
Abstract
Background
Medical management of thyroid eye disease remains controversial due to a paucity of high quality evidence on long-term treatment outcomes. Glucocorticoids are known to be effective initially but have significant side-effects with long-term use and recrudescence can occur on cessation. Current evidence is conflicting on the efficacy of radiotherapy and non-steroid systemic immunosuppression, and the majority of previous studies have been retrospective, uncontrolled, small or poorly designed.
The Combined Immunosuppression and Radiotherapy in Thyroid Eye Disease (CIRTED) trial was designed to investigate the efficacy of radiotherapy and azathioprine in combination with a standard course of oral prednisolone in patients with active thyroid eye disease.
Methods/design
Patients with active thyroid eye disease will be randomised to receive (i) azathioprine or oral placebo and (ii) radiotherapy or sham-radiotherapy in this multi-centre, factorial randomised control trial. The primary outcome is improvement in disease severity (assessed using a composite binary measure) at 12 months and secondary end-points include quality of life scores and health economic measures.
Discussion
The CIRTED trial is the first study to evaluate the role of radiotherapy and azathioprine as part of a long-term, combination immunosuppressive treatment regime for Thyroid Eye Disease. It will provide evidence for the role of radiotherapy and prolonged immunosuppression in the management of this condition, as well as pilot data on their use in combination. We have paid particular attention in the trial design to establishing (a) robust placebo controls and masking protocols which are effective and safe for both radiotherapy and the systemic administration of an antiproliferative drug; (b) constructing effective inclusion and exclusion criteria to select for active disease; and (c) selecting pragmatic outcome measures.
Trial registration
Current controlled trials ISRCTN22471573
Similar content being viewed by others

Background
Thyroid eye disease (TED) can be a visually disabling and cosmetically disfiguring condition which significantly impairs health-related quality of life [1–3]. A wide variety of immunosuppressive therapies have been used to target the early active inflammatory phase of the disease, including glucocorticoids (GC's), radiotherapy, antiproliferative agents, T-cell inhibitors and, more recently, biologics. The goal of these interventions is to suppress the autoimmune inflammatory phase of the disease, thereby altering the course of the disease and reducing the severity of residual changes in the extraocular muscles, orbital fat and other periocular soft tissues which result in permanent visual and cosmetic dysfunction [1, 4, 5].
GC's have been used to treat TED for over forty years, and several studies have demonstrated their efficacy with an overall response rate of 63–77% [1]. However, GC treatment is typically discontinued after 3 to 5 months [6–9] because of the side-effects associated with their long-term use, and subsequent disease recrudescence is a common problem [10]. This recurrence may be prevented by the concomitant use of ciclosporin [11] (which continues after GC treatment stops), and combinations of ciclosporin and prednisolone also achieve a better initial treatment response than either agent alone [6]. However, the routine use of such second-line immunosuppressive drugs in the management of TED has been limited by fears about their potential toxicity, and glucocorticoid monotherapy remains the mainstay of conventional treatment.
Radiotherapy is also well established in the treatment of TED [12, 13], and its efficacy was affirmed in 1993 by the results of a prospective double-blind randomised control trial (RCT) which reported that orbital radiotherapy was as effective as oral prednisolone[7]. However, the same authors subsequently found that radiotherapy was no better than placebo (except in a subgroup of patients with motility impairment) [14], and a recent trial from the Mayo Clinic in the USA also could not demonstrate a beneficial therapeutic effect [15]. As a result, many clinicians, particularly in North America, have abandoned its use.
The Mayo study has been widely criticised [16]and European groups argue that the balance of evidence remains in favour of radiotherapy [17]. In support of this, the most recent randomised control trial (in patients with relatively mild disease) found that radiotherapy was better than placebo when outcome was assessed by clinical measures [18]. However, this was not associated with quality-of-life or health economic gain, and the role of radiotherapy as monotherapy for TED remains the subject of heated debate [19–28].
Older RCTs have shown that radiotherapy is more effective in combination with steroids than when either agent is used alone [29, 30]. However, this distinction between monotherapy and combination therapy is rarely highlighted, and the confusion which has resulted from the inconsistent recent evidence has caused many ophthalmologists to discontinue using radiotherapy as part of a combined treatment regime. Consequently there is a pressing need for a definitive study to investigate radiotherapy's role in combination with steroids.
The data supporting the use of ciclosporin combined with steroids [6, 11], and radiotherapy combined with steroids [30, 31] are not surprising given current knowledge about the benefits of combination therapies in other autoimmune ocular and systemic conditions [32, 33]. Manipulation of an autoimmune response is more effective when more than one mechanism of immunosuppression is used, and this can be especially so early in the disease [32]. Combined drug treatments can also be continued long-term, preventing the disease reactivation which is commonly seen at the end of typical short-term low dose steroid monotherapy regimes, and enabling the duration of an individual's treatment to be tailored to the length of the active phase of their disease.
Azathioprine is a low-cost second-line immunosuppressive agent which is widely used in the management of other autoimmune conditions. It is better tolerated than ciclosporin [34] and does not cause the renal toxicity and cardiovascular side-effects [35] (including hypertension and hypercholesterolaemia) associated with ciclosporin. However, it can cause bone marrow suppression [36] and hepatotoxicity [37]. This risk has been greatly reduced with the advent of a laboratory assay for the enzyme thiopurine methyltransferase (TPMT), which regulates a key step in azathioprine metabolism. Now individuals with low TPMT activity, who would otherwise be at particular risk of azathioprine toxicity, can be identified and their treatment modified or withheld [38]. Hence, azathioprine has safety, tolerability and cost advantages over ciclosporin.
Retrospective data suggests that azathioprine, when used in combination with oral prednisolone and radiotherapy, can significantly reduce the long-term severity of TED and the need for rehabilitative surgery [39, 40]. However, the use of azathioprine for TED remains highly controversial; in part because it has previously been proven ineffective as monotherapy [41]. As with radiotherapy, there is no clear evidence base and clinical practice consequently varies widely.
The current trial is designed to investigate whether (i) radiotherapy and (ii) azathioprine are effective when used in combination with steroids for the treatment of active TED. It will also provide pilot data on the possible benefits of triple therapy (steroids, azathioprine and radiotherapy). Particular attention in the trial design has been given to (a) establishing robust placebo controls and masking protocols which are effective and safe for both radiotherapy and the systemic administration of an antiproliferative drug; (b) constructing effective inclusion and exclusion criteria to select for active disease; and (c) selecting pragmatic outcome measures. Health economic and carefully designed quality of life/disfigurement analyses also accompany the trial.
Methods/design
Trial design
Factorial design, double-masked, randomised controlled trial. Study recruits receive either azathioprine or placebo plus either radiotherapy or sham-radiotherapy, in combination with a standard oral prednisolone regime (Figure 1).

Trial Design.
Objectives
Primary
To test the hypotheses that in patients being treated with prednisolone for active TED:
-
1.
Radiotherapy (compared with placebo) induces early remission and reduces long-term disease severity.
-
2.
Azathioprine (compared with placebo) reduces long-term disease severity.
Secondary
-
1.
To test the hypotheses that in patients being treated with prednisolone for active TED, radiotherapy and azathioprine improve health-related quality of life, and are cost-effective.
-
2.
To validate the use of the GO-QoL (a new TED specific quality of life score) in the UK population.
-
3.
To improve understanding of the extent and type of psychosocial distress experienced by TED patients.
-
4.
To conduct an economic evaluation of the cost of TED and its treatment to patients, the National Health Service (NHS) and Society.
-
5.
To report the safety and tolerability of radiotherapy and azathioprine in the study cohort.
Eligibility
Inclusion criteria
-
Clinical Activity Score (Table 1) ≥ 4 (worst eye) OR ≥ 2 (worst eye) with a history of proptosis or motility restriction of less than 6 months duration.
-
Past or present history of abnormal Thyroid Gland Function OR a clinical diagnosis of TED made and confirmed by ≥ 2 muscle involvement on Computed Tomography (CT) or Magnetic Resonance Imaging (MRI) scan.
Definitions
-
Proptosis: Either subjective unilateral proptosis confirmed by asymmetry in exophthalmometry of ≥ 2 mm or subjective bilateral proptosis.
-
Motility restriction: Intermittent, inconstant or constant diplopia [42].
Exclusion criteria
-
Age < 20 or > 75 yrs
-
Dysthyroid optic neuropathy
-
No proptosis or motility restriction (see above for definitions)
-
Use of radioiodine within the last 3 months
-
Previous orbital irradiation
-
Pre-existing glaucoma with a characteristic optic disc appearance and associated visual field defect
-
Pre-existing Diabetes Mellitus (not simply steroid induced disease from recent therapy)
-
Previous adverse event associated with, or contraindication to, either prednisolone or azathioprine
-
Within 6/12 of pregnancy, women planning pregnancy
-
Lactation
-
Haemoglobin Concentration > 1 g/dl below the local laboratory's reference range
-
Platelet Count < 130 × 109/L
-
White Cell Count below the local laboratory's reference range
-
Lymphocyte Count < 0.8 × 109/L
-
Abnormal Thiopurine Methyltransferase (TPMT) activity
-
Abnormal renal function (estimated Glomerular Filtration Rate (eGFR) < 60 ml/min/1.73 m2)
-
Abnormal liver function, specifically: bilirubin, alanine aminotransferase or alkaline phosphatase concentrations > 1.5× above the local laboratory's reference range
-
Malignant or pre-malignant (dysplastic) condition within the past 5 years
-
Previous tuberculosis
-
Shingles within the past 3/12
-
HIV/AIDS
-
Concurrent use of other immunosuppressive agents or allopurinol
-
Live vaccines within the past 3 months
All. patients referred to the Trial Centres with TED are screened, and those meeting the eligibility criteria are invited to enrol. The study is conducted according to ICH-GCP (International Conference on Harmonisation for Good Clinical Practice in clinical research), as set out in the European Union Clinical Trials Directive (2001) and associated UK Regulations (2004), which adhere to the principles of the Helsinki Declaration. At the first trial visit informed written consent is obtained and baseline characteristics measured, including completion of a quality of life questionnaire. Recruits then receive a standard course of high dose tapering prednisolone (Table 2 &3). If they either have a < 6 month history of TED (defined as time since first symptom) or report improvement in any item of the Clinical Activity Score (CAS) 2 weeks after starting the trial prednisolone regime, they are considered to have 'active' disease and are randomised at the second trial visit (Table 4).
Interventions
Radiotherapy
A dose of 20 Gy is administered to the retrobulbar orbit in 12 fractions over 2 to 3 weeks. The protocols used for treatment planning, verification, field arrangement and patient immobilisation vary according to the usual practice of each Trial Radiotherapy Centre.
In Bristol (Bristol Oncology Centre), patients are marked up on the treatment machine (a 6-MV linear accelerator) and immobilised with tape across their forehead. Isodose plans are not produced as a routine, but verification is carried out with Electronic Portal Imaging Devices (EPIDs) on days 1 and 2. Lasers are used to orientate the patient and radiation is delivered to 4 cm × 4 cm lateral opposed fields, using asymmetric jaws to eliminate divergence towards the contra-lateral lens.
In London (St Bartholomew's Hospital) and Manchester (Christie Hospital), all patients have an orbital CT scan prior to treatment as well as a dosimetric plan. Patients are immobilised with a full head shell, which is individually made for each trial subject, and radiation is delivered to 5 cm × 5 cm lateral opposed fields using a 6-MV linear accelerator. Thermoluminescence dosimetry (TLD) is then used for both eyes during the first treatment to check the lens dose.
Azathioprine
Treatment dose varies between 100 mg and 200 mg daily (dispensed as 50 mg tablets), depending on body weight (Table 5).
Masking
Trial participants, the clinical investigators responsible for patient assessments, and data analysts are masked to treatment allocation. Masking success is evaluated at the end of the study with a questionnaire asking participants and investigators to state whether they think they know which treatments they received, and if so why.
Placebo treatments
Sham – radiotherapy
In Bristol, treatment is planned as for active therapy, which does not involve radiation. However, in London and Manchester, patients having active treatment are exposed to radiation during both their CT scan and dosimetric planning. To avoid this in the sham-treatment group, sham-CT scans and a generic dosimetric plan with zero downloaded monitor units (ie no radiation exposure) are used.
Radiotherapy machines make a loud buzzing noise when switched on and this is the only indicator patients have that they are receiving treatment. In order to mimic this for patients allocated to receive sham-radiotherapy, bespoke noise-emitting devices (NEDs) which make a similar sound to that which accompanies normal treatment have been constructed, and are positioned above the gantry of the machines in Bristol, London and Manchester. The NEDs are activated from the radiotherapy suite's control room for the same duration of time that treatment would usually be administered. Hence, patients allocated to the sham-radiotherapy arm of the study have the same planning and treatment experience, but receive no radiation at all.
Placebo azathioprine tablets
These are made for the trial by St Thomas' Hospital Pharmacy Manufacturing Unit in accordance with Good Manufacturing Practice and exactly match the appearance of the active azathioprine tablets, which are removed from their blister-packs and re-packaged in the same bottles as the placebos. Both active and placebo drugs are then labelled as trial-specific Investigational Medicinal Products (IMPs) and distributed to the trial centres.
Placebo azathioprine tablets are also dispensed according to body weight, so patients in the placebo group take the same number of tablets per day as they would if receiving active treatment. (Table 5).
Additional measures to maintain masking
Sham-radiotherapy
The trial radiographers are by necessity aware of each patient's treatment allocation. Their contact with trial recruits is therefore the time at which patients are at greatest risk of being un-masked. In order to minimise this risk, the number of radiographers involved in the administration of radiotherapy to trial subjects has been limited as much as possible. These individuals have all received specific training about the study and are very aware of the importance of maintaining masking, which they achieve by acting out their well-practiced treatment set-up and delivery protocols in a standard fashion to all patients regardless of whether they receive sham or active therapy. The clinical oncologists who consult with patients before and during treatment are not informed of treatment allocation, and the contents of patients' hospital records are specially adapted to keep this information concealed until they complete the study.
The radiotherapy departments involved in the trial have also examined their local facilities to ensure that other potential indicators of treatment allocation, such as the activation of warning lights outside radiotherapy suites when radiotherapy machines are being used, are identified and addressed (for example, taking care to sit patients and relatives out of sight of the warning lights).
Placebo azathioprine tablets
All trial recruits, whether allocated to the azathioprine or placebo arms of the trial, will have regular blood tests to monitor for the potential adverse effects of azathioprine. These results are monitored by an un-masked trial co-ordinator, who arranges for trial subjects to be recalled if abnormalities are revealed and adjusts their treatment in accordance with a standard algorithm (Figure 2).

Azathioprine Dose Adjustment/Blood Test Algorithm.
In order to maintain masking about 5% of the placebo treated group are also being randomly recalled for identical drug 'dose' adjustment, and repeat blood tests. The un-masked trial co-ordinator is never in direct contact with trial recruits, and the instructions issued to the clinical investigators to recall patients and adjust their treatment is standard, regardless of whether they are receiving active or placebo tablets. Hence, safety is ensured for patients on azathioprine, and treatment allocation is not revealed to the trial subjects or investigators.
If the un-masked trial co-ordinator has a query about a blood test result which is not adequately addressed by the standard treatment algorithm (Figure 2), they can reveal a patient's treatment allocation and ask for advice from one of the clinical investigators at another Trial Centre. This clause provides a safety net should an unforeseen clinical scenario arise, and should not introduce bias as the patient and the clinical investigator responsible for their assessment remains masked.
Allocation of trial interventions
Patients who are eligible to continue in the study at the second trial visit (after confirmation of active disease by response to steroids or short disease duration – see above) are allocated to treatment groups by minimisation. This is a dynamic process which reduces the imbalance between trial arms with respect to a range of predefined prognostic variables, and a randomisation schedule is therefore not drawn up in advance. Instead, a form categorising each study recruit according to the minimisation criteria set out in Table 6 is returned to the randomisation service at Moorfields Eye Hospital on enrolment, giving them time to determine the patient's treatment allocation should they be eligible to proceed in the study. The trial pharmacists at each Trial Centre then phone the randomisation service on receipt of a trial IMP prescription at the patient's second visit, and are notified whether they are to dispense azathioprine or placebo. This is confirmed by email, which also states the patient's radiotherapy group – a copy of which is sent to the Trial Radiotherapy Centre radiographers and the un-masked trial co-ordinator.
Further measures to reduce bias
Standard Patient Assessments
An 'atlas' of standard photographs is being used for reference in clinical assessments to reduce inter-observer differences in the measurement of disease activity and severity [43].
Management of other factors which have the potential to influence TED
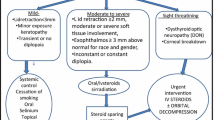
Endocrine Management
The protocol for endocrine treatment is outlined in Figure 3. This will be administered in liaison with the patient's endocrinologist, or if the patient is not under the care of an endocrinologist when enrolled, by the trial investigators. The trial endocrinologist (CMD) will mediate if a patient's endocrinologist does not adhere to the trial protocol, and all protocol deviations will be reported at the end of the study.

Endocrine Treatment Protocol.
Smoking
All trial subjects are advised to stop smoking and changes in smoking habits are recorded.
Follow-up
The timing and content of trial assessments are illustrated in Table 4.
In addition to completing quality of life questionnaires, a subset of patients will also be selected to participate in a semi-structured interview to explore their individual concerns, strengths and difficulties in social functioning, coping strategies, levels of perceived support and the personal financial consequences of their disease. Interviews will be tape recorded and subsequently transcribed, after which the tapes will be erased (transcriptions will also be destroyed at the end of the study).
At the second study visit all patients are issued with a trial diary (see Additional file 1). This records a wide variety of information relevant to their treatment costs, including use of primary and secondary healthcare, prescriptions, travel costs, over-the-counter medications, cosmetics and loss of earnings.
Outcome measures
Primary
1. Binary composite outcome score:
Treatment success and failure will be defined at study completion (1 year) using a system of major and minor criteria modified from others [8, 14, 18] (Figure 4).

Binary Composite Primary Outcome Measure.
2. Ophthalmopathy index [44]:
Treatment response at study completion (1 year) will also be quantified using the Ophthalmopathy Index (OI) as a continuous measure of disease severity.
Co-primary
1. Clinical activity score [45, 46]:
Short-term response to treatment (3 months) will be evaluated using the Clinical Activity Score (CAS, Table 1) as a continuous measure of disease activity.
Secondary
-
1.
Clinical measures:
Total eye score (TES) [6].
-
2.
Psychosocial measures [see Additional file 2]:
-
Hospital Anxiety and Depression Scale (HADS) score [47]
-
Graves' Ophthalmopathy Quality of Life (GO-QoL) score [50, 51]
-
WHOQoL-Brief Quality of Life Assessment Score [52]
-
Open-ended responses to interview questions
-
3.
Health economic measures [see Additional file 1]:
The cost of TED and its treatment will be measured from the perspective of the NHS, the patient and society.
Adverse events
Adverse events will be documented and serious adverse events will be reported to the trial sponsor and to the Medicines and Healthcare Products Regulatory Agency (MHRA). Specific enquiries regarding known side-effects of the active interventions in the trial (as documented below) will be made at each visit and documented. Clinical examination will include monitoring for adverse events.
Prednisolone – gastric irritation/ulceration, weight gain, sleep disturbance, mood
disturbance, increased susceptibility to infection, increase in blood pressure, steroid induced diabetes.
Azathioprine – malaise, nausea, vomiting, diarrhoea, rash, myalgia, hair loss, increased susceptibility to infection. Regular blood tests will be carried out on all patients to monitor for bone marrow suppression and liver impairment.
Radiotherapy – localized redness of skin, cataracts, radiation retinopathy.
As this is a double-masked trial all adverse events will be documented and reviewed by the data monitoring committee who may be unmasked to treatment allocation.
Trial size
Number of patients
A two group continuity corrected chi-squared test with a 0.050 two-sided significance level will have 80% power to detect the difference between a success rate in the placebo arm of 60% and success rate in the treated arm of 87% (odds ratio of 4.462) when the sample size in each group is 48. It is therefore necessary for there to be 96 subjects with complete final data sets to be equally distributed to the 4 treatment groups.
The power for continuous outcome measures is more favourable and a total sample size of 100 patients will yield 92% power to detect a difference of 2.7 in the OI with two-sided 5% alpha, assuming a standard deviation of 3.3.
CAS is not considered as important as the primary outcomes in terms of influencing future clinical practice. However it is of greater interest and importance than the secondary outcome variables, and as such it has been selected as a coprimary endpoint and the power of the trial to detect a clinically important difference is presented here. Assuming a standard deviation of between 1.2 and 2.0, a total sample of 100 patients will yield between 71% and 99% power to detect a difference of 1 point with 5% two-sided alpha.
The above power calculations are based on comparisons 'at the margins' of the factorial design (ie, comparing all azathioprine treated patients with placebo treated patients, and all radiotherapy treated patients with sham-radiotherapy treated patients). There will be low power for comparisons between individual cells of the factorial design (eg azathioprine and sham-radiotherapy vs. placebo and radiotherapy), although these analyses will still be conducted to generate pilot data on the comparative efficacy of the 4 combination regimes under investigation.
Compliance and loss to follow-up
We predict that 33% of eligible patients will choose not to take part in the study [14], 11% will be excluded because of TPMT inactivity [53], 10% of the azathioprine treated group will be intolerant of therapy or suffer an adverse event necessitating withdrawal from the trial [54], and 5% will drop-out for other reasons.
Statistical analyses
Data analysis will proceed according to CONSORT guidelines for randomised controlled trials. The first stage of the analysis will be to use descriptive statistics to describe the group of individuals recruited to the trial in relation to those eligible, and to investigate comparability of the trial arms at baseline. The primary intention-to-treat analyses will comprise comparisons between radiotherapy versus sham-radiotherapy, and azathioprine versus placebo, for each of the two primary outcomes at 12 months follow up. The comparisons will be made using appropriate (that is, logistic or linear) multivariable regression models, adjusting for minimisation variables, the factorial design, and the value of the outcome variable at baseline. Full attention will be paid to the estimates and the confidence intervals for these comparisons as well as the p-values. Secondary analyses will then be conducted using regression models with further adjustment for any prognostic factors that exhibit marked imbalance at baseline. Patients who have no outcome data for the primary analyses will have data imputed using last observation carried forward. Patients who have become non-compliant for any reason will still be invited to produce outcome data such that they can be included in the intention to treat analyses. The assumptions required for the regression models will be investigated using appropriate diagnostic plots, and actions such as transformation of continuous outcome variables taken as necessary.
The co-primary outcome CAS at 3 months follow up, and all secondary outcomes, will be analysed in the same way, using appropriate (linear or logistic) regression models depending on the nature of the outcome measure. Bonferroni corrections for multiple testing will be considered for the secondary outcomes.
Other secondary analyses will involve: (a) investigation of any interaction between the two interventions for each of the two primary outcomes; (b) pre-planned sub-group analyses to ascertain any differential effects of the interventions according to steroid use versus no steroid use in the six months prior to enrolment. These secondary analyses are readily performed as extensions to the multivariable regression models described above, by simply introducing the appropriate interaction terms. However, the precision of the estimates of interaction is very likely to be too poor, and high p-values will most likely reflect low power and so cannot be taken as evidence for no interactions.
Economic evaluation
Costs will be related to the percentage of patients responding to treatment. Incremental cost effectiveness ratios will be formed, which will estimate the extra cost per extra patient responding to (i) radiotherapy; and (ii) azathioprine 12 months after randomisation. A secondary analysis will estimate the cost-of-illness of TED from a societal perspective.
Trial organisation
Trial Centres and Investigators are listed in Table 7. Additional trial centres may be added if recruitment is slower than anticipated.
Trial Steering Committee
The Trial Steering Committee will monitor and supervise the trial and comment on any proposed major protocol amendments (Table 8).
Data Monitoring Committee
The data monitoring committee (DMC) includes an independent ophthalmologist with experience of thyroid eye disease and an independent statistician (Table 8).
No formal interim analysis is planned. The trial statistician will report to an independent DMC which will monitor the trial in all its respects. It will review data from the trial every 12 months, consider the findings from other relevant studies and if considered appropriate may conduct a masked interim analysis. The trial will only be terminated as a result of this analysis if the p value is smaller than 0.0001 and the DMC decides that this is the best course of action to take.
Study co-ordination
This study will be centrally co-ordinated by Moorfields Eye Hospital Clinical Trials Unit (CTU). As well as providing the telephone randomisation service the CTU will be responsible for data management.
Trial documentation and data collection
The trial centres are provided with a Protocol, Standard Operating Procedures [see Additional file 3] (including standardised colour plates for patient assessments [43]), Source Documentation and Case Report Forms [see Additional file 4]. Adverse event forms are completed at each participant's visit. Serious adverse events will be reported to the University of Bristol (the Trial Sponsor) and to the Medicines and Healthcare products Regulatory Agency (MHRA).
Ethics and competent authority review
Applications to UK Main and Local Research Ethics Committee (REC) have received favourable opinions and a Clinical Trials Authorisation has been issued by the MHRA.
Publication policy
The results of this trial will be submitted for publication in peer-reviewed medical journals regardless of whether the outcome is in favour of the trial interventions. Authorship agreements have been signed by the investigators prior to the start of the study.
Proposed trial time-table
Trial start
January 2006
Projected trial end
December 2011
Trial duration
6 Years
Duration of each patient's participation
12 months
Discussion
This randomised controlled trial of azathioprine and radiotherapy (with prednisolone) will be the first to evaluate the role of these interventions as part of a long-term combined immunosuppressive treatment regime for TED. We hope to help resolve the debate which has arisen from recent monotherapy studies [16, 55], and seek not only to establish whether the addition of azathioprine or radiotherapy confers benefit in patients treated with glucocorticoids, but also to test the principle of combination therapies in TED.
The use of a factorial design enables us to evaluate two interventions in a single study and is the most valid means of establishing whether the combination of two or more therapies achieves incremental benefits [56]. It also makes efficient use of patients, which is especially important because of the limited numbers of potential recruits with active TED [56, 57]. However, in using a factorial design we are assuming (but will have insufficient power to test) that there is no interaction between trial interventions, as if a significant biological or statistical interaction between radiotherapy and azathioprine treatment exists, our calculation of their independent effects will be incorrect. The validity of the study design therefore rests on the judgement of the trial investigators and statistical advisors that the potential for an interaction between our interventions to have a 'clinically significant' effect on the interpretation of the trial data is very low (ie ≤ 5%). We believe this to be a reasonable assertion and consider that the risk of generating uninterpretable data is outweighed by the benefits of using a factorial design to assess combination treatments for an uncommon condition. Our small sample size will also limit our sensitivity to small differences in treatment efficacy, which is a compromise we accept as integral to the conduct of a pragmatic study.
In addition to the restricted number of trial subjects, there are many factors in the natural history and clinical evaluation of TED which pose challenges to the objective and scientific comparison of treatment outcomes. For example, the control of systemic thyroid dysfunction reduces disease severity, but delay in TED treatment until a 2 month period of euthyroidism has been maintained (as has been advocated by other investigators [7, 9, 14, 18]) in order to prevent this confounding interpretation of treatment efficacy, potentially misses the opportunity to obtain maximal benefit from immunosuppression in the earliest, most active phase of the disease. A patient's smoking status, previous exposure to steroids and disease severity can also independently influence their response to treatment. Furthermore, measures of disease activity and severity are notoriously subjective [58, 59], and although some outcome measures have been used more often than others in previous studies, there is no single standardised and robustly validated scoring system to use in the assessment of treatment efficacy.
We have sought to address the potential influence of multiple confounding factors through the use of a minimisation strategy to ensure their even distribution across trial arms (Table 6). The absence of validated outcomes is more problematic, particularly with regard to measures of disease severity, which need to incorporate a range of parameters to quantify both cosmetic deformity and visual dysfunction. The available continuous scoring systems [6, 44] can be relatively insensitive to clinically significant treatment differences and depend on large sample sizes. Consequently, recent studies have tended to use binary composite outcome measures, weighted to detect improvement in a selected disease components, such as oculomotility [8, 42, 43, 46]. However, no two studies have used the same criteria to define treatment success, and we have combined elements of each in order to generate our scoring system (Figure 4). There is greater consensus on the measurement of clinical activity and the CAS is an accepted standard which has been widely used in recent studies [8, 14, 15, 18, 60, 61], however it remains limited by its subjectivity [58, 59] and inability to predict response to steroids [45].
In order to ameliorate the problem of subjectivity in clinical assessments with consequent inter-observer variability we are using Dickinson et al.'s [43] reference atlas of colour plates and our standard operating procedures for patient examinations are based on those advocated by the European Group on Graves Ophthalmopathy (EUGOGO). In addition, we are seeking to lay the foundations for fully objective measurement of disease activity and severity through an ancillary Magnetic Resonance Imaging (MRI) study which is being conducted alongside the trial.
We have gone to great lengths to establish placebo controls for the trial interventions, in particular with sham-radiotherapy and decoy dose changes for patients allocated to receive placebo tablets. However, there is a risk that the common side-effects of azathioprine, such as lethargy and nausea, will unmask investigators and patients to their treatment allocation. This may be off-set by the overlap between the side-effect profiles of azathioprine and prednisolone and the success of our masking procedures will ultimately be evaluated by questionnaire when each patient completes the trial.
Referral patterns to the study centres dictate that it is impractical to restrict trial entry to patients who have not been previously exposed to steroids. Hence, all potential recruits who have sufficiently active disease to be eligible for trial entry will be enrolled regardless of their prior steroid exposure, and the potential this has to bias their subsequent immunoresponsiveness is accounted for in the minimisation criteria. In addition, the practicalities of day-case admission for intravenous (IV) prednisolone treatment have prevented the trial centres from adopting this as their usual practice, despite recent evidence in favour of IV administration [62], and we are consequently using oral prednisolone in the trial.
In summary, the CIRTED trial [63] seeks to utilise a high-quality, pragmatic trial design to evaluate combination immunotherapies for the treatment of TED and enhance the evidence base available to inform treatment decisions for both patients and health care professionals.
Abbreviations
- CAS:
-
Clinical Activity Score
- CT:
-
Computed Tomography
- CTU:
-
Clinical Trials Unit
- DMC:
-
Data Monitoring Committee
- EUGOGO:
-
European Group on Graves Ophthalmopathy
- GC:
-
Glucocorticoids
- GO-QoL:
-
Graves Ophthalmopathy Quality of Life
- HADS:
-
Hospital Anxiety and Depression Scale
- IMP:
-
Investigational Medicinal Product
- MHRA:
-
Medicines and Healthcare products Regulatory Agency
- MRI:
-
Magnetic Resonance Imaging
- NED:
-
Noise-emitting device
- NHS:
-
National Health Service
- OI:
-
Ophthalmopathy Index
- RCT:
-
Randomised Control Trial
- REC:
-
Research Ethics Committee
- TED:
-
Thyroid Eye Disease
- TES:
-
Total Eye Score
- TLD:
-
Thermoluminescence Dosimetry
- TPMT:
-
Thiopurine Methyltransferase
References
Bartalena L, Pinchera A, Marcocci C: Management of Graves' ophthalmopathy: reality and perspectives. Endocrine Reviews. 2000, 21: 168-199. 10.1210/er.21.2.168.
Terwee C, Wakelkamp I, Tan S, Dekker F, Prummel MF, Wiersinga W: Long-term effects of Graves' ophthalmopathy on health-related quality of life. European Journal of Endocrinology. 2002, 146: 751-757. 10.1530/eje.0.1460751.
Farid M, Roch-Levecq A-C, Levi L, Brody BL, Granet DB, Kikkawa DO: Psychological Disturbance in Graves Ophthalmopathy. Arch Ophthalmol. 2005, 123: 491-496. 10.1001/archopht.123.4.491.
Cawood T, Moriarty P, O'Shea D: Recent developments in thyroid eye disease. BMJ. 2004, 329: 385-390. 10.1136/bmj.329.7462.385.
Rundle F: Development and course of exophthalmos and ophthalmoplegia in Graves' disease with special reference to the effect of thyroidectomy. Clin Sci. 1945, 5: 177-194.
Prummel MF, Mourits MP, Berghout A, Krenning EP, van der Gaag R, Koornneef L, Wiersinga WM: Prednisone and cyclosporine in the treatment of severe Graves' ophthalmopathy. New England Journal of Medicine. 1989, 321: 1353-1359.
Prummel MF, Mourits MP, Blank L, Berghout A, Koornneef L, Wiersinga WM: Randomized double-blind trial of prednisone versus radiotherapy in Graves' ophthalmopathy. Lancet. 1993, 342: 949-954. 10.1016/0140-6736(93)92001-A.
Marcocci C, Bartalena L, Tanda ML, Manetti L, Dell'Unto E, Rocchi R, Barbesino G, Mazzi B, Bartolomei MP, Lepri P, Cartei F, Nardi M, Pinchera A: Comparison of the effectiveness and tolerability of intravenous or oral glucocorticoids associated with orbital radiotherapy in the management of severe Graves' ophthalmopathy: results of a prospective, single-blind, randomized study. Journal of Clinical Endocrinology & Metabolism. 2001, 86 (8): 3562-3567. 10.1210/jc.86.8.3562.
Kahaly G, Pitz S, Muller-Forell W, Hommel G: Randomized trial of intravenous immunoglobulins versus prednisolone in Graves' ophthalmopathy. Clinical & Experimental Immunology. 1996, 106 (2): 197-202. 10.1046/j.1365-2249.1996.d01-854.x.
Bartalena L, Marcocci C, Pinchera A: Treating severe Graves' ophthalmopathy. Baillieres Clinical Endocrinology & Metabolism. 1997, 11: 521-536. 10.1016/S0950-351X(97)80738-0.
Kahaly G, Schrezenmeir J, Krause U, Schweikert B, Meuer S, Muller W, Dennebaum R, Beyer J: Ciclosporin and prednisone v. prednisone in treatment of Graves' ophthalmopathy: a controlled, randomized and prospective study. European Journal of Clinical Investigation. 1986, 16: 415-422.
Bartalena L, Marcocci C, Tanda ML, Rocchi R, Mazzi B, Barbesino G, Pinchera A: Orbital radiotherapy for Graves' ophthalmopathy. Thyroid. 2002, 12: 245-250. 10.1089/105072502753600223.
Pinchera ABL, Chiovato LCM: The Eye and Orbit in Thyroid Disease. The Eye and Orbit in Thyroid Disease. Edited by: Gorman CAWR, JA D. 1984, New York: Raven Press, 301-316.
Mourits MP, van Kempen-Harteveld ML, Garcia MB, Koppeschaar HP, Tick L, Terwee CB: Radiotherapy for Graves' orbitopathy: randomised placebo-controlled study. Lancet. 2000, 355: 1505-1509. 10.1016/S0140-6736(00)02165-6.
Gorman CA, Garrity JA, Fatourechi V, Bahn RS, Petersen IA, Stafford SL, Earle JD, Forbes GS, Kline RW, Bergstralh EJ, Offord KP, Rademacher DM, Stanley NM, Bartley GB: A prospective, randomized, double-blind, placebo-controlled study of orbital radiotherapy for Graves' ophthalmopathy. 2004 Jul; 111(7):1306. Ophthalmology. 2001, 108: 1523-1534. 10.1016/S0161-6420(01)00632-7.
Feldon SE: Radiation therapy for Graves' ophthalmopathy: trick or treat?. Ophthalmology. 2001, 108: 1521-1522. 10.1016/S0161-6420(01)00729-1.
Bartalena L, Marcocci C, Pinchera A: Orbital Radiotherapy for Graves' Ophthalmopathy. J Clin Endocrinol Metab. 2004, 89: 13-14. 10.1210/jc.2003-031769.
Prummel MF, Terwee CB, Gerding MN, Baldeschi L, Mourits MP, Blank L, Dekker FW, Wiersinga WM: A randomized controlled trial of orbital radiotherapy versus sham irradiation in patients with mild Graves' ophthalmopathy. Journal of Clinical Endocrinology & Metabolism. 2004, 89 (1): 15-20. 10.1210/jc.2003-030809.
Haddad HM: Orbital radiation therapy for Graves' ophthalmopathy. [comment]. Ophthalmology. 2003, 110: 450-10.1016/S0161-6420(02)01842-0. author reply 450
Bartalena L, Marcocci C, Gorman CA, Wiersinga WM, Pinchera A: Orbital radiotherapy for Graves' ophthalmopathy: useful or useless? Safe or dangerous?. Journal of Endocrinological Investigation. 2003, 26: 5-16.
Bartalena L: Orbital radiation therapy for Graves' ophthalmopathy. [comment]. Ophthalmology. 2003, 110: 452-453. 10.1016/S0161-6420(02)01848-1.
Steinsapir KD, Goldberg RA: Orbital radiation therapy for Graves' ophthalmopathy. [comment]. Ophthalmology. 2003, 110: 451-452. 10.1016/S0161-6420(02)01846-8. author reply 452
Meyer DR: Orbital radiation therapy for Graves' ophthalmopathy. [comment]. Ophthalmology. 2003, 110: 450-451. 10.1016/S0161-6420(02)01844-4. author reply 451
Ainbinder DJ, Halligan JB: Orbital radiation therapy for Graves' ophthalmopathy. [comment]. Ophthalmology. 2003, 110: 449-10.1016/S0161-6420(02)01840-7. author reply 449–450
Wiersinga WM: Perspective – Part III: retrobulbar irradiation in Graves orbitopathy: the Dutch experience. Ophthalmic Plastic & Reconstructive Surgery. 2002, 18 (3): 175-176. 10.1097/00002341-200205000-00004.
Kazim M: Perspective – Part II: radiotherapy for Graves Orbitopathy: the Columbia University experience. Ophthalmic Plastic & Reconstructive Surgery. 2002, 18 (3): 173-174. 10.1097/00002341-200205000-00003.
Bartley GB, Gorman CA: Perspective – Part I: the Mayo Orbital Radiotherapy for Graves Ophthalmopathy (ORGO) study: lessons learned. Ophthalmic Plastic & Reconstructive Surgery. 2002, 18 (3): 170-172. 10.1097/00002341-200205000-00002.
Feldon SE: Radiation therapy for Graves' ophthalmopathy: trick or treat? [comment]. Ophthalmology. 2001, 108: 1521-1522. 10.1016/S0161-6420(01)00729-1.
Marcocci C, Bartalena L, Bogazzi F, Bruno-Bossio G, Lepri A, Pinchera A: Orbital radiotherapy combined with high dose systemic glucocorticoids for Graves' ophthalmopathy is more effective than radiotherapy alone: results of a prospective randomized study. Journal of Endocrinological Investigation. 1991, 14: 853-860.
Bartalena L, Marcocci C, Chiovato L, Laddaga M, Lepri G, Andreani D, Cavallacci G, Baschieri L, Pinchera A: Orbital cobalt irradiation combined with systemic corticosteroids for Graves' ophthalmopathy: comparison with systemic corticosteroids alone. Journal of Clinical Endocrinology & Metabolism. 1983, 56 (6): 1139-1144.
Marcocci C, Bartalena L, Panicucci M, Marconcini C, Cartei F, Cavallacci G, Laddaga M, Campobasso G, Baschieri L, Pinchera A: Orbital cobalt irradiation combined with retrobulbar or systemic corticosteroids for Graves' ophthalmopathy: a comparative study. Clinical Endocrinology. 1987, 27: 33-42.
O'Dell JR: Therapeutic strategies for rheumatoid arthritis. New England Journal of Medicine. 2004, 350: 2591-2602. 10.1056/NEJMra040226.
Dunn JP: Review of immunosuppressive drug therapy in uveitis. Current Opinion in Ophthalmology. 2004, 15: 293-298. 10.1097/00055735-200408000-00003.
Ahern MJ, Harrison W, Hollingsworth P, Bradley J, Laing B, Bayliss C: A randomised double-blind trial of cyclosporin and azathioprine in refractory rheumatoid arthritis. Australian & New Zealand Journal of Medicine. 1991, 21 (6): 844-849.
MacPhee IA, Bradley JA, Briggs JD, Junor BJ, MacPherson SG, McMillan MA, Rodger RS, Watson MA: Long-term outcome of a prospective randomized trial of conversion from cyclosporine to azathioprine treatment one year after renal transplantation. Transplantation. 1998, 66: 1186-1192. 10.1097/00007890-199811150-00013.
Connell WR, Kamm MA, Ritchie JK, Lennard-Jones JE: Bone marrow toxicity caused by azathioprine in inflammatory bowel disease: 27 years of experience. Gut. 1993, 34: 1081-1085. 10.1136/gut.34.8.1081.
Seidman EG: Clinical use and practical application of TPMT enzyme and 6-mercaptopurine metabolite monitoring in IBD. Reviews in Gastroenterological Disorders. 2003, 3 (Suppl 1): S30-38.
Clunie GPR, Lennard L: Relevance of thiopurine methyltransferase status in rheumatology patients receiving azathioprine. Rheumatology. 2004, 43: 13-18. 10.1093/rheumatology/keg442.
Claridge KG, Ghabrial R, Davis G, Tomlinson M, Goodman S, Harrad RA, Potts MJ: Combined radiotherapy and medical immunosuppression in the management of thyroid eye disease. [see comment]. Eye. 1997, 11: 717-722.
Beigi B: The management of Thyroid Eye Disease. Focus. 2003, The Royal College of Ophthalmologists
Perros P, Weightman DR, Crombie AL, Kendall-Taylor P: Azathioprine in the treatment of thyroid-associated ophthalmopathy. Acta Endocrinologica. 1990, 122: 8-12.
Bahn RS, Gorman CA: Choice of therapy and criteria for assessing treatment outcome in thyroid-associated ophthalmopathy. Endocrinology & Metabolism Clinics of North America. 1987, 16 (2): 391-407.
Dickinson AJ, Perros P: Controversies in the clinical evaluation of active thyroid-associated orbitopathy: use of a detailed protocol with comparative photographs for objective assessment. Clinical Endocrinology. 2001, 55: 283-303. 10.1046/j.1365-2265.2001.01349.x.
Perros P, Crombie AL, Matthews JN, Kendall-Taylor P: Age and gender influence the severity of thyroid-associated ophthalmopathy: a study of 101 patients attending a combined thyroid-eye clinic. Clinical Endocrinology. 1993, 38: 367-372.
Mourits MP, Prummel MF, Wiersinga WM, Koornneef L: Clinical activity score as a guide in the management of patients with Graves' ophthalmopathy.[see comment][erratum appears in Clin Endocrinol (Oxf) 1997 Nov;47(5):632. Clinical Endocrinology. 1997, 47: 9-14. 10.1046/j.1365-2265.1997.2331047.x.
Haggerty H, Richardson S, Mitchell KW, Dickinson AJ: A modified method for measuring uniocular fields of fixation: reliability in healthy subjects and in patients with Graves orbitopathy. Archives of Ophthalmology. 2005, 123: 356-362. 10.1001/archopht.123.3.356.
Zigmond AS, Snaith RP: The hospital anxiety and depression scale. Acta Psychiatrica Scandinavica. 1983, 67: 361-370. 10.1111/j.1600-0447.1983.tb09716.x.
Carr TMT, Harris D: The DAS24: A short form of the Derriford Appearance Scale DAS59 to measure individual responses to living with problems of appearance. British Journal of Health Psychology. 2005, 10: 285-298. 10.1348/135910705X27613.
Moss T: Individual Variation in adjusting to visible differences. Visibly Different: Coping with Disfigurement. Edited by: Partridge J. 1997, London: Butterworth Heinemann, 122-130.
Terwee CB, Gerding MN, Dekker FW, Prummel MF, Wiersinga WM: Development of a disease specific quality of life questionnaire for patients with Graves' ophthalmopathy: the GO-QOL. British Journal of Ophthalmology. 1998, 82: 773-779.
Terwee CB, Dekker FW, Mourits MP, Gerding MN, Baldeschi L, Kalmann R, Prummel MF, Wiersinga WM: Interpretation and validity of changes in scores on the Graves' ophthalmopathy quality of life questionnaire (GO-QOL) after different treatments. Clinical Endocrinology. 2001, 54: 391-398. 10.1046/j.1365-2265.2001.01241.x.
WHOQOLGroup: Development of the World Health Organization WHOQOL-BREF quality of life assessment. Psychological Medicine. 1998, 28: 551-558. 10.1017/S0033291798006667.
Reuther LO, Vainer B, Sonne J, Larsen N-E: Thiopurine methyltransferase (TPMT) genotype distribution in azathioprine-tolerant and -intolerant patients with various disorders. The impact of TPMT genotyping in predicting toxicity. European Journal of Clinical Pharmacology. 2004, 59: 797-801. 10.1007/s00228-003-0698-8.
Pearson DC, May GR, Fick GH, Sutherland LR: Azathioprine and 6-mercaptopurine in Crohn disease. A meta-analysis. Annals of Internal Medicine. 1995, 123: 132-142.
McNab AA, Cockerham KP, Kennerdell JS, Mourits MP: Does radiotherapy have a role in the management of thyroid orbitopathy?. Br J Ophthalmol. 2002, 86: 102-107. 10.1136/bjo.86.1.102.
McAlister FA, Straus SE, Sackett DL, Altman DG: Analysis and reporting of factorial trials: a systematic review. Jama. 2003, 289: 2545-2553. 10.1001/jama.289.19.2545.
Montgomery AA, Peters TJ, Little P: Design, analysis and presentation of factorial randomised controlled trials. BMC Medical Research Methodology. 2003, 3: 24-10.1186/1471-2288-3-26.
Rose GE: Clinical activity score as a guide in the management of patients with Graves' orbitopathy. [comment]. Clinical Endocrinology. 1997, 47: 15-10.1046/j.1365-2265.1997.2070991.x.
Boboridis KG, Konstas AGP, Krassas GE, Georgiadis NS: Viral conjunctivitis interfering with the clinical activity score and management of thyroid ophthalmopathy. Orbit. 2006, 25: 27-29. 10.1080/01676830500505871.
Kahaly GJ, Rosler H-P, Pitz S, Hommel G: Low-Versus High-Dose Radiotherapy for Graves' Ophthalmopathy: A Randomized, Single Blind Trial. J Clin Endocrinol Metab. 2000, 85: 102-108. 10.1210/jc.85.1.102.
Dickinson AJ, Vaidya B, Miller M, Coulthard A, Perros P, Baister E, Andrews CD, Hesse L, Heverhagen JT, Heufelder AE, Kendall-Taylor P: Double-Blind, Placebo-Controlled Trial of Octreotide Long-Acting Repeatable (LAR) in Thyroid-Associated Ophthalmopathy. J Clin Endocrinol Metab. 2004, 89: 5910-5915. 10.1210/jc.2004-0697.
Kahaly GJPS, Hommel G, Dittmar M: Randomized, single blind trial of intravenous versus oral steroid monotherapy in Graves' orbitopathy. J Clin Endocrinol Metab. 2005, 90: 5234-5240. 10.1210/jc.2005-0148.
CIRTED website. http://www.cirted.org
Acknowledgements
We wish to thank:
▪ Dr Alan Montgomery (Department of Community Based Medicine, University of Bristol) and Rosemary Greenwood (Research and Development Support Unit, United Bristol Healthcare Trust) for their statistical support and advice on trial design.
▪ The clinical research teams at Bristol Eye Hospital (Annie Hinchcliffe and Cherry Daly), the Western Eye Hospital (Maryam Zamani and Avril Charnock), University College Hospital London (Helen Armstrong) and Manchester Royal Eye Hospital (Sophie Perry), for establishing and managing each trial centre.
▪ Nicola Harris (Moorfields Eye Hospital Research and Development department) for co-ordinating the trial's administration across study sites and Suzanne Cabral (Moorfields Eye Hospital Research and Development department) for facilitating the trial's multi-centre set up.
▪ Emma Dawson (Moorfields Eye Hospital) and Sarah Schofield (Bristol Eye Hospital) for their work on the development of the orthoptic assessment protocols.
▪ Dr Sally Goodman for her contribution to setting up the trial radiotherapy protocols at Bristol Oncology Centre
▪ The Radiotherapy teams in Bristol Oncology Centre (Sue Yarrow and Sue Cowley), St Bartholomew's Hospital, London (Christine Usher, Christine Bradley, Margaret Sharp) and the Christie Hospital, Manchester (Libby Mills)
This trial was funded by the following charitable bodies:
▪ The National Eye Research Centre
▪ Moorfields Eye Hospital Special Trustees
▪ Medical Research Committee of the Charitable Trusts for the United Bristol Hospitals
The following author receives funding related to the trial:
▪ RR receives a half-time salary from Moorfields Eye Hospital Special Trustees for her participation in the trial.
▪ RL receives funding from the National Eye Research Centre
The following authors receive funding unrelated to the trial:
▪ CD receives funding from- Diabetes Vaccine Development Centre, Jules Thorn Trust, Diabetes UK, Diabetes Research and Wellness Foundation.
▪ RL receives funding from the James Tudor Foundation and the Charles Hayward Foundation
The funding bodies did not have a role in study design or the collection of data. They also did not have a role in the writing of the manuscript or the decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
RR: Participated in development of the trial protocol. Coordinated the trial's set-up at Moorfields Eye Hospital and facilitated the set-up of the other study sites in London. Prepared the trial's standard operating procedures, study documentation and publicity. Drafted the manuscript.
RWJL: Conceived and designed the trial. Secured trial funding and coordinated its multi-centre management. Led the trial's set-up at Bristol Eye Hospital. Prepared the trial's standard operating procedures, study documentation and publicity. Drafted the manuscript.
MJP: Participated in development of the trial protocol.
GER: Contributed to trial design and participated in development of the trial protocol.
RJ and JMO: Led the trial's set-up and coordination at the Western Eye Hospital, London.
FB, SH: Led the trial's set-up and coordination at University College London Hospitals.
AC: Led the trial's set-up and coordination at Manchester Royal Eye Hospital.
RG: Led the set-up of trial radiotherapy at the Christie Hospital, Manchester.
MT: Responsible for trial radiotherapy at Bristol Oncology Centre.
NP: Led the trial's radiotherapy set-up at St Bartholomew's Hospital, London.
CB: Participated in development of the trial protocol. Set-up the trial's randomisation procedure. Coordinates statistical analyses.
SPH: Designed and coordinates the trial's health economic evaluation.
ADD: Helped to secure trial funding and facilitated its set-up at Bristol Eye Hospital.
NR, SJ and LK: Designed, implement and analyse the trial's psychosocial assessments.
OCM: Leads coordination at Moorfields Eye Hospital.
CMD: Conceived and designed the trial. Secured trial funding and coordinated its multi-centre management. Designed and coordinates the trial's endocrine management protocol. Drafted the manuscript.
JMU: Conceived and designed the trial. Secured trial funding and led its set-up at Moorfields Eye Hospital. Helped to draft the manuscript.
All authors read and approved the final manuscript.
Electronic supplementary material
13063_2007_219_MOESM1_ESM.pdf
Additional file 1: Patient Diary: Resource use, Personal Costs and Health Record. A diary for patients to keep their appointments in and to document travel and other related expenses. (PDF 521 KB)
13063_2007_219_MOESM2_ESM.pdf
Additional file 2: Quality of Life Questionnaires. Questionnaire to assess the patent's quality of life. This material uses the WHOQOL-UK and the assistance of the University of Bath and the World Health Organisation is acknowledged. (PDF 229 KB)
13063_2007_219_MOESM3_ESM.pdf
Additional file 3: Standard Operating Procedures. Standard operating procedures for the combined immunosuppression & radiotherapy in thyroid eye disease (CIRTED) trial. This material is a modification of the Standard Operating Procedures advocated by the European Group on Graves Ophthalmopathy (EUGOGO), and utilises Dickinson & Perros's reference atlas of colour plates [43]. (PDF 257 KB)
13063_2007_219_MOESM4_ESM.pdf
Additional file 4: Case Report Form. Case report form used in the combined immunosuppression & radiotherapy in thyroid eye disease (CIRTED) trial. (PDF 177 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rajendram, R., Lee, R.W., Potts, M.J. et al. Protocol for the combined immunosuppression & radiotherapy in thyroid eye disease (CIRTED) trial: A multi-centre, double-masked, factorial randomised controlled trial. Trials 9, 6 (2008). https://doi.org/10.1186/1745-6215-9-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1745-6215-9-6