine residues in anti-nuclear activity, this information supports the conclusion that somatic hypermutation is importantly involved in inducing ANA-specific B cells in a T cell-dependent manner. Only 1 out of 10 clones (J7.13 using a Jk1 gene segment) retained ANA-binding activity after reversion to germline sequences consistent with the possibility that it either escaped all tolerance checkpoints in B-cell development or may have defects in receptor editing. The data indicate that the majority of autoreactive B cells emerge as a result of somatic hypermutation, although a minority of autoantigen-binding B cells can manage to escape tolerance consistent with the contention that some autoimmunity may arise from checkpoint abnormalities permitting an expanded autoimmune repertoire in the pre-immune B-cell compartment. As mentioned above, the importance of somatic hypermutation in the development of autoimmunity has been demonstrated for autoreactive monoclonal antibodies that were derived from lupus patients and that lost autoantigen binding when back-mutated to the germline configuration [44, 45]. These independent data obtained by different technologies indicate that somatic hypermutation is an almost universal factor establishing autoreactivity in SLE.
Additional results also have focused on the role of somatic hypermutation in the generation of autoanti-bodies. Plasma cells from subjects with active SLE manifest extensively mutated VH and VL genes, and the pattern of mutation is comparable to that noted following T cell-dependent antigen stimulation [14–17, 44–46, 68] (Figure 2). The Ig gene rearrangements of such plasma cells show an increased replacement-to-silent (R/S) mutation ratio within complementarity-determining regions (CDRs) 1 and 2 compared with the framework regions (FRs) as well as exhibiting mutational targeting into RGYW/WRCY mutation hot spots. The available data are all consistent with a GC-type molecular mutation pattern.

Potential pathways involved in autoantibody generation in systemic lupus erythematosus (SLE). SHM, somatic hypermutation.
A comparison of the molecular characteristics of VH gene rearrangements of antigen-experienced B cells from healthy subjects and those circulating in SLE patients (Figure 3) shows that one difference is a higher R/S ratio within CDRs versus FRs in SLE compared with healthy volunteers (Figure 3b) undergoing tetanus booster vaccination. In recent years, mutations targeted especially to the Gs and Cs of RGYW/WRCY motifs have become of special interest since they are thought to be the direct consequence of AIDCA deamination [69] occurring typically within GCs, as this pattern is specifically reduced in CD154-deficient individuals, virtually lacking GCs [70]. It should be noted that the frequency of somatic hypermutations and the length of CDR3, both of which are considered to be markers of the influence of antigen, were comparable between healthy subjects and subjects with SLE (Figure 3a, c, d). Thus, the higher R/S ratio in CDRs in patients with SLE suggests that defects of selection after the GC reaction likely play a major role in SLE. Possibly, the negative selection within GCs could be overridden or exhausted by the greatly increased immune activity in GCs so that the emergence of autoreactive B cells is permitted.

Comparative analysis of molecular germinal center signatures between antigen-experienced cells obtained from vaccinated controls versus systemic lupus erythematosus (SLE). VH sequences of individual cells sorted as recombinant C-fragment of tetanus toxin (TT)-specific plasma cells (TT+ PCs) and TT-specific memory B cells (TT+ mBCs) were pooled from three healthy donors after tetanus booster [81] and plasma cells from one patient with SLE (SLE PCs) [14]. TT+ PCs and TT+ mBCs serve as effector cells generated as a result of prototypic T cell-dependent responses. (a) Mutation frequency. Each dot represents the value for one individual cell. (b) Ratios of replacement (R) to silent (S) mutations within complementarity-determining regions (CDRs) 1 and 2 and framework regions (FRs), respectively. (c) Frequency of mutations located within the two motifs RGYW and WRCY (R = purine, Y = pyrimidine, and W = adenine/thymine). (d) CDR3 length of individual B cells related to the underlying total number of mutations per sequence. Sequences of each cell type were divided into three categories according to their VH region mutations (that is, 0 to 5 mutations, 6 to 10 mutations, and more than 10 mutations) and are plotted against their respective CDR3 lengths. Bar indicates median.
2e. Selection
A variety of results in human and murine SLE are consistent with the possibility that selection is critically impaired in censoring autoreactive B cells after GC reactions. One possibility to consider is that an abnormality in selection may relate to defects in apoptosis characteristic of SLE. In this regard, deficiencies in the clearance of apoptotic material in SLE [41, 71] appear to play an important role. Herrmann and colleagues [72] demonstrated in vitro that phagocytes in about 50% of patients with SLE show reduced phagocytotic activity of autologous apoptotic material. With regard to GCs in vivo, tingible body macrophages, which are phagocytes that most efficiently engulf apoptotic cells and locate in close proximity to FDCs, were reduced in total numbers in a subgroup of patients with SLE. Furthermore, those tingible body macrophages rarely contained apoptotic material but instead apoptotic debris was found to be uningested outside these phagocytes [73]. Consequently, DCs may present self-antigens via MHC molecules on their surfaces [74] and may lead to activation of auto-reactive T cells. Moreover, apoptotic material has been visualized on the surface of FDC within GCs [75], suggesting that they may be capable of acting to positively select autoreactive memory B cells that emerge from GCs. Evidence that these 'censoring units' are apparently abnormal and may positively select autoreactive B cells comes from a study showing that B cells expressing the anti-DNA encoding 9G4 idiotype were detectable in tonsils of patients with SLE but are not found in normal GCs [26]. These data support the view that malfunction of the clearance of apoptotic material may not only induce but also select autoreactive B cells in GCs. Thus, there is a possibility that autoantigens derived from inefficient clearance of apoptotic remnants and presented by FDCs are of central importance in the selection of autoantibodies (Figure 4).

Characteristics of somatic hypermutation and generation of autoantibodies. AID, activation-induced cytidine deaminase; CDR, complementarity-determining region; Ig, immuonglobulin; RGYW, purine (R), guanine (G), pyrimidine (Y), adenine/thymine (W); S → R, silent-to-replacement; SHM, somatic hypermutation; WRCY, adenine/thymine (W), purine (R), cytosine (C), pyrimidine (Y).
Clinical aspects of therapeutic strategies targeting B cells
Instructive data have been obtained from a number of recent clinical trials of targeted B-cell therapy. Immunotherapies using the chimeric anti-CD20 antibody rituximab [9], tocilizumab blocking IL-6 effects [37], blocking CD154-CD40 interactions [36], and autologous stem cell transplantation [18] in severe SLE demonstrated that the expanded memory B-cell and plasma cell populations could be normalized and resulted in predominantly naïve B cells after successful interventions. In contrast, the BAFF/BLyS neutralizing antibody, belimumab, caused a decrease in naïve B cells and plasma cells but an increase in circulating memory B cells [76]. Despite changes in B-cell phenotype, two trials investigating rituximab in SLE did not meet their clinical endpoints [77], whereas two trials of belimumab showed significant clinical benefit. The disparate results of these trials raise questions about the pharmacodynamic differences of these reagents and their most relevant effects. While no data on lymphocyte subsets are available from trials studying abatacept in SLE [78], blockade of co-stimulation between CD154/CD40 in an early study and blockade of ICOS/ICOS-L in preclinical studies [39] are consistent with the idea that inhibition of certain GC processes are prime candidates for successful interventions.
Interestingly, blocking BAFF/BLys by the humanized monoclonal belimumab has been reported to preferen-tially reduce transitional naïve B cells and plasma cells [76], indicating a possible involvement of this cytokine in early and late B-cell differentiation. Another monoclonal antibody directed against CD22 expressed on B cells, epratuzumab, was shown to preferentially reduce peripheral CD27- B cells [79]. A recent observation of higher expression of CD22 on CD27- naïve versus CD27+ memory B cells may provide an explanation for this disparity. Moreover, enhanced binding of epratuzumab to naïve B cells is apparently linked with enhanced migration toward CXCL12 in vitro along with reduced expression of CD62L and β7 integrin [80]. These two possibilities imply that B cells are involved in SLE, although it remains to be shown whether they interfere directly or indirectly with GC activation versus positive selection on FDCs.
Conclusions
Although the underlying causes of B-cell over-reactivity in SLE remain to be fully delineated, the data indicate that this increased activity contributes to the emergence of autoimmunity. Two major abnormalities of B cells in lupus are defective central checkpoints with a resultant partially autoreactive pre-immune repertoire, on the one hand, and enhanced T cell-dependent B-cell activation in the periphery with autoimmunity arising from somatic hypermutation and selected by apoptotic material displayed on FDC on the other. Although both may contribute to the emergence of autoimmunity, most of the data favor the latter as a major cause of the development of pathogenic autoantibodies. As such, this hypothesis would indicate that a substantial part of autoimmunity is not strictly initiated by autoantigen stimulating a specific response directed to the initiating self-antigen but rather by autoantigen selecting B cells for survival that have acquired autoreactivity by somatic hypermutation. Moreover, the results indicate that therapies aimed at decreasing B-cell reactivity during T cell-dependent immune response in GCs may have a positive role in treating subjects with SLE.
Note
Autoimmune Basis of Rheumatic Diseases
This article is part of a series on Systemic lupus erythematosus, edited by David Pisetsky, which can be found online at http://arthritis-research.com/series/lupus
This series forms part of a special collection of reviews covering major autoimmune rheumatic diseases, available at: http://arthritis-research.com/series/abrd
Abbreviations
- ANA:
-
anti-nuclear antibody
- APC:
-
antigen-presenting cell
- APRIL:
-
a proliferation-inducing ligand
- BAFF:
-
B cell-activating factor
- BCR:
-
B-cell receptor
- BLyS:
-
B lymphocyte stimulator
- CDR:
-
complementarity-determining region
- DC:
-
dendritic cell
- dsDNA:
-
double-stranded DNA
- FDC:
-
follicular dendritic cell
- FR:
-
framework region
- GC:
-
germinal center
- ICOS:
-
inducible T-cell co-stimulator
- Ig:
-
immunoglobulin
- IL:
-
interleukin
- MHC:
-
major histocompatibility complex
- NF-κB:
-
nuclear factor-kappa-B
- R/S:
-
replacement-to-silent
- SLE:
-
systemic lupus erythematosus
- TFH:
-
T follicular helper
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor.
References
Townsend MJ, Monroe JG, Chan AC: B-cell targeted therapies in human autoimmune diseases: an updated perspective. Immunol Rev. 2010, 237: 264-283. 10.1111/j.1600-065X.2010.00945.x.
Taylor KE, Chung SA, Graham RR, Ortmann WA, Lee AT, Langefeld CD, Jacob CO, Kamboh MI, Alarcón-Riquelme ME, Tsao BP, Moser KL, Gaffney PM, Harley JB, Petri M, Manzi S, Gregersen PK, Behrens TW, Criswell LA: Risk alleles for systemic lupus erythematosus in a large case-control collection and associations with clinical subphenotypes. Plos Genetics. 2011, 7: e1001311-10.1371/journal.pgen.1001311.
Lee HS, Bae SC: What can we learn from genetic studies of systemic lupus erythematosus? Implications of genetic heterogeneity among populations in SLE. Lupus. 2010, 19: 1452-1459. 10.1177/0961203310370350.
Ozaki K, Spolski R, Ettinger R, Kim HP, Wang G, Qi CF, Hwu P, Shaffer DJ, Akilesh S, Roopenian DC, Morse HC, Lipsky PE, Leonard WJ: Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of blimp-1 and bcl-61. J Immunol. 2004, 173: 5361-5371.
Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L, Barrera J, Lowell CA, Utz PJ, Malynn BA, Ma A: The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010, 33: 181-191. 10.1016/j.immuni.2010.07.017.
Edwards JC, Cambridge G: B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006, 6: 394-403. 10.1038/nri1838.
Dörner T, Isenberg D, Jayne D, Wiendl H, Zillikens D, Burmester G, International Roundtable on B cells as Therapeutic Target for Intervention: Current status on B-cell depletion therapy in autoimmune diseases other than rheumatoid arthritis. Autoimmun Rev. 2009, 9: 82-89. 10.1016/j.autrev.2009.08.007.
Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, Utset TO, Gordon C, Isenberg DA, Hsieh HJ, Zhang D, Brunetta PG: Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62: 222-233. 10.1002/art.27233.
Anolik JH, Barnard J, Owen T, Zheng B, Kemshetti S, Looney RJ, Sanz I: Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis Rheum. 2007, 56: 3044-3056. 10.1002/art.22810.
Dorner T, Jacobi AM, Lipsky PE: B cells in autoimmunity. Arthritis Res Ther. 2009, 11: 247-10.1186/ar2780.
Lee J, Kuchen S, Fischer R, Chang S, Lipsky PE: Identification and characterization of a human CD5(+) pre-naive B cell population. J Immunol. 2009, 182: 4116-4126. 10.4049/jimmunol.0803391.
Yurasov S, Tiller T, Tsuiji M, Velinzon K, Pascual V, Wardemann H, Nussenzweig MC: Persistent expression of autoantibodies in SLE patients in remission. J Exp Med. 2006, 203: 2255-2261. 10.1084/jem.20061446.
Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, Nussenzweig MC: Defective B cell tolerance checkpoints in systemic lupus erythematosus. J Exp Med. 2005, 201: 703-711. 10.1084/jem.20042251.
Odendahl M, Jacobi A, Hansen A, Feist E, Hiepe F, Burmester GR, Lipsky PE, Radbruch A, Dörner T: Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol. 2000, 165: 5970-5979.
Dorner T, Foster SJ, Farner NL, Lipsky PE: Immunoglobulin kappa chain receptor editing in systemic lupus erythematosus. J Clin Invest. 1998, 102: 688-694. 10.1172/JCI3113.
Dorner T, Farner NL, Foster SJ, Lipsky PE: Immunoglobulin light chain usage in early SLE: Indications for receptor editing of both V kappa and V lambda light chains. Arthritis Rheum. 1999, 42: S52-
Dorner T, Lipsky PE: Immunoglobulin variable-region gene usage in systemic autoimmune diseases. Arthritis Rheum. 2001, 44: 2715-2727. 10.1002/1529-0131(200112)44:12<2715::AID-ART458>3.0.CO;2-L.
Alexander T, Thiel A, Rosen O, Massenkeil G, Sattler A, Kohler S, Mei H, Radtke H, Gromnica-Ihle E, Burmester GR, Arnold R, Radbruch A, Hiepe F: Depletion of autoreactive immunologic memory followed by autologous hematopoietic stem cell transplantation in patients with refractory SLE induces long-term remission through de novo generation of a juvenile and tolerant immune system. Blood. 2009, 113: 214-223. 10.1182/blood-2008-07-168286.
Nimmerjahn F, Ravetch JV: Fc-receptors as regulators of immunity. Adv Immunol. 2007, 96: 179-204.
Ettinger R, Kuchen S, Lipsky PE: Interleukin 21 as a target of intervention in autoimmune disease. Ann Rheum Dis. 2008, 67 (Suppl 3): iii83-iii86.
Jacobi AM, Reiter K, Mackay M, Aranow C, Hiepe F, Radbruch A, Hansen A, Burmester GR, Diamond B, Lipsky PE, Dörner T: Activated memory B cell subsets correlate with disease activity in systemic lupus erythematosus-Delineation by expression of CD27, IgD, and CD95. Arthritis Rheum. 2008, 58: 1762-1773. 10.1002/art.23498.
Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, Lee EH, Milner EC, Sanz I: A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol. 2007, 178: 6624-6633.
Klein U, Rajewsky K, Kuppers R: Human immunoglobulin (Ig)M(+)IgD(+) peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998, 188: 1679-1689. 10.1084/jem.188.9.1679.
Fecteau JF, Cote G, Neron S: A new memory CD27(-)IgG(+) B cell population in peripheral blood expressing V-H genes with low frequency of somatic mutation. J Immunol. 2006, 177: 3728-3736.
Ma CS, Pittaluga S, Avery DT, Hare NJ, Maric I, Klion AD, Nichols KE, Tangye SG: Selective generation of functional somatically mutated IgM(+)CD27(+), but not Ig isotype-switched, memory B cells in X-linked lymphoproliferative disease. J Clin Invest. 2006, 116: 322-333. 10.1172/JCI25720.
Cappione A, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, Sanz I: Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J Clin Invest. 2005, 115: 3205-3216. 10.1172/JCI24179.
Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF: A regulatory B cell subset with a unique CD1d(hi)CD5(+) phenotype controls T celldependent inflammatory responses. Immunity. 2008, 28: 639-650. 10.1016/j.immuni.2008.03.017.
Bouaziz JD, Yanaba K, Venturi GM, Wang Y, Tisch RM, Poe JC, Tedder TF: Therapeutic B cell depletion impairs adaptive and autoreactive CD4(+) T cell activation in mice. Proc Natl Acad Sci USA. 2007, 104: 20878-20883. 10.1073/pnas.0709205105.
Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, Mauri C: CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010, 32: 129-140. 10.1016/j.immuni.2009.11.009.
Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, Szabolcs PM, Bernstein SH, Magro CM, Williams AD, Hall RP, St Clair EW, Tedder TF: Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood. 2011, 117: 530-541. 10.1182/blood-2010-07-294249.
Odendahl M, Keitzer R, Wahn U, Hiepe F, Radbruch A, Dörner T, Bunikowski R: Perturbations of peripheral B lymphocyte homoeostasis in children with systemic lupus erythematosus. Ann Rheum Dis. 2003, 62: 851-858. 10.1136/ard.62.9.851.
Medina F, Segundo C, Campos-Caro A, Gonzalez-Garcia I, Brieva JA: The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood. 2002, 99: 2154-2161. 10.1182/blood.V99.6.2154.
Odendahl M, Mei H, Hoyer BF, Jacobi AM, Hansen A, Muehlinghaus G, Berek C, Hiepe F, Manz R, Radbruch A, Dörner T: Generation of migratory antigenspecific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood. 2005, 105: 1614-1621. 10.1182/blood-2004-07-2507.
Mei HE, Yoshida T, Sime W, Hiepe F, Thiele K, Manz RA, Radbruch A, Dörner T: Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood. 2009, 113: 2461-2469. 10.1182/blood-2008-04-153544.
Arce E, Jackson DG, Gill MA, Bennett LB, Banchereau J, Pascual V: Increased frequency of pre-germinal center B cells and plasma cell precursors in the blood of children with systemic lupus erythematosus. J Immunol. 2001, 167: 2361-2369.
Grammer AC, Slota R, Fischer R, Gur H, Girschick H, Yarboro C, Illei GG, Lipsky PE: Abnormal germinal center reactions in systemic lupus erythematosus demonstrated by blockade of CD154-CD40 interactions. J Clin Invest. 2003, 112: 1506-1520.
Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, Fleisher T, Balow JE, Lipsky PE: Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum. 2010, 62: 542-552. 10.1002/art.27221.
Jacobi AM, Odendahl M, Reiter K, Bruns A, Burmester GR, Radbruch A, Valet G, Lipsky PE, Dörner T: Correlation between circulating CD27(high) plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2003, 48: 1332-1342. 10.1002/art.10949.
Hutloff A, Büchner K, Reiter K, Baelde HJ, Odendahl M, Jacobi A, Dörner T, Kroczek RA: Involvement of inducible costimulator in the exaggerated memory B cell and plasma cell generation in systemic lupus erythematosus. Arthritis Rheum. 2004, 50: 3211-3220. 10.1002/art.20519.
Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG: Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005, 435: 590-597. 10.1038/nature03724.
Meffre E, Wardemann H: B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol. 2008, 20: 632-638. 10.1016/j.coi.2008.09.001.
Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC: Predominant autoantibody production by early human B cell precursors. Science. 2003, 301: 1374-1377. 10.1126/science.1086907.
Yurasov SV, Wardemann H, Hammersen J, Pascual V, Meffre E, Nussenzweig MC: Autoreactive B cells and immune tolerance alterations in peripheral blood of patients with systemic autoimmunity. Pediatric Research. 2004, 55: 292A-
Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, Gonzalez JB, Pascual V, Stichweh D, Wardemann H, Nussenzweig MC: Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci USA. 2008, 105: 9727-9732. 10.1073/pnas.0803644105.
Winkler TH, Fehr H, Kalden JR: Analysis of immunoglobulin variable region genes from human IgG anti-DNA hybridomas. Eur J Immunol. 1992, 22: 1719-1728. 10.1002/eji.1830220709.
Davidson A, Manheimerlory A, Aranow C, Peterson R, Hannigan N, Diamond B: Molecular characterization of a somatically mutated anti-DNA antibody bearing two systemic lupus erythematosus-related idiotypes. J Clin Invest. 1990, 85: 1401-1409. 10.1172/JCI114584.
Guo WZ, Smith D, Aviszus K, Detanico T, Heiser RA, Wysocki LJ: Somatic hypermutation as a generator of antinuclear antibodies in a murine model of systemic autoimmunity. J Exp Med. 2010, 207: 2225-2237. 10.1084/jem.20092712.
Breden F, Lepik C, Longo NS, Montero M, Lipsky PE, Scott JK: Comparison of antibody repertoires produced by HIV-1 infection, other chronic and acute infections, and systemic autoimmune disease. PLoS One. 2011, 6: e16857-10.1371/journal.pone.0016857.
Harley JB, Moser KL, Gaffney PM, Behrens TW: The genetics of human systemic lupus erythematosus. Curr Opin Immunol. 1998, 10: 690-696. 10.1016/S0952-7915(98)80090-3.
Lulli P, Sebastiani GD, Trabace S, Passiu G, Cappellacci S, Porzio F, Morellini M, Cutrupi F, Galeazzi M: HLA antigens in Italian patients with systemic lupus erythematosus: evidence for the association of DQw2 with the autoantibody response to extractable nuclear antigens. Clin Exp Rheumatol. 1991, 9: 475-479.
Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dörner T, Hiepe F: Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. 2006, 6: 741-750. 10.1038/nri1886.
International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, Guthridge JM, Cobb BL, Mirel DB, Marion MC, Williams AH, Divers J, Wang W, Frank SG, Namjou B, Gabriel SB, Lee AT, Gregersen PK, Behrens TW, Taylor KE, Fernando M, Zidovetzki R, Gaffney PM, Edberg JC, Rioux JD, Ojwang JO, James JA, Merrill JT, Gilkeson GS, Seldin MF, Yin H, Baechler EC, Li QZ, Wakeland EK, Bruner GR, Kaufman KM, Kelly JA: Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008, 40: 204-210. 10.1038/ng.81.
Jacobi AM, Mei H, Hoyer BF, Mumtaz IM, Thiele K, Radbruch A, Burmester GR, Hiepe F, Dörner T: HLA-DRhigh/CD27(high) plasmablasts indicate active disease in patients with systemic lupus erythematosus. Ann Rheum Dis. 2010, 69: 305-308. 10.1136/ard.2008.096495.
Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, Kroczek RA: ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature. 1999, 397: 263-266. 10.1038/16717.
Tokunaga M, Saito K, Kawabata D, Imura Y, Fujii T, Nakayamada S, Tsujimura S, Nawata M, Iwata S, Azuma T, Mimori T, Tanaka Y: Efficacy of rituximab (anti-CD20) for refractory systemic lupus erythematosus involving the central nervous system. Ann Rheum Dis. 2007, 66: 470-475.
O'Neill SK, Cao YX, Hamel KM, Doodes PD, Hutas G, Finnegan A: Expression of CD80/86 on B cells is essential for autoreactive T cell activation and the development of arthritis. J Immunol. 2007, 179: 5109-5116.
Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, Yu D, Domaschenz H, Whittle B, Lambe T, Roberts IS, Copley RR, Bell JI, Cornall RJ, Goodnow CC: A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005, 435: 452-458. 10.1038/nature03555.
Bubier JA, Sproule TJ, Foreman O, Spolski R, Shaffer DJ, Morse HC, Leonard WJ, Roopenian DC: A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc Natl Acad Sci USA. 2009, 106: 1518-1523. 10.1073/pnas.0807309106.
Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, Chan TD, Palendira U, Bustamante J, Boisson-Dupuis S, Choo S, Bleasel KE, Peake J, King C, French MA, Engelhard D, Al-Hajjar S, Al-Muhsen S, Magdorf K, Roesler J, Arkwright PD, Hissaria P, Riminton DS, Wong M, Brink R, Fulcher DA, Casanova JL, Cook MC, Tangye SG: B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. 2010, 207: 155-171. 10.1084/jem.20091706.
Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S: Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009, 325: 1006-1010. 10.1126/science.1175870.
Webb R, Wren JD, Jeffries M, Kelly JA, Kaufman KM, Tang Y, Frank MB, Merrill J, Kimberly RP, Edberg JC, Ramsey-Goldman R, Petri M, Reveille JD, Alarcón GS, Vilá LM, Alarcón-Riquelme ME, James JA, Vyse TJ, Moser KL, Gaffney PM, Gilkeson GS, Harley JB, Sawalha AH: Variants within MECP2, a key transcription regulator, are associated with increased susceptibility to lupus and differential gene expression in patients with systemic lupus erythematosus. Arthritis Rheum. 2009, 60: 1076-1084. 10.1002/art.24360.
Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, Jacques S, Ba N, Ittah M, Lepajolec C, Labetoulle M, Ardizzone M, Sibilia J, Fournier C, Chiocchia G, Mariette X: Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren's syndrome. Proc Natl Acad Sci USA. 2006, 103: 2770-2775. 10.1073/pnas.0510837103.
Lindh E, Lind SM, Lindmark E, Hässler S, Perheentupa J, Peltonen L, Winqvist O, Karlsson MC: AIRE regulates T-cell-independent B-cell responses through BAFF. Proc Natl Acad Sci USA. 2008, 105: 18466-18471. 10.1073/pnas.0808205105.
Bernasconi NL, Traggiai E, Lanzavecchia A: Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002, 298: 2199-2202. 10.1126/science.1076071.
Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ: Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity. 2010, 33: 967-978. 10.1016/j.immuni.2010.11.025.
Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ: A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999, 189: 1639-1647. 10.1084/jem.189.10.1639.
Rönnblom L, Alm GV: A pivotal role for the natural interferon alphaproducing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus. J Exp Med. 2001, 194: F59-F63. 10.1084/jem.194.12.f59.
Sfikakis PP, Karali V, Lilakos K, Georgiou G, Panayiotidis P: Clonal expansion of B-cells in human systemic lupus erythematosus: evidence from studies before and after herapeutic B-cell depletion. Clin Immunol. 2009, 132: 19-31. 10.1016/j.clim.2009.02.010.
Petersen-Mahrt SK, Harris RS, Neuberger MS: AID mutates E-coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002, 418: 99-103.
Longo NS, Lugar PL, Yavuz S, Zhang W, Krijger PH, Russ DE, Jima DD, Dave SS, Grammer AC, Lipsky PE: Analysis of somatic hypermutation in X-linked hyper-IgM syndrome shows specific deficiencies in mutational targeting. Blood. 2009, 113: 3706-3715. 10.1182/blood-2008-10-183632.
Muñoz LE, Janko C, Schulze C, Schorn C, Sarter K, Schett G, Herrmann M: Autoimmunity and chronic inflammation - two clearance-related steps in the etiopathogenesis of SLE. Autoimmun Rev. 2010, 10: 38-42. 10.1016/j.autrev.2010.08.015.
Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR: Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998, 41: 1241-1250. 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H.
Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, Kirchner T, Kalden JR, Herrmann M: Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002, 46: 191-201. 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K.
Rovere P, Vallinoto C, Bondanza A, Crosti MC, Rescigno M, Ricciardi-Castagnoli P, Rugarli C, Manfredi AA: Bystander apoptosis triggers dendritic cell maturation and antigen-presenting function. J Immunol. 1998, 161: 4467-4471.
Gaipl US, Munoz LE, Grossmayer G, Lauber K, Franz S, Sarter K, Voll RE, Winkler T, Kuhn A, Kalden J, Kern P, Herrmann M: Clearance deficiency and systemic lupus erythematosus (SLE). J Autoimmun. 2007, 28: 114-121. 10.1016/j.jaut.2007.02.005.
Jacobi AM, Huang W, Wang T, Freimuth W, Sanz I, Furie R, Mackay M, Aranow C, Diamond B, Davidson A: Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus: extension of a phase II, doubleblind, placebo-controlled, dose-ranging study. Arthritis Rheum. 2010, 62: 201-210. 10.1002/art.27189.
Merrill JT, Buyon JP: Rituximab: wanted dead or alive. Arthritis Rheum. 2010, 62: 2188-2191. 10.1002/art.27544.
Merrill JT, Burgos-Vargas R, Westhovens R, Chalmers A, D'Cruz D, Wallace DJ, Bae SC, Sigal L, Becker JC, Kelly S, Raghupathi K, Li T, Peng Y, Kinaszczuk M, Nash P: The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, doubleblind, placebo-controlled trial. Arthritis Rheum. 2010, 62: 3077-3087. 10.1002/art.27601.
Jacobi AM, Goldenberg DM, Hiepe F, Radbruch A, Burmester GR, Dorner T: Differential effects of epratuzumab on peripheral blood B cells of patients with systemic lupus erythematosus versus normal controls. Ann Rheum Dis. 2008, 67: 450-457.
Daridon C, Blassfeld D, Reiter K, Mei HE, Giesecke C, Goldenberg DM, Hansen A, Hostmann A, Frölich D, Dörner T: Epratuzumab targeting of CD22 affects adhesion molecule expression and migration of B-cells in systemic lupus erythematosus. Arthritis Res Ther. 2010, 12: R204-10.1186/ar3179.
Frölich D, Giesecke C, Mei HE, Reiter K, Daridon C, Lipsky PE, Dorner T: Secondary immunization generates clonally related antigen-specific plasma cells and memory B cells. J Immunol. 2010, 185: 3103-3110. 10.4049/jimmunol.1000911.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Dörner, T., Giesecke, C. & Lipsky, P.E. Mechanisms of B cell autoimmunity in SLE . Arthritis Res Ther 13, 243 (2011). https://doi.org/10.1186/ar3433
Published:
DOI: https://doi.org/10.1186/ar3433