
Abstract
Macrophages play a crucial role in innate and adaptative immunity in response to microorganisms and are an important cellular target during HIV-1 infection. Recently, the heterogeneity of the macrophage population has been highlighted. Classically activated or type 1 macrophages (M1) induced in particular by IFN-γ display a pro-inflammatory profile. The alternatively activated or type 2 macrophages (M2) induced by Th-2 cytokines, such as IL-4 and IL-13 express anti-inflammatory and tissue repair properties. Finally IL-10 has been described as the prototypic cytokine involved in the deactivation of macrophages (dM). Since the capacity of macrophages to support productive HIV-1 infection is known to be modulated by cytokines, this review shows how modulation of macrophage activation by cytokines impacts the capacity to support productive HIV-1 infection. Based on the activation status of macrophages we propose a model starting with M1 classically activated macrophages with accelerated formation of viral reservoirs in a context of Th1 and proinflammatory cytokines. Then IL-4/IL-13 alternatively activated M2 macrophages will enter into the game that will stop the expansion of the HIV-1 reservoir. Finally IL-10 deactivation of macrophages will lead to immune failure observed at the very late stages of the HIV-1 disease.
Similar content being viewed by others


Introduction
Macrophages (Ms) are the first line of defence of the organism against pathogens and, in response to the microenvironment, become differentially activated. The classical pathway of interferon-γ-dependent activation of macrophages (M1) by T helper 1 (Th1)-type responses is a well-established feature of cellular immunity to infection with HIV-1. In the presence of cytokines that are produced in a Th-2 type response, such as IL-4 and IL-13, macrophages become differentially activated (M2) and play an important role in HIV-1 pathogenesis. Although it is superficially similar to a Th2-type cytokine and is often co-induced with Th2 cytokines in the course of an immune response, it is not appropriate to classify IL-10 together with IL-4 and IL-13 as an alternative activator of macrophages. IL-10 acts on a distinct plasma membrane receptor to those for IL-4 and IL-13 [1], and its effects on macrophage gene expression are different, involving a more profound inhibition of a range of antigen-presenting and effector functions, leading to a deactivation stage of macrophages [2]. Following this line of reasoning, it seems appropriate to classify macrophages in IFN-γ classically activated macrophages (M1), IL-4/IL-13 alternatively activated macrophages (M2), and IL-10 deactivated macrophages (dM). In addition, T cells themselves are more heterogeneous than was thought originally [3, 4], including not only Th0, Th1 and Th2 type cells, but also among other regulatory (Treg) and Th17 cells [5]. In addition, a wide variety of stimuli, both endogenous and exogenous, influence the susceptibility of macrophages to infection by HIV-1. The differentiation stage of monocytes/macrophages also modulates permissiveness to HIV-1: primary monocytes are less susceptible to the virus than differentiated macrophages [6–9]. The localization of macrophages in different tissues results in cells with distinct activation status and susceptibility to HIV-1 infection. Addressing the effects of macrophage differentiation and/or activation on HIV-1 replication provides some insight into the impact of specific microenvironments on macrophage infection in vivo. Modulation of HIV-1 replication induced by diverse stimuli have however been addressed using monocytic cell lines, primary monocytes or macrophages differentiated in vitro from primary monocytes. Keeping these data in mind, the present review will focus on the distinctive patterns of macrophage activation (classically activated M1, alternatively activated M2, and deactivated dM) in HIV-1 pathogenesis.
Classical Activation of Macrophages and HIV-1 Infection
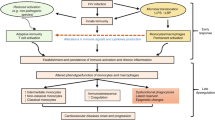
Classically activated or type 1 macrophages induced in particular by IFN-γ [10], display a pro-inflammatory profile (Figure 1). In addition pro-inflammatory cytokines modulate HIV-1 replication in macrophages and could depend on the maturation and/or activation stages of monocytes/macrophages [7, 8]. High levels of proinflammatory cytokines, such as tumor necrosis factor α (TNFα), interleukin (IL)-1β and IL-6 in both plasma and lymph nodes are observed from the early stages of HIV-1 infection [11–15]. The secretion of chemokines such as macrophage inflammatory protein (MIP)-1α, MIP-1β and RANTES (CCL3, CCL4 and CCL5 respectively) is increased in these patients [16, 17]. Immune activation also reflects the mounting of antiviral immunity with enhanced Th1 activity and increased levels of IFNγ, IL-12, IL-2 and IL-18, especially in lymph nodes of HIV-infected subjects [18]. In addition these cytokines and their receptors have validated the importance of this pathway in cellular immunity, immunodeficiency syndromes, delayed hypersensitivity responses and tissue damage [2]. In classically activated macrophages, the following steps of the HIV-1 life cycle are modulated (Table 1).

Classical activation (M1), alternative activation (M2) and deactivation of macrophages. Classical activation is mediated by the priming stimulus IFN-γ, followed by a microbial trigger (lipopolysaccharide, LPS). Alternative activation is mediated by IL-4 and IL-13, acting through a common receptor chain (IL-4Rα). Deactivation can be innate or acquired in origin. The uptake of apoptotic cells or lysosomal storage of host molecules generates anti-inflammatory responses. Cytokines (IL-10, TGF-β, M-CSF, IFNα/β) and glucocorticoids are potent modulators of activation. Pathogens can deactivate macrophages by various mechanisms.
Entry
HIV-1 infects monocytes/macrophages via interaction of gp120 with CD4 and either coreceptor CXCR4 or CCR5 which determines the cellular tropism [19–31]. HIV-1 envelope glycoprotein gp120 down-regulates CD4 expression in primary human macrophages through induction of endogenous TNFα [32–37]. TNFα, IL-1β and IFN-γ down-regulate both surface and total CD4 expression in primary human macrophages at the level of transcription [36, 38–41]. TNFα, IFN-β, and IFN-γ inhibit R5 and R5/X4 HIV-1 entry into primary macrophages via down-regulation of both cell surface CD4 and CCR5 and via enhanced secretion of C-C chemokines, MIP-1α, MIP-1β, and RANTES [37, 38, 40, 42–46]. An iterative pre-treatment of primary macrophages with TNFα prior to HIV infection inhibits HIV-1 replication [43]. The inhibition of HIV-1 entry into primary macrophages by TNFα involves the 75-kDa TNFR2 [43]. Another explain could be that TNFα triggers the release of granulocyte-macrophage colony-stimulating factor (GM-CSF) that has been reported to down-regulate CCR5 and subsequently block entry of R5 HIV into macrophages [47]. Interestingly, TNFR2 stimulation triggers GM-CSF secretion that has been shown to block R5 HIV-1 entry via CCR5 downregulation [47]. The inhibition of HIV-1 entry into macrophages observed following TNFα pre-treatment could be mediated via the secretion of C-C chemokines, such as RANTES, MIP-1α and MIP-1β. TNFα induces the production of RANTES, MIP-1α, and MIP-1β which in turn down-regulate cell surface CCR5 expression on primary macrophages resulting in inhibition of R5 HIV-1 entry [48–53]. In agreement with this observation, RANTES inhibits HIV-1 envelope-mediated membrane fusion in primary macrophages [54] and the activity of RANTES promoter that contains four NF-kB binding sites is up-regulated by TNFα [55]. Nevertheless, some authors report an enhancement of HIV-1 replication by RANTES in primary macrophages [27, 56]. The enhancing effect of RANTES on HIV-1 infectivity may be independent of the route of virus-cell fusion and could involve two different mechanisms: one mediated via cellular activation, and the other mediated via increased virion attachment to target cells [56]. Another explanation for this discrepancy is the activation and/or differentiation status of macrophages with a more potent inhibitory effect of RANTES on monocyte-derived macrophages cultivated in vitro in absence of additional cytokines such as M-CSF [57].
The monocyte chemotactic protein-2 (MCP-2), but not MCP-1, has been shown to bind to CCR1, CCR2b, and CCR5 and to inhibit CD4/CCR5-mediated HIV-1 entry/replication [58]. Pretreatment of macrophages with IL-16 also inhibits R5 and R5/X4 HIV-1 replication in primary macrophages at the level of entry, although the secretion of CC-chemokines does not seem to be involved in this phenomenon [59].
IL-2 has been reported to inhibit HIV-1 replication in macrophages by down-regulating CD4 and CCR5 expression [60]. IL-15 is a Th1 cytokine produced by mononuclear phagocytes and shares many activities with IL-2, such as T-cell proliferation and activation. In addition IL-15 is more potent than IL-2 in stimulating NK cell function, including secretion of IFN-γ and of CCR5-binding chemokines [61]. Ex vivo, increased levels of IL-15 were detected in histocultures established from lymph nodes of individuals who were HIV positive in comparison to their uninfected counterparts [62]. Supernatants of NK cells stimulated with IL-12 and IL-15 inhibited both macrophage-tropic HIV-1NFN-SX and T cell-tropic HIV-1NL4-3 replication in vitro, but not dual-tropic HIV-189.6 due to the use of multiple coreceptors for entry by this latter, including CXCR4, CCR5, but also CCR3 and CCR2b [24, 63]. Importantly, the C-C chemokines MIP-1α, MIP-1β, and RANTES were responsible only for a fraction of the HIV-1-suppressive activity exhibited by NK cell supernatants against macrophage-tropic HIV-1. Collectively these data indicate that NK cells from normal and HIV-1+ donors produce C-C chemokines and other unidentified factors that can inhibit both macrophage- and T cell-tropic HIV-1 replication in vitro [63].
IL-18 is a pro-inflammatory cytokine related to the IL-1 family of cytokines that plays an important role in both innate and adaptative immune responses against viruses [64, 65]. Increased levels of circulating IL-18 from HIV-1 infected patients have been reported especially in the advanced and late stages of the disease [65]. IL-18 reduces cell surface expression of the HIV-1 receptor CD4 [66]. In the advanced stages of the disease, strong activation of IL-18 production along with persistent decreased production of IFN-γ, IL-12 and IL-2 may promote a Th2 immune response, which leads to persistent viral replication [65].
CD40 ligand (CD40L) is a cell surface molecule of CD4+ T cells that interacts with its receptor CD40 on antigen-presenting cells (APC) to mediate thymus-dependent humoral immunity and inflammatory reactions. The stimulation of macrophages by CD40L has been shown to trigger the release of TNFα and CC-chemokines which results in down-regulation of cell surface CD4 and CCR5 and subsequent inhibition of HIV-1 entry into macrophages [17, 67–69]. An in situ hybridization study showed that macrophages in lymph nodes of HIV-1 infected individuals produce MIP-1α and MIP-1β, and to a lesser extent RANTES, suggesting that HIV-1 infection might be modulated in vivo by activated macrophages [70]. It is interesting to note that the CD40/CD40L interaction triggers signalling through TNF receptor-associated factor 6 (TRAF6) in antigen presenting cells. TRAF6 has also been involved in innate immune responses mediated by TLR-4, such as the response to lipopolysaccharide (LPS) [68]. Like CD40L activation, LPS stimulation also induces high secretion of C-C chemokines and TNFα and inhibits infection of macrophages and CD4+ T cells with R5 HIV-1 strains. Thus, during opportunistic infections, LPS might also be produced that, either directly or indirectly via TNFα production, might block HIV-1 entry into macrophages [71, 72]. In human blood monocyte tissue culture-derived macrophages (TCDM), endogenous TNFα and IL-1β induced by LPS, down-regulate surface and total CD4 expression in primary macrophages [41]. Conversely, neither LPS nor TNFα/IL-1β were able to modulate surface CD4 expression on quiescent or PHA-activated lymphocytes [41]. Thus, opportunistic infections during HIV disease can result in a sustained but controlled viral production within infected macrophages.
Transcription
TNFα has been reported to stimulate HIV-1 replication in chronically infected promonocytic U1 cell line through NF-kB activation and subsequent transactivation of the proviral LTR [73–76]. The stimulation of HIV-1 replication in U1 cell line with TNFα is mediated through the TNFR1, and not via TNFR2 [77]. Similarly, IL-1β binding to the IL-1 receptor 1, but not to the IL-1 receptor 2, stimulates HIV-1 transcription through activation of NF-kB or by an independent mechanism [75, 78]. IL-1 can act alone or in synergy with IL-6 to stimulate viral replication in chronically infected promonocytic U1 cell line [78]. In addition IL-6 alone stimulates HIV-1 replication in U1 cells and primary macrophages infected with R5 AD-87 strain, but not in T cell lines [76]. Nuclear factor IL-6 (NF-IL6) is a nuclear factor that activates gene expression in response to IL-6. A consensus binding site for NF-IL6 is present in the LTR of many HIV-1 variants and the regulation of HIV-1 LTR by NF-IL6 and NF-kB/Rel transcription factors has been reported [79–81]. IL-6 stimulates HIV replication by activating viral transcription in synergy with TNFα and also by targeting a post-transcriptional step [76]. In addition, endothelial cells enhance C/EBPbeta binding activity and HIV-1 replication in macrophages. This increase in HIV-1 transcription is due in part to the production of soluble factors, such as IL-6 and also is mediated by ICAM-1 activation [82], indicating that endothelial cells, through the activation of C/EBPβ, provide a microenvironment that supports HIV-1 replication in monocytes/macrophages. The stimulation of HIV-1 replication in primary macrophages by GM-CSF is primarily due to enhanced viral transcription rather than increased viral entry [76]. GM-CSF stimulates HIV-1 replication in promonocytic U1 cells [83] and in primary human macrophages infected with the R5 HIV-1 JR-FL strain [84] by targeting HIV LTR at a site different from NF-κB [76].
In vitro, both acute HIV infection and incubation of the THP-1 monocytoid cell line with the accessory viral protein Nef induced expression of IL-18 [85]. Like most proinflammatory cytokines, IL-18 induces HIV expression in chronically infected monocytic cell lines via induction of the release of endogenous TNFα and IL-6 [86]. IL-18 stimulates HIV-1 replication in the chronically infected U1 monocytic cells, mediated in part via TNFα and IL-6 since the addition of anti-TNFα and anti-IL-6 antibodies reduced IL-18 increased HIV-1 production by 48% and 63%, respectively [86]. IL-18 stimulation of HIV-1 replication in U1 cells involves NF-kB and p38 MAPK activation [86].
Posttranscription
The effect of IFN-γ on HIV-1 replication might be more complex. Pretreatment of human primary macrophages with IFN-γ before viral input has been reported either to stimulate or to inhibit HIV-1 replication [45, 46, 84]. In addition, IL-18 has been reported as an IFN-γ-inducing factor which inhibits HIV-1 production in PBMC through IFN-γ [66].
Altogether classically activated macrophages M1 are in contact with Th1 cytokines (IFN-γ, IL-2, IL-12), proinflammatory cytokines (TNFα, IL-1β, IL-6, IL-18) and chemokines (MIP-1α, MIP-1β, RANTES) that favor the formation of viral reservoirs with inhibition of HIV-1 entry, assembling and budding parallel to increased viral transcription within the infected macrophages (Figure 2).

A model of HIV-1 pathogenesis based on the activation status of macrophages.
Alternative Activation of Macrophages and HIV-1 Infection
The alternatively activated or type 2 macrophages (M2) induced by Th-2 cytokines, express anti-inflammatory and tissue repair properties [2] (Figure 1). Alternative activation of macrophages is induced by IL-4 and IL-13, cytokines that are produced in a Th-2 type response, particularly during allergic, cellular and humoral responses to parasitic and selected pathogen infections. The alternative activation of macrophages is mediated by IL-4 and IL-13, acting through a common receptor chain (IL-4Rα) [87]. IL-4 is a pleiotropic cytokine produced by a subpopulation of CD4+ T cells, designated Th-2 cells, and by basophiles and mast cells. IL-4 modulates other lymphoid cell activities such as regulation of the differentiation of antigen-stimulated T lymphocytes [88, 89] and control of immunoglobulin class switching in B lymphocytes [90–93]. IL-13 is a cytokine secreted by activated T cells which has been shown to be a potent in vitro modulator of human monocytes and B cell functions [94–96]. Among its pleiotropic activities, IL-13 induces significant changes in the phenotype of human monocytes, up-regulating their expression of multiple cell surface molecules and increasing their antigen presenting capabilities. IL-4 and IL-13 upregulate expression of the mannose receptor and MHC class II molecules by macrophages which stimulate endocytosis and antigen presentation, and they induce the expression of macrophage-derived chemokine (MDC, also known as CCL22). IL-4 and IL-13 augment expression of IL-1 decoy receptor and the IL-1 receptor α-chain in vitro and in vivo, thereby counteracting the proinflammatory actions of IL-1 [97, 98]. In alternatively activated macrophages, the following steps of the HIV-1 life cycle are modulated (Table 1).
Entry
Infection of macrophages by primary R5X4 and X4 isolates of HIV-1 is inhibited by IL-4 and IL-13, an effect that is associated with down-regulation of surface CXCR4, CCR5 and CD4 expression [38, 99].
Reverse transcription
Upon cell infection by HIV-1, the reverse transcriptase copies the genomic RNA to generate the proviral DNA flanked by two LTRs [100]. IL-13 has been shown to inhibit HIV-1 replication in blood-derived monocytes and mature lung macrophages, but not in T cells [95, 101]. The mechanism by which IL-13 inhibits HIV-1 is not yet clear. IL-13 has been reported either not to modulate reverse transcription [102] or to block the completion of reverse transcription in macrophages [103].
Transcription
IL-13 has been reported to block HIV-1 replication at the level of transcription in human alveolar macrophages [102]. In fact, the state of maturation of monocytes into macrophages determines the effects of IL-4 and IL-13 on HIV-1 replication. In freshly isolated monocytes, IL-4 up-regulates the expression of both genomic and spliced HIV mRNA [104, 105]. IL-4 stimulates NF-κB translocation and binding resulting in enhanced HIV RNA expression [105]. IL-4 up-regulates the expression of HIV mRNA within the first two days after infection of promonocytic U937 cells and 3 to 4 days after infection of plastic-adherent blood-derived macrophages with HIV-1 [104, 106]. Conversely, IL-13 and IL-4 inhibit HIV-1 replication at the transcriptional level in differentiated macrophages, but not in peripheral blood lymphocytes [95, 104, 105]. In addition, exposure to IL-13 inhibits the transcription of many other cytokines in monocytes, including IL-1α, IL-1β, IL-6, TNF, and GM-CSF [96], all of which have been implicated in enhancing HIV-1 replication in vitro [107–110].
Altogether alternatively activated macrophages are in contact with IL-4/IL-13 producing Th2 cells that will curtail the formation of HIV-1 reservoirs in the macrophages (Figure 2).
Deactivation of Macrophage and HIV-1 Infection
The prototypic cytokine involved in the deactivation of macrophages is IL-10. Although it is superficially similar to a Th2-type cytokine and is often co-induced with Th2 cytokines in the course of an immune response, it is not appropriate to classify IL-10 together with IL-4 and IL-13 as an alternative activator of macrophages [2]. IL-10 acts on a distinct plasma membrane receptor to those for IL-4 and IL-13 [1]. Similar to IL-10, other cytokines such as TGF-β, M-CSF and IFNα/β result in macrophage deactivation [2] with strong anti-inflammatory properties, down-regulation of MHC class II molecules on the plasma membrane (Figure 1). Deactivation of macrophages leads to immune suppression through at least two independent mechanisms: diminished MHC class II expression and increased uptake of apoptotic cells generating an anti-inflammatory response [111–115]. In deactivated macrophages, the following steps of the HIV-1 life cycle are modulated (Table 1).
Entry
IL-10 up-regulates cell surface CCR5 expression on monocytes and thereby enhances viral entry [116]. M-CSF has been shown to favor HIV-1 replication in human macrophages, probably via an increased maturation stage and increased CCR5 expression, also resulting in enhanced viral entry [29, 117]. By contrast, IFN-β inhibit R5 HIV-1 entry into primary macrophages via down-regulation of both cell surface CD4 and CCR5 and via enhanced secretion of C-C chemokines, MIP-1α, MIP-1β, and RANTES [37, 40, 42–46].
Reverse transcription
IL-10 suppresses HIV-1 replication in primary human macrophages by inhibiting the initiation of reverse transcription; therefore, IL-10 mediates a virostatic latent stage in cells of the monocyte/macrophage lineage [118–120]. TGF-β inhibits the synthesis of different viral proteins especially reverse transcriptase in U1 promonocytic cells activated by phorbol ester or IL-6 [121]. Members of the APOBEC (acronym for apolipoprotein B editing catalytic polypeptide) family of cellular cytidine deaminases represent a recently identified group of proteins that provide immunity to infection by retroviruses [122–125]. The cytidine deaminases APOBEC exert anti-HIV-1 activity that is countered by the HIV-1 vif protein [122]. Tripartite motif (TRIM) proteins constitute a family of proteins that share a conserved tripartite architecture [126–128]. Interferons, especially type I IFNα/β bolster innate defence against HIV-1 via the up-regulation of APOBEC/TRIM proteins which blocks retroviral replication, especially reverse transcription [129–131].
Transcription
High concentrations of IL-10 inhibit the production of proinflammatory cytokines such as TNFα, IL-1β, IL-6, and thereby IL-10 inhibits HIV-1 transcription [132]. By contrast, low concentrations of IL-10 have been reported to enhance HIV replication in macrophages induced by TNF-α and IL-6 via an increase in HIV mRNA accumulation and stimulation of phorbol ester-induced LTR-driven transcription that is independent of the NF-κB and Sp1 transcription factors [133].
Posttranscription
Primary macrophages treated with IL-10 after HIV-1 inoculation show an accumulation of Gag protein suggestive of an inhibitory effect at the level of virus assembly [134]. IFNα and IFNβ reduce HIV-1 replication in primary macrophages although inhibition by IFNα has been reported to be more efficient [45, 135]. Anti-HIV effects of IFNα/β are mediated by both inhibition of viral assembly and budding [136, 137]. IL-27 inhibits HIV replication in monocyte-derived macrophages like IFN-α and IFN-β[138]. IL-27 suppresses the transcription of HIV-1 and preferentially inhibits HIV-1 replication in macrophages compared with CD4+ T cells and activates multiple IFN-inducible genes (ISG) in macrophages like IFN-α, suggesting that IL-27 inhibits HIV-1 replication in macrophages via a mechanism similar to that of IFN-α [138–140]. Recently, of the hundred of IFN-inducible genes discovered to date, ISG15 and ISG20 have been reported to inhibit assembly and release of HIV-1 virions [141–144]. In addition the IFN-inducible tripartite motif protein TRIM22 inhibits the budding of HIV-1 with diffuse cytoplasmic distribution of Gag rather than accumulation at the plasma membrane [145]. The effects of TGF-β on the post-transcriptional steps of HIV-1 replication are more complex. In primary human macrophages, both inhibition and stimulation of HIV-1 replication have been reported following a posttreatment with TGF-β[121, 146].
Altogether in deactivated macrophages, HIV-1 replication is strongly blocked at several steps of the viral life cycle especially reverse transcription, transcription and viral budding and assembly (Figure 2).
Activation Status of Macrophages and HIV-1 Pathogenesis
Because of the various behaviours of macrophages reported (classically activated M1, alternatively activated M2, deactivated dM), we would like to present a new model that highlights the role of macrophage activation status in the modulation of viral persistence and T-cell apoptosis and could thereby further enhance our understanding of pathogenesis of HIV-mediated disease (Figure 2). We will first propose a model that applies to the monocytes/macrophages present in the blood and in the lymph nodes of HIV-1-infected patients. We will then discuss this HIV model in light of the different populations of macrophages present in distinct tissues and highlight the critical role of the microenvironment in tissues such as mucosal tissue and the central nervous system (CNS).
Activation status of monocytes/macrophages in peripheral blood and in lymph nodes of HIV-1-infected subjects
Early in the disease, when the levels of proinflammatory cytokines, C-C chemokines and type I IFN are low and chronic immune activation is not yet predominant viral proteins are crucial for establishing a productive infection and for the activation of macrophages [147–149]. Viral proteins expressed early in the viral cycle, such as Nef, Tat, and virion-associated Vpr, activate the TNFR pathway to partially mimic TNFα biological effects, suggesting that these viral proteins can fuel the progression of the disease even in the absence of proinflammatory cytokines, especially in macrophages [9, 148, 150]. These viral proteins play a role in the formation of viral reservoirs in macrophages by activating transcription from the LTR and interfering with apoptotic machinery [6, 151]. The classically activated macrophages M1 are in contact with high levels of Th1 cytokines (IFN-γ, IL-2, IL-12), proinflammatory cytokines (TNFα, IL-1β, IL-6, IL-18) and chemokines (MIP-1α, MIP-1β, RANTES) that favor the formation of viral reservoirs with strongly increased viral transcription and inhibition of HIV-1 entry to block superinfection within infected macrophages. In addition type I interferon production is impaired in primary HIV-1 infection with only limited inhibition of viral assembling and budding [147, 152, 153]. During this stage of the disease M1 macrophages are predominant, tissue injury especially in lymph nodes is observed and the rate of T-cell apoptosis is increasing [148].
At a later stage of the disease, a M1 toward M2 shift is observed with IL-4/IL-13 as pleiotropic modulators of macrophage activation that induce distinctive programmes of altered macrophage gene expression after the engagement of their specific cytokine receptors [154]. At this intermediate stage M2 macrophages appear and will favor tissue repair, the MHC class II-mediated antigen presentation and T-cell activation, the stimulation of bacterial endocytosis via the up-regulation of the mannose receptor on the cell surface [2, 155]. Alternative activation of macrophages might help to favor the clearance of opportunistic infections during HIV-1 disease [156, 157]. Intermediate levels of T-cell apoptosis are observed that does not totally block the production of proinflammatory cytokines [111, 158]. The combination of IL-4/IL-13 cytokines and proinflammatory cytokines in the microenvironment present in the vicinity of infected macrophages will curtail the expansion of macrophage HIV-1 reservoirs [38, 159].
At the onset of AIDS, T-cell apoptosis is dramatically increased and opportunistic infections are very frequent [148, 158, 160], resulting in an enhanced apoptotic cell clearance by IL-10-deactivated macrophages [161, 162]. An imbalance in the TH1-type and TH2-type responses has been proposed to contribute to the immune dysregulation associated with HIV infection, and that progression to AIDS is dependent on a TH1/TH2 shift [163]. This hypothesis was based on the following facts: (1) progression to AIDS is characterized by loss of IL-2- and IFN-gamma production concomitant with increases in IL-10; and (2) many seronegative, HIV-exposed individuals generate strong TH1-type responses to HIV antigens. Recently, haplotypes of the IL-4 and IL-10 genes associated with AIDS progression have been reported [164, 165]. In HIV-infected patients, the amount of IL-10, but not IL-4, increases significantly in patients with AIDS [166]. Opportunistic infections, especially present at the late stages of the disease, trigger IL-10 production [167] and IL-10 production from patients with AIDS has been reported to decrease in vitro HIV-1 replication and TNFα production [168]. In addition, IL-10 has been reported to suppress antiviral T-cell activity during persistent viral infection [169] and Tat-induced IL-10 mediates immune suppression during HIV-1 infection [170]. In addition, the IL-10 deactivated macrophages inhibit the production of proinflammatory cytokines such as TNFα and C-C chemokines that were produced abundantly due to chronic immune stimulation during the previous stages of the disease [171, 172]. IL-10 inhibits HIV-1 LTR-driven gene expression in human macrophages through the induction of cyclin T1 proteolysis [173]. At the late stages of the disease the decreased levels of proinflammatory cytokines result in a strong reduction of viral transcription. In addition high expression of IFNα/β inducible proteins such as APOPEC and TRIM proteins inhibit strongly the HIV-1 reverse transcription and assembly/budding (Table 1). The deactivation of macrophages also results in a profound immune suppression resulting from the decreased expression of MHC class II expression on the plasma membrane of macrophages with diminished Ag-mediated T cell response and the depletion of both CD4+ and CD8+ T cell by accelerated apoptosis. Thus, IL-10 and type I IFN restrict strongly HIV-1 replication in macrophages parallel to the immune failure observed at the very late stages of the HIV-1 disease.
Activation status of macrophages in mucosal tissues and in the CNS
The localization of macrophages in distinct tissues has been reported to modulate their susceptibility to HIV-1 infection. In human and macaque gastrointestinal mucosa, most attention has been focused on the small intestine, where lamina propria CD4+ T cells are prominent HIV-1 and SIV target cells and undergo profound depletion shortly after infection [174–182]. In contrast, macrophages in the gastrointestinal mucosa, unlike monocyte-derived macrophages, are rather resistant to infection with HIV-1 [183–185]. In contrast to monocytes and monocyte-macrophages, intestinal macrophages do not express many innate response receptors [186, 187], are downregulated for triggering receptor expressed on monocytes (i.e., TREM-1) [188, 189] and costimulatory molecules [187, 190], and display markedly reduced CD4 and CCR5 cell surface protein and mRNA [191]. Thus, the striking and well-defined phenotypic and functional differences between blood monocytes and mucosal macrophages, in particular macrophages in the gastrointestinal mucosa [186, 187, 192], preclude the simple extrapolation from findings in HIV-1-infected monocytes to HIV-1 infection of mucosal macrophages. Human vaginal macrophages have been reported recently to support R5 virus entry in explanted vaginal mucosa, and purified vaginal macrophages support substantial levels of R5 HIV-1 replication [193]. Vaginal macrophages display the innate response receptors CD14, CD89, CD16, CD32 and CD64, and the CD4 receptor and CCR5 and CXCR4 coreceptors [193]. The difference in phenotype and HIV-1 permissiveness between vaginal and intestinal macrophages may reflect differences in the local microenvironment, since mucosa-derived cytokines, including TGF-β, regulate the phenotype and function of blood monocytes after their recruitment to the mucosa, at least in the intestinal mucosa [187]. In agreement with this hypothesis, intestinal macrophages are threefold less frequently CD4+ CCR5+ than vaginal macrophages, and yet virus is detected in intestinal macrophages, indicating low-level receptor mediated entry, but intestinal macrophages do not support viral replication suggesting a post-entry block such as described for TGF-β [193].
Macrophages of the central nervous system (CNS) are permissive to HIV-1 infection. Two models have been proposed: the Trojan horse model and the late invasive model [194]. In the Trojan horse model, the virus enters the CNS early, and replicates at low levels as a reservoir separated from the periphery. A viral phenotype that is more virulent in the context of the CNS emerges, leading to the development of disease. In the late invasion model, uncontrolled virus replication and resulting immune deficiency lead to alterations in the myeloid differentiation pathway, promoting the expansion of an activated monocyte subset that is capable of tissue invasion. The hallmark of the brain histopathology is productive infection in macrophages (perivascular macrophages and microglia) [195]. HIV encephalitis (HIVE) is characterized by monocyte/macrophage infiltration into the brain, multinucleated giant cell formation (fusion of several macrophages), and presence of microglial nodules [196]. There is little evidence for infection in neurons, endothelial cells, or macroglia (astrocytes and oligodendrocytes) [197–199]. In the Trojan horse model, it has been hypothesized that the virus enters the CNS mainly through infected monocytes and macrophages destined to become brain-resident macrophages or perivascular macrophages [200]. It is assumed that HIV-1 enters early after primary infection (at a peak of primary viremia), and HIV-1 infection persists at low levels due to the immune-privileged status of the CNS. In addition there is an uniqueness of the brain microenvironment with several anatomic/structural, physiological, and immunoregulatory mechanisms that ensure the immune priviledge of the brain, preventing recognition of foreign antigens, to minimize/deviate and block inflammatory responses [201]. Soluble anti-inflammatory molecules have been shown to play a role in immune privilege in the CNS. TGF-β has the ability to inhibit activation of macrophages, T lymphocytes, and NK cells [202], and TGF-β has been shown to possess neuroprotective capabilities [203]. Upregulation of TGF-β is observed during HIV-1 infection and is correlated with the magnitude of inflammatory responses during HIV-1 brain infection [204]. High concentrations of gangliosides downregulate expression of MHC class II on astrocytes [205] and could contribute to generally low levels of MHC class II on microglia. In contrast, a significant increase in MHC class II has been reported in the context of HIVE on activated microglia [206, 207] and it is considered the best neuropathologic correlate of cognitive impairment [208]. TGF-β, IL-10, and TRAIL have been reported to contribute significantly to the CNS-DC-mediated inhibition of allo-T-cell proliferation [209] and to participate in the control of viral CNS infections [210]. In agreement with this observation, only few DC-like cells were found in perivascular spaces in SIV-infected macaques [211]. Although invasion of the CNS by HIV-1 occurs at the time of primary infection and induces a transitory inflammatory process with increased number of microglial cells, upregulation of MHC class II antigens, and local production of cytokines [212], viral replication remains very low during the asymptomatic stage of HIV-1 infection. Specific immune responses including Th2 cytokines and CTLs continuously inhibit viral replication at this stage of infection [213–216]. While HIV-1 enters the brain early following viral infection [200], detectable productive viral replication and brain macrophage infiltration occur years later and only in some infected patients [217]. The replication of HIV-1 in microglia depends on the microenvironment in the CNS. Recently, it has been reported astrocyte-mediated regulation of microglial function and its influence on the onset and the progression of neuroAIDS [218]. HIV-1, recombinant gp120, and viral transactivator Tat activate astrocytes to secrete pro-inflammatory cytokines TNFα, IL-6, and IL-1β and the pro-inflammatory chemokines MCP-1 and IP-10 [195, 219–224], all of which could contribute to the overall inflammatory environment in the brain. To further contribute to the inflammatory environment in the CNS, microglia and macrophages release proinflammatory cytokines such as IL-1β and TNFα which play a role in CNS injury [225, 226]. In agreement with these data, in vivo expression of proinflammatory cytokines in HIV-1 encephalitis has been reported and the macrophage/microglia lineage is the main cell type reported to release cytokines in HIVE [227]. Altogether, after an early and transitory stage of macrophage/microglia activation at the time of primary infection, a stage of deactivation of macrophage/microglia is observed parallel to the presence of "deactivating" cytokines such as TGF-β and IL-10 in the CNS microenvironment. In some patients, detectable productive viral infection and brain macrophage infiltration occur years later parallel to increased levels of pro-inflammatory cytokines in the context of HIVE.
A M1/M2/Md macrophage polarization model and vice versa
Altogether, in the lymph nodes of HIV-1-infected patients a shift from activated to deactivated macrophages throughout the disease is observed parallel to a Th1 pro-inflammatory/Th2 anti-inflammatory switch. In some tissue such as the intestinal mucosal tissue, the macrophages are mostly in a deactivated stage with a local microenvironment curtailing the viral replication through the release of anti-inflammatory cytokines such as TGF-β. In contrast to the intestinal mucosa, macrophages from the vaginal mucosa are more permissive to HIV-1 replication and are activated by proinflammatory cytokines. In the CNS of HIV-infected patients, the macrophage/microglia are mostly deactivated under the control of cytokines such as TGF-β, although in some cases HIVE occurs parallel to the production of proinflammatory cytokines and high viral production at advanced stage of the disease. Thus the shift of macrophage/microglia from activation to deactivation and vice-versa depends on the tissue infected by HIV-1 and on the local microenvironment. In agreement with this hypothesis, the reversion of M2/Md macrophages to M1 polarization has been recently reported in vitro, and was associated with a renewed capacity to support HIV-1 replication [228]. M1/M2/Md macrophage polarization may represent a mechanism that allows macrophages to cycle between productive and latent HIV-1 infection and vice-versa, parallel to the critical role of the tissue microenvironment which can drive the macrophage polarization either way and thereby can modulate HIV-1 replication specifically in distinct tissues at different stages of the disease.
Conclusion
The concept of macrophage heterogeneity and differentiation has been recently highlighted by the description of at least three types of macrophage activation: M1, M2 and deactivated macrophages. Based on the activation status of macrophages we propose a model starting with M1 classically activated macrophages with accelerated formation of viral reservoirs in a context of Th1 and proinflammatory cytokines. Then IL-4/IL-13 alternatively activated M2 macrophages will enter into the game that will be concomitant to tissue repair, enhanced MHC class II-mediated antigen presentation, increased T-cell activation, and enhanced clearance of opportunistic pathogens via bacterial endocytosis. At this stage of the disease, the expansion of the HIV-1 reservoir in IL-4/IL-13 alternatively activated M2 macrophages will be stopped [228]. The M2 macrophages will be in the vicinity of Th2 cells with the appearance of IL-10 deactivation of macrophages leading to immune failure observed at the very late stages of the HIV-1 disease with diminished Ag-mediated T cell response and accelerated depletion of both CD4+ and CD8+ T cells by apoptosis [229]. A better understanding of the macrophage activation status during the progression of HIV-1 infection could lead to the development of new therapeutic approaches.
References
Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A: Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001, 19: 683-765. 10.1146/annurev.immunol.19.1.683.
Gordon S: Alternative activation of macrophages. Nat Rev Immunol. 2003, 3: 23-35. 10.1038/nri978.
O'Garra A, Arai N: The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. 2000, 10: 542-550. 10.1016/S0962-8924(00)01856-0.
Colonna M: Can we apply the TH1-TH2 paradigm to all lymphocytes?. Nat Immunol. 2001, 2: 899-900. 10.1038/ni1001-899.
Locksley RM: Nine lives: plasticity among T helper cell subsets. J Exp Med. 2009, 206: 1643-1646. 10.1084/jem.20091442.
Coleman CM, Wu L: HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology. 2009, 6: 51-10.1186/1742-4690-6-51.
Rich EA, Chen IS, Zack JA, Leonard ML, O'Brien WA: Increased susceptibility of differentiated mononuclear phagocytes to productive infection with human immunodeficiency virus-1 (HIV-1). J Clin Invest. 1992, 89: 176-183. 10.1172/JCI115559.
Sonza S, Maerz A, Deacon N, Meanger J, Mills J, Crowe S: Human immunodeficiency virus type 1 replication is blocked prior to reverse transcription and integration in freshly isolated peripheral blood monocytes. J Virol. 1996, 70: 3863-3869.
Yu W, Wang Y, Shaw CA, Qin XF, Rice AP: Induction of the HIV-1 Tat co-factor cyclin T1 during monocyte differentiation is required for the regulated expression of a large portion of cellular mRNAs. Retrovirology. 2006, 3: 32-10.1186/1742-4690-3-32.
Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA: Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993, 259: 1739-1742. 10.1126/science.8456300.
Weiss L, Haeffner-Cavaillon N, Laude M, Gilquin J, Kazatchkine MD: HIV infection is associated with the spontaneous production of interleukin-1 (IL-1) in vivo and with an abnormal release of IL-1 alpha in vitro. AIDS. 1989, 3: 695-699. 10.1097/00002030-198911000-00002.
Molina JM, Scadden DT, Byrn R, Dinarello CA, Groopman JE: Production of tumor necrosis factor alpha and interleukin 1 beta by monocytic cells infected with human immunodeficiency virus. J Clin Invest. 1989, 84: 733-737. 10.1172/JCI114230.
Emilie D, Peuchmaur M, Maillot MC, Crevon MC, Brousse N, Delfraissy JF, Dormont J, Galanaud P: Production of interleukins in human immunodeficiency virus-1-replicating lymph nodes. J Clin Invest. 1990, 86: 148-159. 10.1172/JCI114678.
Birx DL, Redfield RR, Tencer K, Fowler A, Burke DS, Tosato G: Induction of interleukin-6 during human immunodeficiency virus infection. Blood. 1990, 76: 2303-2310.
Lafeuillade A, Poizot-Martin I, Quilichini R, Gastaut JA, Kaplanski S, Farnarier C, Mege JL, Bongrand P: Increased interleukin-6 production is associated with disease progression in HIV infection. AIDS. 1991, 5: 1139-1140. 10.1097/00002030-199109000-00014.
Canque B, Rosenzwajg M, Gey A, Tartour E, Fridman WH, Gluckman JC: Macrophage inflammatory protein-1alpha is induced by human immunodeficiency virus infection of monocyte-derived macrophages. Blood. 1996, 87: 2011-2019.
Cotter RL, Zheng J, Che M, Niemann D, Liu Y, He J, Thomas E, Gendelman HE: Regulation of human immunodeficiency virus type 1 infection, beta-chemokine production, and CCR5 expression in CD40L-stimulated macrophages: immune control of viral entry. J Virol. 2001, 75: 4308-4320. 10.1128/JVI.75.9.4308-4320.2001.
Lisziewicz J, Gabrilovich DI, Varga G, Xu J, Greenberg PD, Arya SK, Bosch M, Behr JP, Lori F: Induction of potent human immunodeficiency virus type 1-specific T-cell-restricted immunity by genetically modified dendritic cells. J Virol. 2001, 75: 7621-7628. 10.1128/JVI.75.16.7621-7628.2001.
Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA: CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996, 272: 1955-1958. 10.1126/science.272.5270.1955.
Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J: The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996, 85: 1135-1148. 10.1016/S0092-8674(00)81313-6.
Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P: Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995, 270: 1811-1815. 10.1126/science.270.5243.1811.
Collin M, Herbein G, Montaner L, Gordon S: PCR analysis of HIV1 infection of macrophages: virus entry is CD4-dependent. Res Virol. 1993, 144: 13-19. 10.1016/S0923-2516(06)80006-3.
Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR: Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996, 381: 661-666. 10.1038/381661a0.
Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW: A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996, 85: 1149-1158. 10.1016/S0092-8674(00)81314-8.
Feng Y, Broder CC, Kennedy PE, Berger EA: HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996, 272: 872-877. 10.1126/science.272.5263.872.
Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR: Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996, 86: 367-377. 10.1016/S0092-8674(00)80110-5.
Schmidtmayerova H, Sherry B, Bukrinsky M: Chemokines and HIV replication. Nature. 1996, 382: 767-10.1038/382767a0.
Verani A, Scarlatti G, Comar M, Tresoldi E, Polo S, Giacca M, Lusso P, Siccardi AG, Vercelli D: C-C chemokines released by lipopolysaccharide (LPS)-stimulated human macrophages suppress HIV-1 infection in both macrophages and T cells. J Exp Med. 1997, 185: 805-816. 10.1084/jem.185.5.805.
Wang J, Roderiquez G, Oravecz T, Norcross MA: Cytokine regulation of human immunodeficiency virus type 1 entry and replication in human monocytes/macrophages through modulation of CCR5 expression. J Virol. 1998, 72: 7642-7647.
Rossi F, Querido B, Nimmagadda M, Cocklin S, Navas-Martin S, Martin-Garcia J: The V1-V3 region of a brain-derived HIV-1 envelope glycoprotein determines macrophage tropism, low CD4 dependence, increased fusogenicity and altered sensitivity to entry inhibitors. Retrovirology. 2008, 5: 89-10.1186/1742-4690-5-89.
Dunfee RL, Thomas ER, Gabuzda D: Enhanced macrophage tropism of HIV in brain and lymphoid tissues is associated with sensitivity to the broadly neutralizing CD4 binding site antibody b12. Retrovirology. 2009, 6: 69-10.1186/1742-4690-6-69.
Bergamini A, Faggioli E, Bolacchi F, Gessani S, Cappannoli L, Uccella I, Demin F, Capozzi M, Cicconi R, Placido R, Vendetti S, Colizzi GM, Rocchi G: Enhanced production of tumor necrosis factor-alpha and interleukin-6 due to prolonged response to lipopolysaccharide in human macrophages infected in vitro with human immunodeficiency virus type 1. J Infect Dis. 1999, 179: 832-842. 10.1086/314662.
Choe W, Volsky DJ, Potash MJ: Induction of rapid and extensive beta-chemokine synthesis in macrophages by human immunodeficiency virus type 1 and gp120, independently of their coreceptor phenotype. J Virol. 2001, 75: 10738-10745. 10.1128/JVI.75.22.10738-10745.2001.
Clouse KA, Cosentino LM, Weih KA, Pyle SW, Robbins PB, Hochstein HD, Natarajan V, Farrar WL: The HIV-1 gp120 envelope protein has the intrinsic capacity to stimulate monokine secretion. J Immunol. 1991, 147: 2892-2901.
Herbein G, Keshav S, Collin M, Montaner LJ, Gordon S: HIV-1 induces tumour necrosis factor and IL-1 gene expression in primary human macrophages independent of productive infection. Clin Exp Immunol. 1994, 95: 442-449.
Karsten V, Gordon S, Kirn A, Herbein G: HIV-1 envelope glycoprotein gp120 down-regulates CD4 expression in primary human macrophages through induction of endogenous tumour necrosis factor-alpha. Immunology. 1996, 88: 55-60. 10.1046/j.1365-2567.1996.d01-648.x.
Merrill JE, Koyanagi Y, Chen IS: Interleukin-1 and tumor necrosis factor alpha can be induced from mononuclear phagocytes by human immunodeficiency virus type 1 binding to the CD4 receptor. J Virol. 1989, 63: 4404-4408.
Bailer RT, Lee B, Montaner LJ: IL-13 and TNF-alpha inhibit dual-tropic HIV-1 in primary macrophages by reduction of surface expression of CD4, chemokine receptors CCR5, CXCR4 and post-entry viral gene expression. Eur J Immunol. 2000, 30: 1340-1349. 10.1002/(SICI)1521-4141(200005)30:5<1340::AID-IMMU1340>3.0.CO;2-L.
Faltynek CR, Finch LR, Miller P, Overton WR: Treatment with recombinant IFN-gamma decreases cell surface CD4 levels on peripheral blood monocytes and on myelomonocyte cell lines. J Immunol. 1989, 142: 500-508.
Hariharan D, Douglas SD, Lee B, Lai JP, Campbell DE, Ho WZ: Interferon-gamma upregulates CCR5 expression in cord and adult blood mononuclear phagocytes. Blood. 1999, 93: 1137-1144.
Herbein G, Doyle AG, Montaner LJ, Gordon S: Lipopolysaccharide (LPS) down-regulates CD4 expression in primary human macrophages through induction of endogenous tumour necrosis factor (TNF) and IL-1 beta. Clin Exp Immunol. 1995, 102: 430-437.
Cremer I, Vieillard V, De Maeyer E: Retrovirally mediated IFN-beta transduction of macrophages induces resistance to HIV, correlated with up-regulation of RANTES production and down-regulation of C-C chemokine receptor-5 expression. J Immunol. 2000, 164: 1582-1587.
Herbein G, Montaner LJ, Gordon S: Tumor necrosis factor alpha inhibits entry of human immunodeficiency virus type 1 into primary human macrophages: a selective role for the 75-kilodalton receptor. J Virol. 1996, 70: 7388-7397.
Hewson TJ, Logie JJ, Simmonds P, Howie SE: A CCR5-dependent novel mechanism for type 1 HIV gp120 induced loss of macrophage cell surface CD4. J Immunol. 2001, 166: 4835-4842.
Meylan PR, Guatelli JC, Munis JR, Richman DD, Kornbluth RS: Mechanisms for the inhibition of HIV replication by interferons-alpha, -beta, and -gamma in primary human macrophages. Virology. 1993, 193: 138-148. 10.1006/viro.1993.1110.
Zaitseva M, Lee S, Lapham C, Taffs R, King L, Romantseva T, Manischewitz J, Golding H: Interferon gamma and interleukin 6 modulate the susceptibility of macrophages to human immunodeficiency virus type 1 infection. Blood. 2000, 96: 3109-3117.
Di Marzio P, Tse J, Landau NR: Chemokine receptor regulation and HIV type 1 tropism in monocyte-macrophages. AIDS Res Hum Retroviruses. 1998, 14: 129-138. 10.1089/aid.1998.14.129.
Capobianchi MR, Abbate I, Antonelli G, Turriziani O, Dolei A, Dianzani F: Inhibition of HIV type 1 BaL replication by MIP-1alpha, MIP-1beta, and RANTES in macrophages. AIDS Res Hum Retroviruses. 1998, 14: 233-240. 10.1089/aid.1998.14.233.
Coffey MJ, Woffendin C, Phare SM, Strieter RM, Markovitz DM: RANTES inhibits HIV-1 replication in human peripheral blood monocytes and alveolar macrophages. Am J Physiol. 1997, 272: L1025-1029.
Jiang Y, Jolly PE: Effect of beta-chemokines on human immunodeficiency virus type 1 replication, binding, uncoating, and CCR5 receptor expression in human monocyte-derived macrophages. J Hum Virol. 1999, 2: 123-132.
Lane BR, Markovitz DM, Woodford NL, Rochford R, Strieter RM, Coffey MJ: TNF-alpha inhibits HIV-1 replication in peripheral blood monocytes and alveolar macrophages by inducing the production of RANTES and decreasing C-C chemokine receptor 5 (CCR5) expression. J Immunol. 1999, 163: 3653-3661.
Ylisastigui L, Amzazi S, Bakri Y, Vizzavona J, Vita C, Gluckman JC, Benjouad A: Effect of RANTES on the infection of monocyte-derived primary macrophages by human immunodeficiency virus type 1 and type 2. Biomedicine & Pharmacotherapy. 1998, 52: 447-453. 10.1016/S0753-3322(99)80023-7.
Grainger DJ, Lever AM: Blockade of chemokine-induced signalling inhibits CCR5-dependent HIV infection in vitro without blocking gp120/CCR5 interaction. Retrovirology. 2005, 2: 23-10.1186/1742-4690-2-23.
Stantchev TS, Broder CC: Consistent and significant inhibition of human immunodeficiency virus type 1 envelope-mediated membrane fusion by beta-chemokines (RANTES) in primary human macrophages. J Infect Dis. 2000, 182: 68-78. 10.1086/315700.
Moriuchi H, Moriuchi M, Fauci AS: Nuclear factor-kappa B potently up-regulates the promoter activity of RANTES, a chemokine that blocks HIV infection. J Immunol. 1997, 158: 3483-3491.
Gordon CJ, Muesing MA, Proudfoot AE, Power CA, Moore JP, Trkola A: Enhancement of human immunodeficiency virus type 1 infection by the CC-chemokine RANTES is independent of the mechanism of virus-cell fusion. J Virol. 1999, 73: 684-694.
Amzazi S, Ylisastigui L, Bakri Y, Rabehi L, Gattegno L, Parmentier M, Gluckman JC, Benjouad A: The inhibitory effect of RANTES on the infection of primary macrophages by R5 human immunodeficiency virus type-1 depends on the macrophage activation state. Virology. 1998, 252: 96-105. 10.1006/viro.1998.9452.
Gong W, Howard OM, Turpin JA, Grimm MC, Ueda H, Gray PW, Raport CJ, Oppenheim JJ, Wang JM: Monocyte chemotactic protein-2 activates CCR5 and blocks CD4/CCR5-mediated HIV-1 entry/replication. J Biol Chem. 1998, 273: 4289-4292. 10.1074/jbc.273.8.4289.
Truong MJ, Darcissac EC, Hermann E, Dewulf J, Capron A, Bahr GM: Interleukin-16 inhibits human immunodeficiency virus type 1 entry and replication in macrophages and in dendritic cells. J Virol. 1999, 73: 7008-7013.
Kutza J, Hayes MP, Clouse KA: Interleukin-2 inhibits HIV-1 replication in human macrophages by modulating expression of CD4 and CC-chemokine receptor-5. Aids. 1998, 12: F59-64. 10.1097/00002030-199808000-00001.
Rodriguez AR, Arulanandam BP, Hodara VL, McClure HM, Cobb EK, Salas MT, White R, Murthy KK: Influence of interleukin-15 on CD8+ natural killer cells in human immunodeficiency virus type 1-infected chimpanzees. J Gen Virol. 2007, 88: 641-651. 10.1099/vir.0.82154-0.
Biancotto A, Grivel JC, Iglehart SJ, Vanpouille C, Lisco A, Sieg SF, Debernardo R, Garate K, Rodriguez B, Margolis LB, Lederman MM: Abnormal activation and cytokine spectra in lymph nodes of people chronically infected with HIV-1. Blood. 2007, 109: 4272-4279. 10.1182/blood-2006-11-055764.
Fehniger TA, Herbein G, Yu H, Para MI, Bernstein ZP, O'Brien WA, Caligiuri MA: Natural killer cells from HIV-1+ patients produce C-C chemokines and inhibit HIV-1 infection. J Immunol. 1998, 161: 6433-6438.
Ahmad R, Sindhu ST, Toma E, Morisset R, Ahmad A: Elevated levels of circulating interleukin-18 in human immunodeficiency virus-infected individuals: role of peripheral blood mononuclear cells and implications for AIDS pathogenesis. J Virol. 2002, 76: 12448-12456. 10.1128/JVI.76.24.12448-12456.2002.
Torre D, Pugliese A: Interleukin-18: a proinflammatory cytokine in HIV-1 infection. Curr HIV Res. 2006, 4: 423-430. 10.2174/157016206778559993.
Choi HJ, Dinarello CA, Shapiro L: Interleukin-18 inhibits human immunodeficiency virus type 1 production in peripheral blood mononuclear cells. J Infect Dis. 2001, 184: 560-568. 10.1086/322805.
di Marzio P, Mariani R, Lui R, Thomas EK, Landau NR: Soluble CD40 ligand induces beta-chemokine production by macrophages and resistance to HIV-1 entry. Cytokine. 2000, 12: 1489-1495. 10.1006/cyto.1999.0594.
Kornbluth RS: The emerging role of CD40 ligand in HIV infection. J Leukoc Biol. 2000, 68: 373-382.
Kornbluth RS, Kee K, Richman DD: CD40 ligand (CD154) stimulation of macrophages to produce HIV-1-suppressive beta-chemokines. Proc Natl Acad Sci USA. 1998, 95: 5205-5210. 10.1073/pnas.95.9.5205.
Tedla N, Palladinetti P, Kelly M, Kumar RK, DiGirolamo N, Chattophadhay U, Cooke B, Truskett P, Dwyer J, Wakefield D, Lloyd A: Chemokines and T lymphocyte recruitment to lymph nodes in HIV infection. Am J Pathol. 1996, 148: 1367-1373.
Alfano M, Schmidtmayerova H, Amella CA, Pushkarsky T, Bukrinsky M: The B-oligomer of pertussis toxin deactivates CC chemokine receptor 5 and blocks entry of M-tropic HIV-1 strains. J Exp Med. 1999, 190: 597-605. 10.1084/jem.190.5.597.
Zybarth G, Reiling N, Schmidtmayerova H, Sherry B, Bukrinsky M: Activation-induced resistance of human macrophages to HIV-1 infection in vitro. J Immunol. 1999, 162: 400-406.
Palmieri C, Trimboli F, Puca A, Fiume G, Scala G, Quinto I: Inhibition of HIV-1 replication in primary human monocytes by the IkappaB-alphaS32/36A repressor of NF-kappaB. Retrovirology. 2004, 1: 45-10.1186/1742-4690-1-45.
Duh EJ, Maury WJ, Folks TM, Fauci AS, Rabson AB: Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc Natl Acad Sci USA. 1989, 86: 5974-5978. 10.1073/pnas.86.15.5974.
Osborn L, Kunkel S, Nabel GJ: Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci USA. 1989, 86: 2336-2340. 10.1073/pnas.86.7.2336.
Poli G, Bressler P, Kinter A, Duh E, Timmer WC, Rabson A, Justement JS, Stanley S, Fauci AS: Interleukin 6 induces human immunodeficiency virus expression in infected monocytic cells alone and in synergy with tumor necrosis factor alpha by transcriptional and post-transcriptional mechanisms. J Exp Med. 1990, 172: 151-158. 10.1084/jem.172.1.151.
Butera ST, Roberts BD, Leung K, Nabel GJ, Folks TM: Tumor necrosis factor receptor expression and signal transduction in HIV-1-infected cells. Aids. 1993, 7: 911-918. 10.1097/00002030-199307000-00002.
Poli G, Kinter AL, Fauci AS: Interleukin 1 induces expression of the human immunodeficiency virus alone and in synergy with interleukin 6 in chronically infected U1 cells: inhibition of inductive effects by the interleukin 1 receptor antagonist. Proc Natl Acad Sci USA. 1994, 91: 108-112. 10.1073/pnas.91.1.108.
Ruocco MR, Chen X, Ambrosino C, Dragonetti E, Liu W, Mallardo M, De Falco G, Palmieri C, Franzoso G, Quinto I, Venuta S, Scala G: Regulation of HIV-1 long terminal repeats by interaction of C/EBP(NF-IL6) and NF-kappaB/Rel transcription factors. J Biol Chem. 1996, 271: 22479-22486. 10.1074/jbc.271.37.22479.
Tesmer VM, Rajadhyaksha A, Babin J, Bina M: NF-IL6-mediated transcriptional activation of the long terminal repeat of the human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1993, 90: 7298-7302. 10.1073/pnas.90.15.7298.
Yang Y, Tesmer VM, Bina M: Regulation of HIV-1 transcription in activated monocyte macrophages. Virology. 2002, 299: 256-265. 10.1006/viro.2001.1530.
Lee ES, Zhou H, Henderson AJ: Endothelial cells enhance human immunodeficiency virus type 1 replication in macrophages through a C/EBP-dependent mechanism. J Virol. 2001, 75: 9703-9712. 10.1128/JVI.75.20.9703-9712.2001.
Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS: Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science. 1987, 238: 800-802. 10.1126/science.3313729.
Koyanagi Y, O'Brien WA, Zhao JQ, Golde DW, Gasson JC, Chen IS: Cytokines alter production of HIV-1 from primary mononuclear phagocytes. Science. 1988, 241: 1673-1675. 10.1126/science.3047875.
Pugliese A, Vidotto V, Beltramo T, Torre D: Regulation of interleukin-18 by THP-1 monocytoid cells stimulated with HIV-1 and Nef viral protein. Eur Cytokine Netw. 2005, 16: 186-190.
Shapiro L, Puren AJ, Barton HA, Novick D, Peskind RL, Shenkar R, Gu Y, Su MS, Dinarello CA: Interleukin 18 stimulates HIV type 1 in monocytic cells. Proc Natl Acad Sci USA. 1998, 95: 12550-12555. 10.1073/pnas.95.21.12550.
Zurawski SM, Vega F, Huyghe B, Zurawski G: Receptors for interleukin-13 and interleukin-4 are complex and share a novel component that functions in signal transduction. EMBO J. 1993, 12: 2663-2670.
Hsieh CS, Heimberger AB, Gold JS, O'Garra A, Murphy KM: Differential regulation of T helper phenotype development by interleukins 4 and 10 in an alpha beta T-cell-receptor transgenic system. Proc Natl Acad Sci USA. 1992, 89: 6065-6069. 10.1073/pnas.89.13.6065.
Seder RA, Paul WE, Davis MM, Fazekas de St Groth B: The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J Exp Med. 1992, 176: 1091-1098. 10.1084/jem.176.4.1091.
Coffman RL, Ohara J, Bond MW, Carty J, Zlotnik A, Paul WE: B cell stimulatory factor-1 enhances the IgE response of lipopolysaccharide-activated B cells. J Immunol. 1986, 136: 4538-4541.
Gascan H, Gauchat JF, Roncarolo MG, Yssel H, Spits H, de Vries JE: Human B cell clones can be induced to proliferate and to switch to IgE and IgG4 synthesis by interleukin 4 and a signal provided by activated CD4+ T cell clones. J Exp Med. 1991, 173: 747-750. 10.1084/jem.173.3.747.
Vitetta ES, Ohara J, Myers CD, Layton JE, Krammer PH, Paul WE: Serological, biochemical, and functional identity of B cell-stimulatory factor 1 and B cell differentiation factor for IgG1. J Exp Med. 1985, 162: 1726-1731. 10.1084/jem.162.5.1726.
Varin A, Gordon S: Alternative activation of macrophages: Immune function and cellular biology. Immunobiology. 2009, 214: 630-41. 10.1016/j.imbio.2008.11.009.
de Waal Malefyt R, Figdor CG, Huijbens R, Mohan-Peterson S, Bennett B, Culpepper J, Dang W, Zurawski G, de Vries JE: Effects of IL-13 on phenotype, cytokine production, and cytotoxic function of human monocytes. Comparison with IL-4 and modulation by IFN-gamma or IL-10. J Immunol. 1993, 151: 6370-6381.
Montaner LJ, Doyle AG, Collin M, Herbein G, Illei P, James W, Minty A, Caput D, Ferrara P, Gordon S: Interleukin 13 inhibits human immunodeficiency virus type 1 production in primary blood-derived human macrophages in vitro. J Exp Med. 1993, 178: 743-747. 10.1084/jem.178.2.743.
Zurawski G, de Vries JE: Interleukin 13, an interleukin 4-like cytokine that acts on monocytes and B cells, but not on T cells. Immunol Today. 1994, 15: 19-26. 10.1016/0167-5699(94)90021-3.
Mantovani A, Locati M, Vecchi A, Sozzani S, Allavena P: Decoy receptors: a strategy to regulate inflammatory cytokines and chemokines. Trends Immunol. 2001, 22: 328-336. 10.1016/S1471-4906(01)01941-X.
Fenton MJ, Buras JA, Donnelly RP: IL-4 reciprocally regulates IL-1 and IL-1 receptor antagonist expression in human monocytes. J Immunol. 1992, 149: 1283-1288.
Wang J, Guan E, Roderiquez G, Calvert V, Alvarez R, Norcross MA: Role of tyrosine phosphorylation in ligand-independent sequestration of CXCR4 in human primary monocytes-macrophages. J Biol Chem. 2001, 276: 49236-49243. 10.1074/jbc.M108523200.
Mougel M, Houzet L, Darlix JL: When is it time for reverse transcription to start and go?. Retrovirology. 2009, 6: 24-10.1186/1742-4690-6-24.
Denis M, Ghadirian E: Interleukin 13 and interleukin 4 protect bronchoalveolar macrophages from productive infection with human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 1994, 10: 795-802. 10.1089/aid.1994.10.1619.
Hatch WC, Freedman AR, Boldt-Houle DM, Groopman JE, Terwilliger EF: Differential effects of interleukin-13 on cytomegalovirus and human immunodeficiency virus infection in human alveolar macrophages. Blood. 1997, 89: 3443-3450.
Montaner LJ, Bailer RT, Gordon S: IL-13 acts on macrophages to block the completion of reverse transcription, inhibit virus production, and reduce virus infectivity. J Leukoc Biol. 1997, 62: 126-132.
Mikovits JA, Meyers AM, Ortaldo JR, Minty A, Caput D, Ferrara P, Ruscetti FW: IL-4 and IL-13 have overlapping but distinct effects on HIV production in monocytes. J Leukoc Biol. 1994, 56: 340-346.
Naif HM, Li S, Ho-Shon M, Mathijs JM, Williamson P, Cunningham AL: The state of maturation of monocytes into macrophages determines the effects of IL-4 and IL-13 on HIV replication. J Immunol. 1997, 158: 501-511.
Naif H, Ho-Shon M, Chang J, Cunningham AL: Molecular mechanisms of IL-4 effect on HIV expression in promonocytic cell lines and primary human monocytes. J Leukoc Biol. 1994, 56: 335-339.
Kobayashi N, Hamamoto Y, Koyanagi Y, Chen IS, Yamamoto N: Effect of interleukin-1 on the augmentation of human immunodeficiency virus gene expression. Biochem Biophys Res Commun. 1989, 165: 715-721. 10.1016/S0006-291X(89)80025-7.
Okamoto T, Matsuyama T, Mori S, Hamamoto Y, Kobayashi N, Yamamoto N, Josephs SF, Wong-Staal F, Shimotohno K: Augmentation of human immunodeficiency virus type 1 gene expression by tumor necrosis factor alpha. AIDS Res Hum Retroviruses. 1989, 5: 131-138. 10.1089/aid.1989.5.131.
Farrar WL, Korner M, Clouse KA: Cytokine regulation of human immunodeficiency virus expression. Cytokine. 1991, 3: 531-542. 10.1016/1043-4666(91)90479-W.
Poli G, Fauci AS: The role of monocyte/macrophages and cytokines in the pathogenesis of HIV infection. Pathobiology. 1992, 60: 246-251. 10.1159/000163729.
Gregory CD, Devitt A: The macrophage and the apoptotic cell: an innate immune interaction viewed simplistically?. Immunology. 2004, 113: 1-14. 10.1111/j.1365-2567.2004.01959.x.
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM: Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998, 101: 890-898. 10.1172/JCI1112.
Tassiulas I, Park-Min KH, Hu Y, Kellerman L, Mevorach D, Ivashkiv LB: Apoptotic cells inhibit LPS-induced cytokine and chemokine production and IFN responses in macrophages. Hum Immunol. 2007, 68: 156-164. 10.1016/j.humimm.2006.12.008.
Chung EY, Kim SJ, Ma XJ: Regulation of cytokine production during phagocytosis of apoptotic cells. Cell Res. 2006, 16: 154-161. 10.1038/sj.cr.7310021.
Alfano M, Crotti A, Vicenzi E, Poli G: New players in cytokine control of HIV infection. Curr HIV/AIDS Rep. 2008, 5: 27-32. 10.1007/s11904-008-0005-5.
Sozzani S, Ghezzi S, Iannolo G, Luini W, Borsatti A, Polentarutti N, Sica A, Locati M, Mackay C, Wells TN, Biswas P, Vicenzi E, Poli G, Mantovani A: Interleukin 10 increases CCR5 expression and HIV infection in human monocytes. J Exp Med. 1998, 187: 439-444. 10.1084/jem.187.3.439.
Kutza J, Crim L, Feldman S, Hayes MP, Gruber M, Beeler J, Clouse KA: Macrophage colony-stimulating factor antagonists inhibit replication of HIV-1 in human macrophages. J Immunol. 2000, 164: 4955-4960.
Montaner LJ, Griffin P, Gordon S: Interleukin-10 inhibits initial reverse transcription of human immunodeficiency virus type 1 and mediates a virostatic latent state in primary blood-derived human macrophages in vitro. J Gen Virol. 1994, 75: 3393-3400. 10.1099/0022-1317-75-12-3393.
Saville MW, Taga K, Foli A, Broder S, Tosato G, Yarchoan R: Interleukin-10 suppresses human immunodeficiency virus-1 replication in vitro in cells of the monocyte/macrophage lineage. Blood. 1994, 83: 3591-3599.
Akridge RE, Oyafuso LK, Reed SG: IL-10 is induced during HIV-1 infection and is capable of decreasing viral replication in human macrophages. J Immunol. 1994, 153: 5782-5789.
Poli G, Kinter AL, Justement JS, Bressler P, Kehrl JH, Fauci AS: Transforming growth factor beta suppresses human immunodeficiency virus expression and replication in infected cells of the monocyte/macrophage lineage. J Exp Med. 1991, 173: 589-597. 10.1084/jem.173.3.589.
Malim MH: Natural resistance to HIV infection: The Vif-APOBEC interaction. C R Biol. 2006, 329: 871-875. 10.1016/j.crvi.2006.01.012.
Goila-Gaur R, Strebel K: HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008, 5: 51-10.1186/1742-4690-5-51.
Franca R, Spadari S, Maga G: APOBEC deaminases as cellular antiviral factors: a novel natural host defense mechanism. Med Sci Monit. 2006, 12: RA92-98.
Pillai SK, Wong JK, Barbour JD: Turning up the volume on mutational pressure: is more of a good thing always better? (A case study of HIV-1 Vif and APOBEC3). Retrovirology. 2008, 5: 26-10.1186/1742-4690-5-26.
Carthagena L, Bergamaschi A, Luna JM, David A, Uchil PD, Margottin-Goguet F, Mothes W, Hazan U, Transy C, Pancino G, Nisole S: Human TRIM gene expression in response to interferons. PLoS One. 2009, 4: e4894-10.1371/journal.pone.0004894.
Rajsbaum R, Stoye JP, O'Garra A: Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur J Immunol. 2008, 38: 619-630. 10.1002/eji.200737916.
Towers GJ: The control of viral infection by tripartite motif proteins and cyclophilin A. Retrovirology. 2007, 4: 40-10.1186/1742-4690-4-40.
Huthoff H, Towers GJ: Restriction of retroviral replication by APOBEC3G/F and TRIM5alpha. Trends Microbiol. 2008, 16: 612-619. 10.1016/j.tim.2008.08.013.
Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH: APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4: e1000231-10.1371/journal.ppat.1000231.
Carthagena L, Parise MC, Ringeard M, Chelbi-Alix MK, Hazan U, Nisole S: Implication of TRIM alpha and TRIMCyp in interferon-induced anti-retroviral restriction activities. Retrovirology. 2008, 5: 59-10.1186/1742-4690-5-59.
Weissman D, Poli G, Fauci AS: Interleukin 10 blocks HIV replication in macrophages by inhibiting the autocrine loop of tumor necrosis factor alpha and interleukin 6 induction of virus. AIDS Res Hum Retroviruses. 1994, 10: 1199-1206. 10.1089/aid.1994.10.1199.
Weissman D, Poli G, Fauci AS: IL-10 synergizes with multiple cytokines in enhancing HIV production in cells of monocytic lineage. J Acquir Immune Defic Syndr Hum Retrovirol. 1995, 9: 442-449.
Kootstra NA, van 't Wout A, Huisman HG, Miedema F, Schuitemaker H: Interference of interleukin-10 with human immunodeficiency virus type 1 replication in primary monocyte-derived macrophages. J Virol. 1994, 68: 6967-6975.
Yamamoto JK, Barre-Sinoussi F, Bolton V, Pedersen NC, Gardner MB: Human alpha- and beta-interferon but not gamma-suppress the in vitro replication of LAV, HTLV-III, and ARV-2. J Interferon Res. 1986, 6: 143-152.
Peng G, Lei KJ, Jin W, Greenwell-Wild T, Wahl SM: Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J Exp Med. 2006, 203: 41-46. 10.1084/jem.20051512.
Mangino G, Percario ZA, Fiorucci G, Vaccari G, Manrique S, Romeo G, Federico M, Geyer M, Affabris E: In vitro treatment of human monocytes/macrophages with myristoylated recombinant Nef of human immunodeficiency virus type 1 leads to the activation of mitogen-activated protein kinases, IkappaB kinases, and interferon regulatory factor 3 and to the release of beta interferon. J Virol. 2007, 81: 2777-2791. 10.1128/JVI.01640-06.
Fakruddin JM, Lempicki RA, Gorelick RJ, Yang J, Adelsberger JW, Garcia-Pineres AJ, Pinto LA, Lane HC, Imamichi T: Noninfectious papilloma virus-like particles inhibit HIV-1 replication: implications for immune control of HIV-1 infection by IL-27. Blood. 2007, 109: 1841-1849. 10.1182/blood-2006-02-001578.
Imamichi T, Yang J, Huang DW, Brann TW, Fullmer BA, Adelsberger JW, Lempicki RA, Baseler MW, Lane HC: IL-27, a novel anti-HIV cytokine, activates multiple interferon-inducible genes in macrophages. AIDS. 2008, 22: 39-45. 10.1097/QAD.0b013e3282f3356c.
Greenwell-Wild T, Vazquez N, Jin W, Rangel Z, Munson PJ, Wahl SM: Interleukin-27 inhibition of HIV-1 involves an intermediate induction of type I interferon. Blood. 2009, 114: 1864-1874. 10.1182/blood-2009-03-211540.
Harty RN, Pitha PM, Okumura A: Antiviral Activity of Innate Immune Protein ISG15. J Innate Immun. 2009, 1: 397-404. 10.1159/000226245.
Okumura A, Lu G, Pitha-Rowe I, Pitha PM: Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc Natl Acad Sci USA. 2006, 103: 1440-1445. 10.1073/pnas.0510518103.
Wahl SM, Greenwell-Wild T, Vazquez N: HIV accomplices and adversaries in macrophage infection. J Leukoc Biol. 2006, 80: 973-983. 10.1189/jlb.0306130.
Espert L, Degols G, Lin YL, Vincent T, Benkirane M, Mechti N: Interferon-induced exonuclease ISG20 exhibits an antiviral activity against human immunodeficiency virus type 1. J Gen Virol. 2005, 86: 2221-2229. 10.1099/vir.0.81074-0.
Barr SD, Smiley JR, Bushman FD: The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 2008, 4: e1000007-10.1371/journal.ppat.1000007.
Lazdins JK, Klimkait T, Woods-Cook K, Walker M, Alteri E, Cox D, Cerletti N, Shipman R, Bilbe G, McMaster G: In vitro effect of transforming growth factor-beta on progression of HIV-1 infection in primary mononuclear phagocytes. J Immunol. 1991, 147: 1201-1207.
Kamga I, Kahi S, Develioglu L, Lichtner M, Maranon C, Deveau C, Meyer L, Goujard C, Lebon P, Sinet M, Hosmalin A: Type I interferon production is profoundly and transiently impaired in primary HIV-1 infection. J Infect Dis. 2005, 192: 303-310. 10.1086/430931.
Herbein G, Khan KA: Is HIV infection a TNF receptor signalling-driven disease?. Trends Immunol. 2008, 29: 61-67. 10.1016/j.it.2007.10.008.
Aukrust P, Liabakk NB, Muller F, Lien E, Espevik T, Froland SS: Serum levels of tumor necrosis factor-alpha (TNF alpha) and soluble TNF receptors in human immunodeficiency virus type 1 infection--correlations to clinical, immunologic, and virologic parameters. J Infect Dis. 1994, 169: 420-424.
Jacquot G, Le Rouzic E, David A, Mazzolini J, Bouchet J, Bouaziz S, Niedergang F, Pancino G, Benichou S: Localization of HIV-1 Vpr to the nuclear envelope: impact on Vpr functions and virus replication in macrophages. Retrovirology. 2007, 4: 84-10.1186/1742-4690-4-84.
Kilareski EM, Shah S, Nonnemacher MR, Wigdahl B: Regulation of HIV-1 transcription in cells of the monocyte-macrophage lineage. Retrovirology. 2009, 6: 118-10.1186/1742-4690-6-118.
Herbeuval JP, Nilsson J, Boasso A, Hardy AW, Kruhlak MJ, Anderson SA, Dolan MJ, Dy M, Andersson J, Shearer GM: Differential expression of IFN-alpha and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc Natl Acad Sci USA. 2006, 103: 7000-7005. 10.1073/pnas.0600363103.
Meyers JH, Justement JS, Hallahan CW, Blair ET, Sun YA, O'Shea MA, Roby G, Kottilil S, Moir S, Kovacs CM, Chun TW, Fauci AS: Impact of HIV on cell survival and antiviral activity of plasmacytoid dendritic cells. PLoS One. 2007, 2: e458-10.1371/journal.pone.0000458.
Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE: The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999, 17: 701-738. 10.1146/annurev.immunol.17.1.701.
Papasavvas E, Sun J, Luo Q, Moore EC, Thiel B, MacGregor RR, Minty A, Mounzer K, Kostman JR, Montaner LJ: IL-13 acutely augments HIV-specific and recall responses from HIV-1-infected subjects in vitro by modulating monocytes. J Immunol. 2005, 175: 5532-5540.
Benoit M, Desnues B, Mege JL: Macrophage polarization in bacterial infections. J Immunol. 2008, 181: 3733-3739.
Goldmann O, von Kockritz-Blickwede M, Holtje C, Chhatwal GS, Geffers R, Medina E: Transcriptome analysis of murine macrophages in response to infection with Streptococcus pyogenes reveals an unusual activation program. Infect Immun. 2007, 75: 4148-4157. 10.1128/IAI.00181-07.
Mahlknecht U, Herbein G: Macrophages and T-cell apoptosis in HIV infection: a leading role for accessory cells?. Trends Immunol. 2001, 22: 256-260. 10.1016/S1471-4906(01)01898-1.
Varin A, Mukhopadhyay S, Herbein G, Gordon S: Alternative activation of macrophages by IL-4 impairs phagocytosis of pathogens but potentiates microbial-induced signalling and cytokine secretion. Blood. 2009, 115: 353-62. 10.1182/blood-2009-08-236711.
Herbein G, Mahlknecht U, Batliwalla F, Gregersen P, Pappas T, Butler J, O'Brien WA, Verdin E: Apoptosis of CD8+ T cells is mediated by macrophages through interaction of HIV gp120 with chemokine receptor CXCR4. Nature. 1998, 395: 189-194. 10.1038/26026.
Ogden CA, Pound JD, Batth BK, Owens S, Johannessen I, Wood K, Gregory CD: Enhanced apoptotic cell clearance capacity and B cell survival factor production by IL-10-activated macrophages: implications for Burkitt's lymphoma. J Immunol. 2005, 174: 3015-3023.
Byrne A, Reen DJ: Lipopolysaccharide induces rapid production of IL-10 by monocytes in the presence of apoptotic neutrophils. J Immunol. 2002, 168: 1968-1977.
Clerici M, Shearer GM: A TH1-->TH2 switch is a critical step in the etiology of HIV infection. Immunol Today. 1993, 14: 107-111. 10.1016/0167-5699(93)90208-3.
Oleksyk TK, Shrestha S, Truelove AL, Goedert JJ, Donfield SM, Phair J, Mehta S, O'Brien SJ, Smith MW: Extended IL10 haplotypes and their association with HIV progression to AIDS. Genes Immun. 2009, 10: 309-322. 10.1038/gene.2009.9.
Vasilescu A, Heath SC, Ivanova R, Hendel H, Do H, Mazoyer A, Khadivpour E, Goutalier FX, Khalili K, Rappaport J, Lathrop GM, Matsuda F, Zagury JF: Genomic analysis of Th1-Th2 cytokine genes in an AIDS cohort: identification of IL4 and IL10 haplotypes associated with the disease progression. Genes Immun. 2003, 4: 441-449. 10.1038/sj.gene.6363983.
Sandanger O, Ryan L, Bohnhorst J, Iversen AC, Husebye H, Halaas O, Landro L, Aukrust P, Froland SS, Elson G, Visintin A, Øktedalen O, Damås JK, Sundan A, Golenbock D, Espevik T: IL-10 enhances MD-2 and CD14 expression in monocytes and the proteins are increased and correlated in HIV-infected patients. J Immunol. 2009, 182: 588-595.
Creery D, Angel JB, Aucoin S, Weiss W, Cameron WD, Diaz-Mitoma F, Kumar A: Nef protein of human immunodeficiency virus and lipopolysaccharide induce expression of CD14 on human monocytes through differential utilization of interleukin-10. Clin Diagn Lab Immunol. 2002, 9: 1212-1221.
Andrade RM, Lima PG, Filho RG, Hygino J, Milczanowski SF, Andrade AF, Lauria C, Brindeiro R, Tanuri A, Bento CA: Interleukin-10-secreting CD4 cells from aged patients with AIDS decrease in-vitro HIV replication and tumour necrosis factor alpha production. AIDS. 2007, 21: 1763-1770. 10.1097/QAD.0b013e3282ca83fa.
Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB: IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci USA. 2008, 105: 20428-20433. 10.1073/pnas.0811139106.
Gupta S, Boppana R, Mishra GC, Saha B, Mitra D: HIV-1 Tat suppresses gp120-specific T cell response in IL-10-dependent manner. J Immunol. 2008, 180: 79-88.
Fantuzzi L, Belardelli F, Gessani S: Monocyte/macrophage-derived CC chemokines and their modulation by HIV-1 and cytokines: a complex network of interactions influencing viral replication and AIDS pathogenesis. J Leukoc Biol. 2003, 74: 719-725. 10.1189/jlb.0403175.
Rajasingh J, Bord E, Luedemann C, Asai J, Hamada H, Thorne T, Qin G, Goukassian D, Zhu Y, Losordo DW, Kishore R: IL-10-induced TNF-alpha mRNA destabilization is mediated via IL-10 suppression of p38 MAP kinase activation and inhibition of HuR expression. FASEB J. 2006, 20: 2112-2114. 10.1096/fj.06-6084fje.
Wang Y, Rice AP: Interleukin-10 inhibits HIV-1 LTR-directed gene expression in human macrophages through the induction of cyclin T1 proteolysis. Virology. 2006, 352: 485-492. 10.1016/j.virol.2006.05.013.
Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC: CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. 2004, 200: 749-759. 10.1084/jem.20040874.
Guadalupe M, Reay E, Sankaran S, Prindiville T, Flamm J, McNeil A, Dandekar S: Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J Virol. 2003, 77: 11708-11717. 10.1128/JVI.77.21.11708-11717.2003.
Guadalupe M, Sankaran S, George MD, Reay E, Verhoeven D, Shacklett BL, Flamm J, Wegelin J, Prindiville T, Dandekar S: Viral suppression and immune restoration in the gastrointestinal mucosa of human immunodeficiency virus type 1-infected patients initiating therapy during primary or chronic infection. J Virol. 2006, 80: 8236-8247. 10.1128/JVI.00120-06.
Li Q, Duan L, Estes JD, Ma ZM, Rourke T, Wang Y, Reilly C, Carlis J, Miller CJ, Haase AT: Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature. 2005, 434: 1148-1152.
Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M: Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005, 434: 1093-1097. 10.1038/nature03501.
Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, Boden D, Racz P, Markowitz M: Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med. 2004, 200: 761-770. 10.1084/jem.20041196.
Mehandru S, Poles MA, Tenner-Racz K, Manuelli V, Jean-Pierre P, Lopez P, Shet A, Low A, Mohri H, Boden D, Racz P, Markowitz M: Mechanisms of gastrointestinal CD4+ T-cell depletion during acute and early human immunodeficiency virus type 1 infection. J Virol. 2007, 81: 599-612. 10.1128/JVI.01739-06.
Smit-McBride Z, Mattapallil JJ, McChesney M, Ferrick D, Dandekar S: Gastrointestinal T lymphocytes retain high potential for cytokine responses but have severe CD4(+) T-cell depletion at all stages of simian immunodeficiency virus infection compared to peripheral lymphocytes. J Virol. 1998, 72: 6646-6656.
Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig M, Johnson RP, Desrosiers RC, Lackner AA: Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998, 280: 427-431. 10.1126/science.280.5362.427.
Li L, Meng G, Graham MF, Shaw GM, Smith PD: Intestinal macrophages display reduced permissiveness to human immunodeficiency virus 1 and decreased surface CCR5. Gastroenterology. 1999, 116: 1043-1053. 10.1016/S0016-5085(99)70007-7.
Meng G, Sellers MT, Mosteller-Barnum M, Rogers TS, Shaw GM, Smith PD: Lamina propria lymphocytes, not macrophages, express CCR5 and CXCR4 and are the likely target cell for human immunodeficiency virus type 1 in the intestinal mucosa. J Infect Dis. 2000, 182: 785-791. 10.1086/315790.
Gorry PR, Bristol G, Zack JA, Ritola K, Swanstrom R, Birch CJ, Bell JE, Bannert N, Crawford K, Wang H, Schols D, De Clercq E, Kunstman K, Wolinsky SM, Gabuzda D: Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. J Virol. 2001, 75: 10073-10089. 10.1128/JVI.75.21.10073-10089.2001.
Smith PD, Smythies LE, Mosteller-Barnum M, Sibley DA, Russell MW, Merger M, Sellers MT, Orenstein JM, Shimada T, Graham MF, Kubagawa H: Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J Immunol. 2001, 167: 2651-2656.
Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD: Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest. 2005, 115: 66-75.
Schenk M, Bouchon A, Birrer S, Colonna M, Mueller C: Macrophages expressing triggering receptor expressed on myeloid cells-1 are underrepresented in the human intestine. J Immunol. 2005, 174: 517-524.
Schenk M, Bouchon A, Seibold F, Mueller C: TREM-1--expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J Clin Invest. 2007, 117: 3097-3106. 10.1172/JCI30602.
Rugtveit J, Bakka A, Brandtzaeg P: Differential distribution of B7.1 (CD80) and B7.2 (CD86) costimulatory molecules on mucosal macrophage subsets in human inflammatory bowel disease (IBD). Clin Exp Immunol. 1997, 110: 104-113. 10.1111/j.1365-2249.1997.507-ce1404.x.
Smith PD, Meng G, Salazar-Gonzalez JF, Shaw GM: Macrophage HIV-1 infection and the gastrointestinal tract reservoir. J Leukoc Biol. 2003, 74: 642-649. 10.1189/jlb.0503219.
Smith PD, Ochsenbauer-Jambor C, Smythies LE: Intestinal macrophages: unique effector cells of the innate immune system. Immunol Rev. 2005, 206: 149-159. 10.1111/j.0105-2896.2005.00288.x.
Shen R, Richter HE, Clements RH, Novak L, Huff K, Bimczok D, Sankaran-Walters S, Dandekar S, Clapham PR, Smythies LE, Smith PD: Macrophages in vaginal but not intestinal mucosa are monocyte-like and permissive to human immunodeficiency virus type 1 infection. J Virol. 2009, 83: 3258-3267. 10.1128/JVI.01796-08.
Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J: Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. J Neurovirol. 2008, 14: 318-326. 10.1080/13550280802132857.
Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS: Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986, 233: 1089-1093. 10.1126/science.3016903.
Cherner M, Masliah E, Ellis RJ, Marcotte TD, Moore DJ, Grant I, Heaton RK: Neurocognitive dysfunction predicts postmortem findings of HIV encephalitis. Neurology. 2002, 59: 1563-1567.
Nuovo GJ, Alfieri ML: AIDS dementia is associated with massive, activated HIV-1 infection and concomitant expression of several cytokines. Mol Med. 1996, 2: 358-366. 10.1007/s0089460020358.
Saito Y, Sharer LR, Epstein LG, Michaels J, Mintz M, Louder M, Golding K, Cvetkovich TA, Blumberg BM: Overexpression of nef as a marker for restricted HIV-1 infection of astrocytes in postmortem pediatric central nervous tissues. Neurology. 1994, 44: 474-481.
Ghafouri M, Amini S, Khalili K, Sawaya BE: HIV-1 associated dementia: symptoms and causes. Retrovirology. 2006, 3: 28-10.1186/1742-4690-3-28.
Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, Young SA, Mills RG, Wachsman W, Wiley CA: Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992, 42: 1736-1739.
Niederkorn JY: See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol. 2006, 7: 354-359. 10.1038/ni1328.
Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA: Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006, 24: 99-146. 10.1146/annurev.immunol.24.021605.090737.
Boche D, Cunningham C, Gauldie J, Perry VH: Transforming growth factor-beta 1-mediated neuroprotection against excitotoxic injury in vivo. J Cereb Blood Flow Metab. 2003, 23: 1174-1182. 10.1097/01.WCB.0000090080.64176.44.
Persidsky Y, Poluektova L: Immune privilege and HIV-1 persistence in the CNS. Immunol Rev. 2006, 213: 180-194. 10.1111/j.1600-065X.2006.00440.x.
Massa PT: Specific suppression of major histocompatibility complex class I and class II genes in astrocytes by brain-enriched gangliosides. J Exp Med. 1993, 178: 1357-1363. 10.1084/jem.178.4.1357.
Achim CL, Morey MK, Wiley CA: Expression of major histocompatibility complex and HIV antigens within the brains of AIDS patients. AIDS. 1991, 5: 535-541. 10.1097/00002030-199105000-00009.
An SF, Ciardi A, Giometto B, Scaravilli T, Gray F, Scaravilli F: Investigation on the expression of major histocompatibility complex class II and cytokines and detection of HIV-1 DNA within brains of asymptomatic and symptomatic HIV-1-positive patients. Acta Neuropathol. 1996, 91: 494-503. 10.1007/s004010050457.
Persidsky Y, Ghorpade A, Rasmussen J, Limoges J, Liu XJ, Stins M, Fiala M, Way D, Kim KS, Witte MH, Weinand M, Carhart L, Gendelman HE: Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999, 155: 1599-1611.
Suter T, Biollaz G, Gatto D, Bernasconi L, Herren T, Reith W, Fontana A: The brain as an immune privileged site: dendritic cells of the central nervous system inhibit T cell activation. Eur J Immunol. 2003, 33: 2998-3006. 10.1002/eji.200323611.
Fassnacht U, Ackermann A, Staeheli P, Hausmann J: Immunization with dendritic cells can break immunological ignorance toward a persisting virus in the central nervous system and induce partial protection against intracerebral viral challenge. J Gen Virol. 2004, 85: 2379-2387. 10.1099/vir.0.80115-0.
Schwartz AJ, Alvarez X, Lackner AA: Distribution and immunophenotype of DC-SIGN-expressing cells in SIV-infected and uninfected macaques. AIDS Res Hum Retroviruses. 2002, 18: 1021-1029. 10.1089/08892220260235380.
Gray F, Lescs MC, Keohane C, Paraire F, Marc B, Durigon M, Gherardi R: Early brain changes in HIV infection: neuropathological study of 11 HIV seropositive, non-AIDS cases. J Neuropathol Exp Neurol. 1992, 51: 177-185. 10.1097/00005072-199203000-00007.
Potula R, Poluektova L, Knipe B, Chrastil J, Heilman D, Dou H, Takikawa O, Munn DH, Gendelman HE, Persidsky Y: Inhibition of indoleamine 2,3-dioxygenase (IDO) enhances elimination of virus-infected macrophages in an animal model of HIV-1 encephalitis. Blood. 2005, 106: 2382-2390. 10.1182/blood-2005-04-1403.
McCrossan M, Marsden M, Carnie FW, Minnis S, Hansoti B, Anthony IC, Brettle RP, Bell JE, Simmonds P: An immune control model for viral replication in the CNS during presymptomatic HIV infection. 2006, 129: 503-516.
Gray F, Scaravilli F, Everall I, Chretien F, An S, Boche D, Adle-Biassette H, Wingertsmann L, Durigon M, Hurtrel B, Chiodi F, Bell J, Lantos P: Neuropathology of early HIV-1 infection. Brain Pathol. 1996, 6: 1-15. 10.1111/j.1750-3639.1996.tb00775.x.
Wingertsmann L, Chretien F, Authier FJ, Paraire F, Durigon M, Gray F: [Central nervous system lesions in the early stages of HIV infection]. Arch Anat Cytol Pathol. 1997, 45: 106-117.
Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, Stern Y, Albert S, Palumbo D, Kieburtz K, De Marcaida JA, Cohen B, Epstein L: HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirol. 2002, 8: 136-142. 10.1080/13550280290049615.
Wang T, Gong N, Liu J, Kadiu I, Kraft-Terry SD, Mosley RL, Volsky DJ, Ciborowski P, Gendelman HE: Proteomic modeling for HIV-1 infected microglia-astrocyte crosstalk. PLoS One. 2008, 3: e2507-10.1371/journal.pone.0002507.
Takahashi K, Wesselingh SL, Griffin DE, McArthur JC, Johnson RT, Glass JD: Localization of HIV-1 in human brain using polymerase chain reaction/in situ hybridization and immunocytochemistry. Ann Neurol. 1996, 39: 705-711. 10.1002/ana.410390606.
Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, Rothstein JD, Volsky DJ: Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology. 2003, 312: 60-73. 10.1016/S0042-6822(03)00181-8.
Fine SM, Angel RA, Perry SW, Epstein LG, Rothstein JD, Dewhurst S, Gelbard HA: Tumor necrosis factor alpha inhibits glutamate uptake by primary human astrocytes. Implications for pathogenesis of HIV-1 dementia. J Biol Chem. 1996, 271: 15303-15306. 10.1074/jbc.271.26.15303.
Zhou BY, Liu Y, Kim B, Xiao Y, He JJ: Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol Cell Neurosci. 2004, 27: 296-305.
Garden GA: Microglia in human immunodeficiency virus-associated neurodegeneration. Glia. 2002, 40: 240-251. 10.1002/glia.10155.
McArthur JC, Brew BJ, Nath A: Neurological complications of HIV infection. Lancet Neurol. 2005, 4: 543-555. 10.1016/S1474-4422(05)70165-4.
Tyor WR, Glass JD, Griffin JW, Becker PS, McArthur JC, Bezman L, Griffin DE: Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol. 1992, 31: 349-360. 10.1002/ana.410310402.
Wesselingh SL, Takahashi K, Glass JD, McArthur JC, Griffin JW, Griffin DE: Cellular localization of tumor necrosis factor mRNA in neurological tissue from HIV-infected patients by combined reverse transcriptase/polymerase chain reaction in situ hybridization and immunohistochemistry. J Neuroimmunol. 1997, 74: 1-8. 10.1016/S0165-5728(96)00160-9.
Xing HQ, Moritoyo T, Mori K, Sugimoto C, Ono F, Izumo S: Expression of proinflammatory cytokines and its relationship with virus infection in the brain of macaques inoculated with macrophage-tropic simian immunodeficiency virus. Neuropathology. 2009, 29: 13-19. 10.1111/j.1440-1789.2008.00929.x.
Cassol E, Cassetta L, Rizzi C, Alfano M, Poli G: M1 and M2a polarization of human monocyte-derived macrophages inhibits HIV-1 replication by distinct mechanisms. J Immunol. 2009, 182: 6237-6246. 10.4049/jimmunol.0803447.
Boasso A, Shearer GM, Chougnet C: Immune dysregulation in human immunodeficiency virus infection: know it, fix it, prevent it?. J Intern Med. 2009, 265: 78-96. 10.1111/j.1365-2796.2008.02043.x.
Acknowledgements
The work of the authors is supported by institutional funds from the Franche-Comte University and from the Association for Macrophage and Infection Research (AMIR).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
GH was responsible for drafting and revising the manuscript as well as organizing the content. AV assisted in revising the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Herbein, G., Varin, A. The macrophage in HIV-1 infection: From activation to deactivation?. Retrovirology 7, 33 (2010). https://doi.org/10.1186/1742-4690-7-33
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-4690-7-33