
Abstract
In recent years, tyrosine kinases (TKs) have been recognized as central players and regulators of cancer cell proliferation, apoptosis, and angiogenesis, and are therefore considered suitable potential targets for anti-cancer therapies. Several strategies for targeting TKs have been developed, the most successful being monoclonal antibodies and small molecule tyrosine kinase inhibitors. However, increasing evidence of acquired resistance to these drugs has been documented, and extensive preclinical studies are ongoing to try to understand the molecular mechanisms by which cancer cells are able to bypass their inhibitory activity.
This review intends to present the most recently identified molecular mechanisms that mediate acquired resistance to tyrosine kinase inhibitors, identified through the use of in vitro models or the analysis of patient samples. The knowledge obtained from these studies will help to design better therapies that prevent and overcome resistance to treatment in cancer patients.
Similar content being viewed by others


Introduction
The most common type of pharmacological anticancer treatment has been, for decades, conventional chemotherapy. This type of treatment does not discriminate between rapidly dividing normal cells and tumor cells, thus leading to severe systemic side effects, while attempting to reduce the tumor mass. In the last decade, the use of novel molecular targeted therapies has raised interest of both patients and clinicians. These treatments inhibit specific molecules that have a role in tumor growth or progression, and that are frequently altered in tumors but not in normal cells; thus, being more specific toward tumor cells, they are accompanied by reduced systemic toxicity [1]. Nowadays, targeted therapies represent an integrative approach to cancer therapy that has already led to important clinical results [2, 3].
Tyrosine Kinases
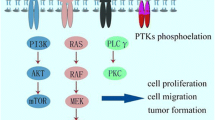
Tyrosine kinases have been identified as signaling molecules and prototypic oncogenes, and shown to play an important role in the development of many diseases, including cancer [4]. There is strong evidence that during tumor progression, the hyperactivation of tyrosine kinases leads to the continuous activation of downstream signaling cascades that block cellular apoptosis, promote cellular proliferation, and increase the nutrient/waste interchange by enhancing angiogenesis.
Receptor Tyrosine Kinases (RTKs) are single pass transmembrane proteins that account for almost two thirds of the genes coding for tyrosine kinases. RTKs possess a common functional kinase domain that is able to translate extracellular signals into active intracellular cues. Under physiological conditions, these receptors are activated only upon ligand binding [5]. Activation of the kinase is achieved by ligand-binding to the extracellular domain, which induces homo/hetero-dimerization of the receptors [6]. Activated receptors phosphorylate tyrosine residues outside their catalytic domain via cross-phosphorylation. This phosphorylation stabilizes the receptor conformation in an active state and creates phosphotyrosine docking sites for proteins which transduce signals within the cell [7, 8].
In cancer, this mechanism of ligand-dependent activation can be bypassed by (i) overexpression of the RTK, which increases the dynamics of receptor homo/heterodimerization in the absence of the ligand [9–11]; (ii) by activating mutations, which stabilize the receptor active conformation [12]; or (iii) by autocrine stimulation. These mechanisms lead to cell autonomous activation of RTKs that drive proliferative and anti-apoptotic signals, contributing to transformation [7].
Non-Receptor Tyrosine Kinases (NRTKs), the second class of TKs, account for the remaining third of the approximately 90 known TKs and are critical signal transducers. Some examples include the well-known and well-characterized NRTKs Src, JAK, c-Abl and FAK. Interestingly, NRTKs were the first tyrosine kinases discovered [13–16]. Their involvement in cancer can occur through various mechanisms such as overexpression, mutation, and translocation; and therefore, many compounds have been developed attempting to inhibit their activity [17].
Treatments with tyrosine kinase inhibitors (TKIs), in some cases, have given promising results. However, most tumors treated with TKIs became resistant to treatment in a short time [18]. In other words, just as bacteria develop resistance to antibiotics, neoplastic cells can acquire new traits that render them more aggressive and able to survive in the presence of molecular inhibitors.
Clinical experience has shown that only a percentage of patients respond to targeted therapies, even if their tumor expresses the altered target. This primary resistance to treatment is often due to constitutive activation of downstream signal transducers [19–21]. Recently, many reports have evidenced that patients carrying activating mutations in effectors downstream of the targeted molecule account for the majority of the non-responsive patients [22, 23].
Given that many patients are starting to benefit from tyrosine kinase inhibitors, including monoclonal antibodies and small molecule inhibitors, clinicians and basic researchers are now trying to unveil and understand the mechanisms through which neoplastic cells loose their ability to respond to these drugs (also known as secondary resistance or acquired resistance). Luckily, it appears that the majority of the resistance models developed in vitro are predictive of what is observed in vivo and can thus help researchers in identifying and studying this crucial clinical problem.
This review will attempt to provide an updated compendium of cellular modifications that contribute to acquired resistance to TKIs, highlighting the importance of preclinical studies of these drugs.
Targeting Tyrosine Kinases
Many research groups, including ours, have shown that the inhibition of RTKs in neoplastic cells - by administration of monoclonal antibodies, interfering RNAs, and/or small kinase inhibitors (TKIs) - impairs cell proliferation and survival, inducing arrest of cell growth and apoptosis [24–28]. Based on these findings, many pharmaceutical companies have invested in designing or identifying new methods of inhibiting tyrosine kinases.
Small molecule tyrosine kinase inhibitors
Pharmaceutical companies have focused their research on the development of small TKIs, some of which have received the approval of governmental drug administration agencies. Additional file 1 lists some TKIs currently approved or undergoing clinical trials. TKIs are small molecules that inhibit the enzymatic activity of the target protein. Most of these molecules can be categorized into four groups: (i) ATP-competitive inhibitors, which bind predominantly to the ATP-binding site of the kinase when this site is in the active conformation; (ii) inhibitors that recognize and bind to the non-active conformation of the ATP-binding site of the kinase, thus making activation energetically unfavorable; (iii) allosteric inhibitors, that bind outside of the ATP-binding site, modifying the tridimensional structure of the receptor and disrupting the interaction between the ATP and the kinase pocket; and (iv) covalent inhibitors, that bind irreversibly by covalently bonding to the ATP-binding site of the target kinase (reviewed in [29]).
While monoclonal antibody (mAb) therapy is particularly suited for extracellular (membrane-bound or secreted) targets, small-molecule kinase inhibitors are effective against both membrane-bound and intracellular targets. While both therapies have advantages and disadvantages when compared to each other, the major differences between monoclonal antibodies and small TKIs are the modality of administration, the bioavailability and half-life, and the mechanisms of resistance to the therapeutic agents [30–32]. (See comparison table 1).
Monoclonal Antibodies
Immunotherapy is based on the production of humanized monoclonal antibodies (mAbs) that bind with high specificity to secreted proteins or to the extracellular domain of membrane-bound proteins. The use of mAbs relies on the principle that most of the targeted molecules are expressed at higher levels on neoplastic cells, when compared to normal cells, where they play an important role in sustaining cancer progression. So far, there are several mechanisms described by which they exert their therapeutic effects; among them are: binding to the ligand or to the receptor, thus preventing ligand-receptor interaction [33, 34]; disrupting receptor internalization [35], promoting receptor internalization [36], shedding of the extracellular portion of the receptor [36, 37], preventing receptor dimerization and activation [38], and induction of apoptosis [39, 40]. However, it is believed that each mAb acts through more than one mechanism. In addition, evidence has shown that activation of the immune response against the targeted tumor cells, upon recognition of the bound antibody, can also account for their biological activity [41]. Table 2 lists monoclonal antibodies directed again tyrosine kinases currently used in preclinical and clinical studies.
Monoclonal antibodies have been widely used in the clinic and have shown promising results, but unfortunately many patients relapse due to development of mechanisms of resistance. Information obtained from cellular models and relapsed patients has provided insights on how cells adapt to the treatment, by reducing the expression or modifying the structure of the target protein or activating alternative survival pathways [42].
Mechanisms of resistance to TKIs
Genetic modifications
Clinical and in vitro evidence have shown that cells treated with TKIs tend to acquire genetic modifications to overcome the inhibitory effects of these agents. Common mechanisms of resistance include, but are not limited to: point mutations, deletions and amplifications of genomic areas. A schematic summary of the main molecular mechanisms of acquired resistance to small molecules is represented in Figure 1.

Schematic summary of the main molecular mechanisms of acquired resistance to TKIs.
Mutations
Mutations are common and occur frequently in rapidly dividing cancer cells. Point mutations are the most common mechanism of resistance to TKIs. The most frequent types of mutations are those that decrease the affinity of the drug for the target kinase domain, while maintaining its catalytic activity. Other mutations alter the amino acids surrounding the binding site of the drug and decrease the availability of the target region towards the inhibitor, without interfering with ATP binding [29]. Finally, some mutations increase the affinity of the kinase for ATP, decreasing the effectiveness of the ATP-competitive inhibitors [43].
The strongest evidence comes from imatinib, a small tyrosine kinase inhibitor that was found to bind with high affinity to c-Abl kinase. Imatinib is used to treat Chronic Myeloid Leukemia (CML) patients who express a constitutively active c-Abl tyrosine kinase, the BCR-ABL fusion protein. Imatinib abrogates the oncogenic function of BCR-ABL by binding the protein in its inactive state, thus preventing its autophosphorylation and, therefore, blocking the activation of downstream signal transducers. The use of imatinib has improved the life expectancy of CML patients, but major concerns have been raised for this and other TKIs by the rapid development of mechanisms of resistance. The majority of the CML patients in advanced stage (66%) and some in the chronic phase (5%) relapse after imatinib treatment, developing c-Abl dependent and independent mechanisms of resistance [44]. Approximately 30-50% of the relapsed patients acquire point mutations (around 90 distinct point mutations identified so far) that change the conformation of the c-Abl kinase, reducing or abrogating the ability of the compound to bind the c-Abl kinase domain [45–47]. This molecular mechanism of resistance has been supported also by structural studies which have shown that imatinib cannot efficiently interact with the ATP binding pocket in the mutated forms of BCR-ABL. When reports started to show that mutations in the kinase domain of c-Abl were present in relapsed patients, and experimental work showed that the mutant kinase was no longer inhibited by imatinib, second generation inhibitors, such as dasatinib [48], nilotinib [48, 49], sunitinib [50], and bosutinib [51] were designed. These new molecules are able to recognize and bind BCR-ABL in different conformations, and are thus suitable for imatinib-relapsed patients. Dasatinib and nilotinib are able to interact with most of the mutated imatinib-resistant c-Abl forms, with the exception of the T315I mutant that changes the kinase and modifies several contact points between the drug and the kinase, while preserving the kinase activity [43, 52, 53]. The only inhibitor so far that has been proven to inhibit this mutant is the multikinase inhibitor KW-2449 [54]. However, CML patients who used these second generation inhibitors developed resistance by acquiring new mutations in the kinase domain [55].
Why do patients develop these mutations during treatment? There are reports that support the idea that the appearance of mutations in tumors after treatment with a specific TKI is the result of a process of selection of a pre-existing cell population. This theory implies that a small population of the tumor bulk a priori contains the mutation, which confers a primary resistance to these cells, therefore giving them a selective advantage. The bulk tumor mass is thus killed by the drug, allowing cells resistant to the TKI to grow. This theory is supported by the fact that some of these "resistance related mutations" can be found in a small percentage of tumor cells in patients that have not yet undergone targeted therapy [56–59]. On the other hand, other researchers believe that the high dependence of a cell on a specific oncogenic survival pathway forces genomic instability, allowing the induction of mutations that confer resistance to the inhibitor. This genomic instability can induce mutations either in the drug target or in other signal transducers that activate alternative pathways able to sustain cell viability. This theory has been supported by groups who have induced resistance to TKIs in imatinib-sensible CML cell lines cloned by limiting dilution; they have found the appearance of BCR-ABL gene amplification and of point mutations in the kinase domain that were not present in the original cells [60].
Further studies revealed that imatinib also binds with high affinity to the cKIT and PDGFR kinases, frequently activated in Gastrointestinal Stromal Tumors (GIST) [61]. GISTs are the first solid tumors in which a tyrosine kinase inhibitor was used as standard care. As these tumors often display mutations in the tyrosine kinase receptors cKIT and PDGFR, imatinib was used to inhibit their activity [62]. Like CML patients, 50-70% of GIST patients treated with imatinib develop secondary mutations within the cKIT gene, conferring a reduced drug binding affinity but still retaining the kinase activity [63, 64]. To suppress the kinase activity of the resistant cKIT mutants, sunitinib was developed. As previously observed in patients treated with other inhibitors of second generation, imatinib-resistant GIST patients treated with sunitinib developed new mutations that made them again resistant to the new drug [65].
Gefitinib and erlotinib are small molecule TKIs targeting the Epidermal Growth Factor Receptor (EGFR) that have been used to treat tumors where this RTK is known to be altered. In particular, they have been used to treat non-small cell lung carcinomas (NSCLC) where EGFR is frequently overexpressed or activated due to point mutations [66]. According to a compendium of studies that include 1170 patients, more than 70% of NSCLCs with EGFR mutations respond to EGFR-TKIs, whereas only 10% of tumors without EGFR mutations do so. Unfortunately, upon treatment of these patients with gefitinib and erlotinib, two major mechanisms of resistance have been observed. The first is the appearance of a "resistance" point mutation in the kinase domain (T790M), observed in 50% of the gefitinib-resistant patients [67]. This mutation increases the affinity for ATP and weakens the affinity for ATP-competitive inhibitors [22, 68]. On the other hand, the second mechanism is the activation of an alternative oncogene able to compensate for the inhibited signaling pathways [69, 70].
Interestingly, in vitro models of acquired resistance to gefitinib, obtained by exposing gefitinib-sensitive cells to increasing concentrations of the drug, led to the appearance of the same mutations identified in patients. This has allowed scientists to study the mechanisms through which these mutations modulate sensitivity to the drug [71–75]. Lapatinib is another EGFR inhibitor, recently approved for treatment of breast cancer. This inhibitor has been designed to block receptor signaling by binding to the ATP-binding pocket of EGFR and ERBB2 kinase domains, thus preventing phosphorylation and subsequent downstream signaling from these two receptors [76]. Using a randomly mutagenized ERBB2 library in vitro, Trowe et al. were able to identify 12 mutations in the kinase domain of ERBB2 that could confer resistance to the inhibitor [77]. Moreover, this same work showed that a new generation inhibitor, EXEL-7647, is still active on all the mutants.
Similarly, activating mutations in the FLT3 RTK occur frequently in Acute Myelogenous Leukemia (AML). When AML patients were treated with PKC412, a staurosporin derivative able to inhibit FLT3's kinase activity, patients rapidly developed point mutations in the kinase domain of FLT3 that rendered the kinase less accessible to the inhibitor [78]. These same mutations had been previously foreseen by a computational predictive analysis and confirmed by in vitro data when Cools et al. identified possible mutations in residues conferring a high level of resistance to small molecules [79]. Recently, another cellular model has predicted new point mutations that confer resistance to FLT3 inhibitors such as SU5614, PKC412, and sorafenib. As the different FLT3 kinase inhibitors generated distinct, non-overlapping mutational profiles, the authors propose that a combination of FLT3 inhibitors might be useful to prevent the appearance of FLT3 resistance mutations [80].
As previously mentioned, another clinically approved TKI currently in use is sorafenib. This small molecular multikinase inhibitor, primarily targets BRAF and can inhibit several other TKs such as PDGFR, VEGFR 1-2, FLT3, and cKIT [81]. This multi-target drug possesses anti-tumoral and anti-angiogenic properties due to its broad blocking activity. The use of sorafenib, just as with other small molecule inhibitors, has caused so far a variety of mutations in PDGFR [82], FLT3 [80], and BRAF [83] that confer resistance to the treatment.
Gene Amplification
Gene amplification is a major mechanism of oncogenic activation [84]. Preclinical and clinical data have shown that the presence of either activating mutations in the kinase domain or gene amplification correlate with the best response to TKI [84, 85]. Unfortunately, the selective pressure of the drug can drive further amplification of the target gene, thus leading to additional overexpression of the encoded protein. This idea originates from in vitro studies that have shown that highly amplified oncogenes are located in extrachromosomal acentromeric double minutes, and such cells undergoing "oncogenic stress" may undergo further gains due to advantageous unsymmetrical nuclear division [86]. These gains alter the stoichiometry of the drug-target interaction in favor of the target and result in its inefficient inhibition. This event has been observed in CML relapsed patients treated with imatinib, who displayed an increase in the BCR-ABL gene copy number [87]. In these patients, an increase in drug dosage is usually sufficient to restore responsiveness to the treatment. This same mechanism of resistance had been observed in an in vitro model where a CML cell line was treated for a long period of time with imatinib [42]. Likewise, the emergence of amplification of the target gene as a mechanism of resistance has been observed in two other cases where resistance cells amplified EGFR [88] or FTL3 [89] in response to inhibitors.
Another way through which gene amplification can mediate resistance to treatment is via amplification of genes that encode for critical transducers driving signaling pathways that can compensate for the signals lost due to target inhibition [69]. A notable example is the amplification of the MET gene, encoding for the receptor tyrosine kinase for hepatocyte growth factor, in a percentage of gefitinib-relapsed patients affected by NSCLC. These results perfectly correlated with those obtained in in vitro studies after treating sensitive NSCLC cell lines with progressively increasing doses of gefitinib or other EGFR inhibitors [57, 58, 70, 90, 91]. In these experiments, MET overexpression led to its constitutive activation by a ligand-independent mechanism, which later resulted in advantageous interactions with other EGFR family members, mainly ERBB3, and activation of downstream signals. Inhibition of MET, in this context, restored sensitivity to EGFR inhibitors [70].
Genomic Deletions
Other genomic alterations frequently observed upon TKI treatment are deletions. Khorashad and collaborators performed a genome-wide study comparing DNA samples from CML patients prior to imatinib treatment and after relapse. CGH analyses for all patients revealed that 28% of the copy number alterations were genomic deletions. Among the genes that were most frequently altered were those involved in the control of the MAPK signaling pathway [92].
Among the genes that are frequently deleted in human cancers are those encoding microRNAs (miRNAs). MiRNAs have emerged as a novel class of regulatory genes involved in human cancer [93, 94]. Lacking the ability to encode a protein, these single-stranded miRNAs bind to imperfectly complementary sequences of encoding mRNAs, causing these mRNA sequences to be silenced or degraded, resulting in reduced levels of the protein encoded by the mRNA. Many reports have highlighted the relevance of these non-coding RNA's in human cancer, where they are frequently altered, more often as consequence of their deletion [95]. Various groups have reported cases where the deletion of miRNA regions has led to overexpression of the targeted RTKs, due to lack of down-regulation [95–97]. In this context, Seike and collaborators recently correlated high EGFR activation with high expression of mir-21 both in NSCLC patient samples and cell lines. They report that inhibition of EGFR by the small molecule AG1478 reduced the levels of this miRNA, concluding that the activation status of the receptor modulates the expression of this anti-apoptotic miRNA [98]. As it considered a growing field of interest, various groups have reported that miRNA expression can mediate resistance to different types of chemotherapy [99–102] (reviewed in [103]), and it is very likely that quite soon miRNAs will also be found to play a role in mediating resistance to TKIs.
Modifications of protein expression
Cells seem to possess a broad repertoire of adaptive reactions that enable them to survive in many adverse conditions. One of the adaptive traits is the overexpression or the repression of genes that sustain cell viability [104]. Mahon et al. recently demonstrated that nilotinib-resistant CML cell lines were able to upregulate the expression of BCR-ABL, thus overcoming the inhibitory threshold of nilotinib [105]. Although this and other similar works lack evidence that the overexpression of the target protein is not due to gene amplification (also known mechanism of resistance to BCR-ABL TKI), this can be considered as a new mechanism of resistance.
This last response does not involve genetic alterations, but simply changes in gene expression, due to microenvironmental stress or to epigenetic modifications. It is known that the use of TKIs can lead to reduced blood flow, which in turn increases the incidence of hypoxic areas [106]. Moreover, hypoxia is known to upregulate HIF-1a, a protein that can promote the expression of many genes including the RTK MET, which is capable of sustaining the MAPK and PI3K survival pathways [107].
Likewise, epigenetic changes can also contribute to TKI resistance. For example, Noro et al. reported an in vitro model where lung cancer cells resistant to gefitinib displayed hypermethylation of the PTEN gene promoter; exogenous re-expression of this enzyme restores senstivity to the EGFR inhibitor [108].
Activation of alternative pathways
Some cells can replace the lack of signal due to target inhibition by activating alternative pathways. The EGFR family of receptors has been shown to develop mechanisms of resistance by modifying the expression of several downstream effectors. For example, Pandya and collaborators developed a cellular model where colorectal carcinoma HCT116 cells, which depend on ERBB2 activity, lose their sensitivity to lapatinib. The major mechanism of resistance observed was the increased expression of MCL-1, and the decreased expression and activity of BAX and BAK [109], altogether leading to decreased apoptotic responses. Another proposed mechanism of resistance was reported by Xia et al. who showed that lapatinib-resistant breast cancer cells and lapatinib-treated patients displayed an increased level of the Estrogen Receptor and the transcription factor FoxoA3 [110]. Another example was recently reported by Turke et al. where EGFR-dependent cells stimulated with MET's ligand, HGF, were resistant both in vivo and in vitro, and such effect could be blocked by the use of MET inhibitors [57]. In a similar manner, McDermott et al. reported that MET-dependent NSCLC cells activate EGFR as a mechanism of resistance to PF2341066 (an irreversible MET kinase inhibitor) using an increasing dose resistant cellular model [111].
Another mechanism of resistance that was reported in NSCLC patients and in cell lines resistant to gefitinib treatment is the cross-talk between the EGFR/ERBB2 receptors and the IGF-1R receptor [112–114]. This mechanism of resistance relies on the fact that cells utilize IGF-1R to activate survival pathways that are able to promote growth [115]. One report shows that a prostate cancer cell line which became resistant to gefitinib displayed an increase of IGFII mRNA and IGF-1R protein phosphorylation [112, 113]. Moreover, it was also published that a gefitinib-resistant lung squamous carcinoma cell line lost the production of IGFBP3-4 when compared to the parental cells; re-expression of these proteins restored the sensitivity to gefitinib's cytostatic effect [116].
The activation of an alternative kinase is known to overcome the inhibitory effects of small molecules. For example, GIST cells resistant to imatinib exhibited increased levels of the AXL receptor, that could in turn activate the AKT pathway and thus overcome c-KIT inhibition [97, 117]. Two different groups have recently shown that in a cellular model of CML, TKI-resistant cells display activation of the Src kinase LYN; inhibition of this kinase by the use of dasatinib restores sensitivity to imatinib or nilotinib [105, 118].
In a similar manner, the human myelomonoblastic cell line MV4-11, generated to be resistant to PKC412, displayed an up-regulation of anti-apoptotic genes and down-regulation of proapoptotic signals as well as genes that are involved in normal and malignant hematopoiesis [89].
Recently, Huang et al. reported that tumor xenografts resistant to sunitinib secreted higher amounts of IL-8 (proangiogenic factor known to be induced by several key regulators of cell survival and hypoxia) which at the same time positively correlated with a higher tumor vessel density [119, 120].
Another commonly observed mechanism of resistance to TKI is the overexpression of survivin a member of the inhibitor of apoptosis family, encoded by the BIRC5 gene [110, 121]. This cancer therapy candidate gene is overexpressed in a large variety of human tumors [122–125] and its expression is absent in terminally differentiated [126, 127]. Survivin is known to inhibit caspase activation, and therefore, leading to negatively regulate apoptosis or programmed cell death, and it has been correlated with both accelerated relapse and chemotherapy resistance [128]. Xia et al. have demonstrated that overexpression of surviving can mediate resistance to lapatinib; such finding was observed by generating lapatinib-resistant breast cancer cells in vitro and correlating clinical observations [110].
Mechanisms of Resistance related to drug influx/efflux
There are many mechanisms implicated in the decrease of the effective intracellular concentration of a drug, leading to lack of response to treatment. Among the most important resistance mechanisms are: increased drug influx/efflux and drug plasma sequestration. Other factors that can contribute to decreased drug delivery to tumors include irregular blood flow, defects in the structure and permeability of tumor vasculature and drug diffusion in the interstitium.
The occurrence of multidrug resistance (MDR) is a very frequent cause of failure of chemotherapeutic treatment in cancer patients. MDR proteins are transmembrane pumps responsible for the active efflux of a broad range of structurally unrelated molecules. This efflux can occur despite considerable concentration gradients at the expense of ATP depletion, resulting in decreased intracellular drug accumulation [129]. It is conceivable that TKIs may inhibit the function of ATP-binding cassette (ABC) transporters by recognizing their ATP-binding sites. In fact, some of these small molecules such as cediranib, lapatinib, and sunitinib have proven to be effective in reversing MDR associated to chemotherapeutics, by directly inhibiting the transport function of some ABC members. This ability renders them useful options for cancer combinational therapy [130, 131]. The initial success of molecularly targeted therapies raised hope that newly developed agents would evade the general mechanisms of resistance that have reduced the efficacy of traditional anticancer drugs. However, ABC transporters related to MDR have emerged as key factors that regulate the intracellular concentrations of many small-molecule inhibitors. Drug transporters may be overexpressed in cancer cells, reducing intracellular drug concentrations, and may allow the evolution of point mutations that confer stronger drug resistance [132].
Mahone and collaborators demonstrated that imatinib-resistant cell lines overexpressed the P-glycoprotein (P-gp) efflux pump [133]. This concept was reinforced when imatinib sensitivity was restored when P-gp pumps were blocked by different inhibitors [134, 135], or silenced using RNAi [136, 137]. All this data indicates that P-gp is a likely candidate contributing to imatinib resistance, and some in vitro data suggests that this may also be true for resistance to nilotinib [105]. Dasatinib and sunitinib have been shown to be a substrate of both efflux proteins, ABCB1 and ABCG2 [138, 139]. ABCG2 has also been shown to bind gefitinib with high affinity, causing an active extrusion of the inhibitor and thus preventing its biological activity [140].
In addition, multiple reports have provided evidence that deregulation of the organic cation transporter hOCT1 can impede the influx of imatinib. Using hOCT inhibitors on different imatinib-sensitive CML cells caused a reduced uptake of imatinib [141]. This finding was further supported by clinical data showing that patients who display a minimal response to imatinib also express a significantly lower amount of hOCT [142, 143]. Therefore, intracellular drug levels depend in part on the differential expression of influx and efflux transporters, which are determinants of TKI resistance.
Another method by which tumors bypass the inhibitory effects of TKI is by the sequestration of such drugs by plasma proteins, such as the plasma protein-1 acid glycoprotein (AGP). It has been shown in vitro and in vivo that AGP binds to imatinib, and this binding decreases imatinib's ability to inhibit c-ABL in a dose-dependent manner [144], findings supported by clinical data [145, 146].
Mechanisms of resistance to monoclonal antibodies
Although monoclonal antibodies have given very good results in the clinic, the emergence of resistance is also frequently observed upon treatment with these agents. Several mechanisms of resistance have been observed in preclinical and clinical studies, mostly with antibodies that have already undergone FDA approval. In the case of monotherapy, preexistence of mutations in the MAPK or PI3K signaling pathways is one of the major causes of primary or intrinsic resistance. In 2009, the American Society of Clinical Oncology suggested that metastatic colorectal cancer (CRC) patients who displayed an alteration in codon 12 or 13 of KRAS should not be considered for monoclonal therapy [147]. This decision was based on multiple studies that have shown that activating mutations in KRAS [148–150], PIK3CA [19], BRAF [151] and loss of expression of PTEN [152–156] correlated negatively with cetuximab or panitumumab response (reviewed in [157]).
Patients undergoing monotherapy are also prone to develop secondary or acquired resistance to such treatment. So far, no mAb therapy has given rise to any point mutation in the target receptor or rearrangements in genomic regions. The mechanisms described up to now typically involve variations in protein expression. At least five modifications of this type have been shown to contribute to resistance to mAbs:
-
(i)
Overexpression and aberrant phosphorylation of alternative RTKs attempting to overcome the inhibition of the targeted protein. In 2008, Wheeler et al. generated NSCLC and HNSCC cetuximab-resistant cell lines, such resistance was mediated by the increased expression of ERBB2, ERBB3, and MET which can interact with other EGFR family members contributing to their activation [35]. In a similar way, Lu et al. and Shattuck et al. have shown that cells can overcome trastuzumab inhibition by the activation of IGF-1R and MET, respectively [114, 158–161].
-
(ii)
The second known protein modification is expression of receptor variants. Sok and collaborators demonstrated that a mutant variant of EGFR (EGFRvIII), which lacks the ligand binding domain, is expressed in more than 42% of HNSCC. In their experiments, overexpression of EGFRvIII in HNSCC cells decreased in the inhibitory response to cetuximab [162].
-
(iii)
The third protein modification involves the targeted protein; in this type of resistance, cells display an increased expression of the target receptor. Reports have shown that NSCLC cell lines resistant to cetuximab display an increase in EGFR protein levels due to a defective deregulation in the degradation pathways [35, 163].
-
(iv)
Activation of alternative pathways is another mechanism of resistance. It has been observed that cells resistant to either cetuximab or trastuzumab can develop a dependency on new signaling pathways either by triggering the same biological effects by interaction with other EGFR family members [35, 164], or by association with other kinases such as Src [165]. Valabrega et al. reported that TGFα (an EGFR ligand) overexpression can contribute to resistance [166]. It is interesting to note that the overexpression of ligands is not a rare event, since patients and cell lines resistant to bevacizumab (a VEGF blocking antibody) cause tumor cells to secrete additional angiogenic factors (FGF [167], PGF [168], members of the notch ligand/receptor family [169]) to compensate for the lack of VEGF signaling [170, 171].
Lastly, (v) the lack of interaction between the target and the mAb due to steric hindrance caused by the formation of complexes with other cell surface proteins, such as in the case of resistance to trastuzumab. It is known that the expression of MUC4, a membrane-associated mucin that contributes to the masking of membrane proteins, decreases the amount of trastuzumab that can bind to ERBB2 [172] When MUC4 was silenced in trastuzumab resistant cells, cells were once again sensitive to the mAb [173].
Conclusions
New clinical and laboratory studies have suggested that multi-targeting approaches against neoplastic cells could help to increase patient survival and, possibly, reduce the emergence of cells resistant to single-target inhibitors [174]. This increased activity will have to be balanced by the expected increased toxicity due to the association of the drugs. Moreover, combination mAbs and multi-target small molecules could be also a very promising therapeutic approach [175, 176].
Accumulating experimental and clinical evidences have supported the idea that targeted therapy should be reassessed. In particular, we should keep in mind that tumors are the result of multiple genetic lesions. Clinicians and researchers should not underestimate the capacity of tumors to easily adapt to new stress conditions, therefore inducing or selecting those cells that can better survive in the presence of an inhibitor.
Abbreviations
- CML:
-
Chronic myelogenous leukemia
- CRC:
-
colorectal cancer
- EGFR:
-
epidermal growth factor receptor
- GIST:
-
gastrointestinal stromal tumors
- HGF:
-
hepatocyte growth factor
- HNSCC:
-
head and neck squamous cell carcinoma
- mAb:
-
monoclonal antibody
- MET:
-
hepatocyte growth factor receptor
- NSCLC:
-
non-small cell lung carcinoma
- NRTK(s):
-
non-receptor tyrosine kinase(s)
- RTK(s):
-
receptor tyrosine kinase(s)
- TGFα:
-
transforming growth factor alpha
- TKI(s):
-
tyrosine kinase inhibitor(s).
References
Sawyers C: Targeted cancer therapy. Nature. 2004, 432: 294-297. 10.1038/nature03095
Shawver LK, Slamon D, Ullrich A: Smart drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell. 2002, 1: 117-123. 10.1016/S1535-6108(02)00039-9
Hubbard SR: Protein tyrosine kinases: autoregulation and small-molecule inhibition. Curr Opin Struct Biol. 2002, 12: 735-741. 10.1016/S0959-440X(02)00383-4
Robertson SC, Tynan J, Donoghue DJ: RTK mutations and human syndromes: when good receptors turn bad. Trends Genet. 2000, 16: 368- 10.1016/S0168-9525(00)02077-1
Robinson DR, Wu YM, Lin SF: The protein tyrosine kinase family of the human genome. Oncogene. 2000, 19: 5548-5557. 10.1038/sj.onc.1203957
Yarden Y, Schlessinger J: Self-phosphorylation of epidermal growth factor receptor: evidence for a model of intermolecular allosteric activation. Biochemistry. 1987, 26: 1434-1442. 10.1021/bi00379a034
Paul MK, Mukhopadhyay AK: Tyrosine kinase - Role and significance in Cancer. Int J Med Sci. 2004, 1: 101-115.
Bennasroune A, Gardin A, Aunis D, Cremel G, Hubert P: Tyrosine kinase receptors as attractive targets of cancer therapy. Crit Rev Oncol Hematol. 2004, 50: 23-38. 10.1016/j.critrevonc.2003.08.004
Worthylake R, Opresko LK, Wiley HS: ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J Biol Chem. 1999, 274: 8865-8874. 10.1074/jbc.274.13.8865
Wang R, Kobayashi R, Bishop JM: Cellular adherence elicits ligand-independent activation of the Met cell-surface receptor. Proc Natl Acad Sci USA. 1996, 93: 8425-8430. 10.1073/pnas.93.16.8425
Giordano S, Ponzetto C, Di Renzo MF, Cooper CS, Comoglio PM: Tyrosine kinase receptor indistinguishable from the c-met protein. Nature. 1989, 339: 155-156. 10.1038/339155a0
Weiner DB, Liu J, Cohen JA, Williams WV, Greene MI: A point mutation in the neu oncogene mimics ligand induction of receptor aggregation. Nature. 1989, 339: 230-231. 10.1038/339230a0
Chang YM, Kung HJ, Evans CP: Nonreceptor tyrosine kinases in prostate cancer. Neoplasia. 2007, 9: 90-100. 10.1593/neo.06694
Johnson FM, Gallick GE: SRC family nonreceptor tyrosine kinases as molecular targets for cancer therapy. Anticancer Agents Med Chem. 2007, 7: 651-659. 10.2174/187152007784111278
Murray MJ, Shilo BZ, Shih C, Cowing D, Hsu HW, Weinberg RA: Three different human tumor cell lines contain different oncogenes. Cell. 1981, 25: 355-361. 10.1016/0092-8674(81)90054-4
Shilo BZ, Weinberg RA: Unique transforming gene in carcinogen-transformed mouse cells. Nature. 1981, 289: 607-609. 10.1038/289607a0
Vlahovic G, Crawford J: Activation of tyrosine kinases in cancer. Oncologist. 2003, 8: 531-538. 10.1634/theoncologist.8-6-531
Engelman JA, Settleman J: Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008, 18: 73-79. 10.1016/j.gde.2008.01.004
Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L: PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009, 69: 1851-1857. 10.1158/0008-5472.CAN-08-2466
de Reynies A, Boige V, Milano G, Faivre J, Laurent-Puig P: KRAS mutation signature in colorectal tumors significantly overlaps with the cetuximab response signature. J Clin Oncol. 2008, 26: 2228-2230. author reply 2230-2221., 10.1200/JCO.2007.15.9186
Bonomi PD, Buckingham L, Coon J: Selecting patients for treatment with epidermal growth factor tyrosine kinase inhibitors. Clin Cancer Res. 2007, 13: s4606-4612. 10.1158/1078-0432.CCR-07-0332
Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE: KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2: e17- 10.1371/journal.pmed.0020017
Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N, Zhang T, Marrano P, Whitehead M, Squire JA: Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2008, 26: 4268-4275. 10.1200/JCO.2007.14.8924
Corso S, Migliore C, Ghiso E, De Rosa G, Comoglio PM, Giordano S: Silencing the MET oncogene leads to regression of experimental tumors and metastases. Oncogene. 2008, 27: 684-693. 10.1038/sj.onc.1210697
Normanno N, Bianco C, Damiano V, de Angelis E, Selvam MP, Grassi M, Magliulo G, Tortora G, Bianco AR, Mendelsohn J: Growth inhibition of human colon carcinoma cells by combinations of anti-epidermal growth factor-related growth factor antisense oligonucleotides. Clin Cancer Res. 1996, 2: 601-609.
McDermott U, Sharma SV, Dowell L, Greninger P, Montagut C, Lamb J, Archibald H, Raudales R, Tam A, Lee D: Identification of genotype-correlated sensitivity to selective kinase inhibitors by using high-throughput tumor cell line profiling. Proc Natl Acad Sci USA. 2007, 104: 19936-19941. 10.1073/pnas.0707498104
Vigna E, Pacchiana G, Mazzone M, Chiriaco C, Fontani L, Basilico C, Pennacchietti S, Comoglio PM: "Active" cancer immunotherapy by anti-Met antibody gene transfer. Cancer Res. 2008, 68: 9176-9183. 10.1158/0008-5472.CAN-08-1688
Montagut C, Settleman J: Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009, 283: 125-134. 10.1016/j.canlet.2009.01.022
Zhang J, Yang PL, Gray NS: Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009, 9: 28-39. 10.1038/nrc2559
Shih YC, Halpern MT: Economic evaluations of medical care interventions for cancer patients: how, why, and what does it mean?. CA Cancer J Clin. 2008, 58: 231-244.
Imai K, Takaoka A: Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006, 6: 714-727. 10.1038/nrc1913
Slamon DJ: The FUTURE of ErbB-1 and ErbB-2 pathway inhibition in breast cancer: targeting multiple receptors. Oncologist. 2004, 9 (Suppl 3): 1-3. 10.1634/theoncologist.9-suppl_3-1
Song JY, Lee SW, Hong JP, Chang SE, Choe H, Choi J: Epidermal growth factor competes with EGF receptor inhibitors to induce cell death in EGFR-overexpressing tumor cells. Cancer Lett. 2009, 283: 135-142. 10.1016/j.canlet.2009.03.034
Guin S, Yao HP, Wang MH: RON Receptor Tyrosine Kinase as a Target for Delivery of Chemodrugs by Antibody Directed Pathway for Cancer Cell Cytotoxicity. Mol Pharm. 2010, 7 (2): 386-97. 10.1021/mp900168v
Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT, Harari PM: Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008, 27: 3944-3956. 10.1038/onc.2008.19
Petrelli A, Circosta P, Granziero L, Mazzone M, Pisacane A, Fenoglio S, Comoglio PM, Giordano S: Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity. Proc Natl Acad Sci USA. 2006, 103: 5090-5095. 10.1073/pnas.0508156103
Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J: Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res. 2001, 61: 4744-4749.
Hudziak RM, Lewis GD, Winget M, Fendly BM, Shepard HM, Ullrich A: p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol Cell Biol. 1989, 9: 1165-1172.
Meira DD, Nobrega I, de Almeida VH, Mororo JS, Cardoso AM, Silva RL, Albano RM, Ferreira CG: Different antiproliferative effects of matuzumab and cetuximab in A431 cells are associated with persistent activity of the MAPK pathway. Eur J Cancer. 2009, 45: 1265-1273. 10.1016/j.ejca.2008.12.012
Petrelli A, Valabrega G: Multitarget drugs: the present and the future of cancer therapy. Expert Opin Pharmacother. 2009, 10: 589-600. 10.1517/14656560902781907
Kurai J, Chikumi H, Hashimoto K, Yamaguchi K, Yamasaki A, Sako T, Touge H, Makino H, Takata M, Miyata M: Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin Cancer Res. 2007, 13: 1552-1561. 10.1158/1078-0432.CCR-06-1726
Nielsen DL, Andersson M, Kamby C: HER2-targeted therapy in breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Cancer Treat Rev. 2009, 35: 121-136. 10.1016/j.ctrv.2008.09.003
Tanaka R, Kimura S: Abl tyrosine kinase inhibitors for overriding Bcr-Abl/T315I: from the second to third generation. Expert Rev Anticancer Ther. 2008, 8: 1387-1398. 10.1586/14737140.8.9.1387
Hochhaus A, La Rosee P: Imatinib therapy in chronic myelogenous leukemia: strategies to avoid and overcome resistance. Leukemia. 2004, 18: 1321-1331. 10.1038/sj.leu.2403426
Melo JV, Chuah C: Resistance to imatinib mesylate in chronic myeloid leukaemia. Cancer Lett. 2007, 249: 121-132. 10.1016/j.canlet.2006.07.010
Litzow MR: Imatinib resistance: obstacles and opportunities. Arch Pathol Lab Med. 2006, 130: 669-679.
Branford S: Chronic myeloid leukemia: molecular monitoring in clinical practice. Hematology Am Soc Hematol Educ Program. 2007, 2007: 376-383.
O'Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, Cowan-Jacob SW, Lee FY, Heinrich MC, Deininger MW, Druker BJ: In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005, 65: 4500-4505. 10.1158/0008-5472.CAN-05-0259
Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E: Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005, 7: 129-141. 10.1016/j.ccr.2005.01.007
O'Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A: SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003, 101: 3597-3605. 10.1182/blood-2002-07-2307
Golas JM, Arndt K, Etienne C, Lucas J, Nardin D, Gibbons J, Frost P, Ye F, Boschelli DH, Boschelli F: SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003, 63: 375-381.
Pricl S, Fermeglia M, Ferrone M, Tamborini E: T315I-mutated Bcr-Abl in chronic myeloid leukemia and imatinib: insights from a computational study. Mol Cancer Ther. 2005, 4: 1167-1174. 10.1158/1535-7163.MCT-05-0101
Lee TS, Potts SJ, Kantarjian H, Cortes J, Giles F, Albitar M: Molecular basis explanation for imatinib resistance of BCR-ABL due to T315I and P-loop mutations from molecular dynamics simulations. Cancer. 2008, 112: 1744-1753. 10.1002/cncr.23355
Shiotsu Y, Kiyoi H, Ishikawa Y, Tanizaki R, Shimizu M, Umehara H, Ishii K, Mori Y, Ozeki K, Minami Y: KW-2449, a novel multi-kinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation. Blood. 2009, 114: 1607-1617. 10.1182/blood-2009-01-199307
Tuma RS: With targeted drugs, chronic myelogenous leukemia therapy may follow HIV's model. J Natl Cancer Inst. 2007, 99: 192-194. 10.1093/jnci/djk073
Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, Lai JL, Philippe N, Facon T, Fenaux P, Preudhomme C: Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002, 100: 1014-1018. 10.1182/blood.V100.3.1014
Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L: Preexistence and Clonal Selection of MET Amplification in EGFR Mutant NSCLC. Cancer Cell. 2010, 17: 77-88. 10.1016/j.ccr.2009.11.022
Bachleitner-Hofmann T, Sun MY, Chen CT, Tang L, Song L, Zeng Z, Shah M, Christensen JG, Rosen N, Solit DB, Weiser MR: HER kinase activation confers resistance to MET tyrosine kinase inhibition in MET oncogene-addicted gastric cancer cells. Mol Cancer Ther. 2008, 7: 3499-3508. 10.1158/1535-7163.MCT-08-0374
Kreuzer KA, Le Coutre P, Landt O, Na IK, Schwarz M, Schultheis K, Hochhaus A, Dorken B: Preexistence and evolution of imatinib mesylate-resistant clones in chronic myelogenous leukemia detected by a PNA-based PCR clamping technique. Ann Hematol. 2003, 82: 284-289. 10.1007/s00277-003-0690-5
Ricci C, Scappini B, Divoky V, Gatto S, Onida F, Verstovsek S, Kantarjian HM, Beran M: Mutation in the ATP-binding pocket of the ABL kinase domain in an STI571-resistant BCR/ABL-positive cell line. Cancer Res. 2002, 62: 5995-5998.
de Silva CM, Reid R: Gastrointestinal stromal tumors (GIST): C-kit mutations, CD117 expression, differential diagnosis and targeted cancer therapy with Imatinib. Pathol Oncol Res. 2003, 9: 13-19. 10.1007/BF03033708
Braconi C, Bracci R, Cellerino R: Molecular targets in Gastrointestinal Stromal Tumors (GIST) therapy. Curr Cancer Drug Targets. 2008, 8: 359-366. 10.2174/156800908785133169
Burgess MR, Sawyers CL: Treating imatinib-resistant leukemia: the next generation targeted therapies. ScientificWorldJournal. 2006, 6: 918-930. 10.1100/tsw.2006.184
Sleijfer S, Wiemer E, Seynaeve C, Verweij J: Improved insight into resistance mechanisms to imatinib in gastrointestinal stromal tumors: a basis for novel approaches and individualization of treatment. Oncologist. 2007, 12: 719-726. 10.1634/theoncologist.12-6-719
Loughrey MB, Waring PM, Dobrovic A, Demetri G, Kovalenko S, McArthur G: Polyclonal resistance in gastrointestinal stromal tumor treated with sequential kinase inhibitors. Clin Cancer Res. 2006, 12: 6205-6206. author reply 6206-6207., 10.1158/1078-0432.CCR-06-1079
Uramoto H, Mitsudomi T: Which biomarker predicts benefit from EGFR-TKI treatment for patients with lung cancer?. Br J Cancer. 2007, 96: 857-863. 10.1038/sj.bjc.6603665
Nguyen KS, Kobayashi S, Costa DB: Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer. 2009, 10: 281-289. 10.3816/CLC.2009.n.039
Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H: Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2: e73- 10.1371/journal.pmed.0020073
Pillay V, Allaf L, Wilding AL, Donoghue JF, Court NW, Greenall SA, Scott AM, Johns TG: The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia. 2009, 11: 448-458.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J: MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007, 316: 1039-1043. 10.1126/science.1141478
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG: Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004, 350: 2129-2139. 10.1056/NEJMoa040938
Mulloy R, Ferrand A, Kim Y, Sordella R, Bell DW, Haber DA, Anderson KS, Settleman J: Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007, 67: 2325-2330. 10.1158/0008-5472.CAN-06-4293
Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, Naumov GN, Yeap BY, Jarrell E, Sun J: Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006, 116: 2695-2706. 10.1172/JCI28656
Haber DA, Bell DW, Sordella R, Kwak EL, Godin-Heymann N, Sharma SV, Lynch TJ, Settleman J: Molecular targeted therapy of lung cancer: EGFR mutations and response to EGFR inhibitors. Cold Spring Harb Symp Quant Biol. 2005, 70: 419-426. 10.1101/sqb.2005.70.043
Godin-Heymann N, Ulkus L, Brannigan BW, McDermott U, Lamb J, Maheswaran S, Settleman J, Haber DA: The T790M "gatekeeper" mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol Cancer Ther. 2008, 7: 874-879. 10.1158/1535-7163.MCT-07-2387
Nelson MH, Dolder CR: Lapatinib: a novel dual tyrosine kinase inhibitor with activity in solid tumors. Ann Pharmacother. 2006, 40: 261-269. 10.1345/aph.1G387
Trowe T, Boukouvala S, Calkins K, Cutler RE, Fong R, Funke R, Gendreau SB, Kim YD, Miller N, Woolfrey JR: EXEL-7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin Cancer Res. 2008, 14: 2465-2475. 10.1158/1078-0432.CCR-07-4367
Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, Brandts C, Serve H, Roesel J, Giles F: Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006, 107: 293-300. 10.1182/blood-2005-06-2469
Cools J, Mentens N, Furet P, Fabbro D, Clark JJ, Griffin JD, Marynen P, Gilliland DG: Prediction of resistance to small molecule FLT3 inhibitors: implications for molecularly targeted therapy of acute leukemia. Cancer Res. 2004, 64: 6385-6389. 10.1158/0008-5472.CAN-04-2148
von Bubnoff N, Engh RA, Aberg E, Sanger J, Peschel C, Duyster J: FMS-Like Tyrosine Kinase 3-Internal Tandem Duplication Tyrosine Kinase Inhibitors Display a Nonoverlapping Profile of Resistance Mutations In vitro. Cancer Res. 2009, 69: 3032. 10.1158/0008-5472.CAN-08-2923
Hiles JJ, Kolesar JM: Role of sunitinib and sorafenib in the treatment of metastatic renal cell carcinoma. Am J Health Syst Pharm. 2008, 65: 123-131. 10.2146/ajhp060661
Lierman E, Michaux L, Beullens E, Pierre P, Marynen P, Cools J, Vandenberghe P: FIP1L1-PDGFRalpha D842V, a novel panresistant mutant, emerging after treatment of FIP1L1-PDGFRalpha T674I eosinophilic leukemia with single agent sorafenib. Leukemia. 2009, 23 (5): 845-51. 10.1038/leu.2009.2
Kloos RT, Ringel MD, Knopp MV, Hall NC, King M, Stevens R, Liang J, Wakely PE, Vasko VV, Saji M: Phase II Trial of Sorafenib in Metastatic Thyroid Cancer. J Clin Oncol. 2009, 27 (10): 1675-84. 10.1200/JCO.2008.18.2717
Lockwood WW, Coe BP, Williams AC, MacAulay C, Lam WL: Whole genome tiling path array CGH analysis of segmental copy number alterations in cervical cancer cell lines. Int J Cancer. 2007, 120: 436-443. 10.1002/ijc.22335
Smolen GA, Sordella R, Muir B, Mohapatra G, Barmettler A, Archibald H, Kim WJ, Okimoto RA, Bell DW, Sgroi DC: Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci USA. 2006, 103: 2316-2321. 10.1073/pnas.0508776103
Hellman A, Zlotorynski E, Scherer SW, Cheung J, Vincent JB, Smith DI, Trakhtenbrot L, Kerem B: A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. 2002, 1: 89-97. 10.1016/S1535-6108(02)00017-X
Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL: Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001, 293: 876-880. 10.1126/science.1062538
Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A, Lifshits E, Brown A, Lee C, Christensen JG: Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene. 2010.
Stolzel F, Steudel C, Oelschlagel U, Mohr B, Koch S, Ehninger G, Thiede C: Mechanisms of resistance against PKC412 in resistant FLT3-ITD positive human acute myeloid leukemia cells. Ann Hematol. 2010.
Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y: Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68: 9479-9487. 10.1158/0008-5472.CAN-08-1643
Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S: MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007, 104: 20932-20937. 10.1073/pnas.0710370104
Khorashad JS, De Melo VA, Fiegler H, Gerrard G, Marin D, Apperley JF, Goldman JM, Foroni L, Reid AG: Multiple sub-microscopic genomic lesions are a universal feature of chronic myeloid leukaemia at diagnosis. Leukemia. 2008, 22: 1806-1807. 10.1038/leu.2008.210
McManus MT: MicroRNAs and cancer. Semin Cancer Biol. 2003, 13: 253-258. 10.1016/S1044-579X(03)00038-5
Gregory RI, Shiekhattar R: MicroRNA biogenesis and cancer. Cancer Res. 2005, 65: 3509-3512. 10.1158/0008-5472.CAN-05-0298
Weiss GJ, Bemis LT, Nakajima E, Sugita M, Birks DK, Robinson WA, Varella-Garcia M, Bunn PA, Haney J, Helfrich BA: EGFR regulation by microRNA in lung cancer: correlation with clinical response and survival to gefitinib and EGFR expression in cell lines. Ann Oncol. 2008, 19: 1053-1059. 10.1093/annonc/mdn006
Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, Shimizu M, Cimmino A, Zupo S, Dono M: MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA. 2004, 101: 11755-11760. 10.1073/pnas.0404432101
Mahadevan D, Cooke L, Riley C, Swart R, Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H: A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene. 2007, 26: 3909-3919. 10.1038/sj.onc.1210173
Seike M, Goto A, Okano T, Bowman ED, Schetter AJ, Horikawa I, Mathe EA, Jen J, Yang P, Sugimura H: MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc Natl Acad Sci USA. 2009, 106: 12085-12090. 10.1073/pnas.0905234106
Pogribny IP, Filkowski JN, Tryndyak VP, Golubov A, Shpyleva SI, Kovalchuk O: Alterations of microRNAs and their targets are associated with acquired resistance of MCF-7 breast cancer cells to cisplatin. Int J Cancer. 2010.
Boren T, Xiong Y, Hakam A, Wenham R, Apte S, Chan G, Kamath SG, Chen DT, Dressman H, Lancaster JM: MicroRNAs and their target messenger RNAs associated with ovarian cancer response to chemotherapy. Gynecol Oncol. 2009, 113: 249-255. 10.1016/j.ygyno.2009.01.014
Sorrentino A, Liu CG, Addario A, Peschle C, Scambia G, Ferlini C: Role of microRNAs in drug-resistant ovarian cancer cells. Gynecol Oncol. 2008, 111: 478-486. 10.1016/j.ygyno.2008.08.017
Yang N, Kaur S, Volinia S, Greshock J, Lassus H, Hasegawa K, Liang S, Leminen A, Deng S, Smith L: MicroRNA microarray identifies Let-7i as a novel biomarker and therapeutic target in human epithelial ovarian cancer. Cancer Res. 2008, 68: 10307-10314. 10.1158/0008-5472.CAN-08-1954
van Jaarsveld MT, Helleman J, Berns EM, Wiemer EA: MicroRNAs in ovarian cancer biology and therapy resistance. Int J Biochem Cell Biol. 2010.
Hanahan D, Weinberg RA: The hallmarks of cancer. Cell. 2000, 100: 57-70. 10.1016/S0092-8674(00)81683-9
Mahon FX, Hayette S, Lagarde V, Belloc F, Turcq B, Nicolini F, Belanger C, Manley PW, Leroy C, Etienne G: Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Res. 2008, 68: 9809-9816. 10.1158/0008-5472.CAN-08-1008
Li L, Lin X, Shoemaker AR, Albert DH, Fesik SW, Shen Y: Hypoxia-inducible factor-1 inhibition in combination with temozolomide treatment exhibits robust antitumor efficacy in vivo. Clin Cancer Res. 2006, 12: 4747-4754. 10.1158/1078-0432.CCR-05-2842
Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM: Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003, 3: 347-361. 10.1016/S1535-6108(03)00085-0
Noro R, Gemma A, Miyanaga A, Kosaihira S, Minegishi Y, Nara M, Kokubo Y, Seike M, Kataoka K, Matsuda K: PTEN inactivation in lung cancer cells and the effect of its recovery on treatment with epidermal growth factor receptor tyrosine kinase inhibitors. Int J Oncol. 2007, 31: 1157-1163.
Yacoub A, Park MA, Hanna D, Hong Y, Mitchell C, Pandya AP, Harada H, Powis G, Chen CS, Koumenis C: OSU-03012 promotes caspase-independent but PERK-, cathepsin B-, BID-, and AIF-dependent killing of transformed cells. Mol Pharmacol. 2006, 70: 589-603. 10.1124/mol.106.025007
Xia W, Bacus S, Hegde P, Husain I, Strum J, Liu L, Paulazzo G, Lyass L, Trusk P, Hill J: A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci USA. 2006, 103: 7795-7800. 10.1073/pnas.0602468103
McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J: Acquired Resistance of Non-Small Cell Lung Cancer Cells to MET Kinase Inhibition Is Mediated by a Switch to Epidermal Growth Factor Receptor Dependency. Cancer Res. 2010, 70: 1625-1634. 10.1158/0008-5472.CAN-09-3620
Jones HE, Goddard L, Gee JM, Hiscox S, Rubini M, Barrow D, Knowlden JM, Williams S, Wakeling AE, Nicholson RI: Insulin-like growth factor-I receptor signalling and acquired resistance to gefitinib (ZD1839; Iressa) in human breast and prostate cancer cells. Endocr Relat Cancer. 2004, 11: 793-814. 10.1677/erc.1.00799
Desbois-Mouthon C, Baron A, Blivet-Van Eggelpoel MJ, Fartoux L, Venot C, Bladt F, Housset C, Rosmorduc O: Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin Cancer Res. 2009, 15: 5445-5456. 10.1158/1078-0432.CCR-08-2980
Nahta R, Yuan LX, Du Y, Esteva FJ: Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol Cancer Ther. 2007, 6: 667-674. 10.1158/1535-7163.MCT-06-0423
Jones HEGJMW, Hutcheson IR, Nicholson RI: Growth factor pathway switching: implications for the use of gefitinib and trastuzumab. Breast Cancer Online. 2006, 9: e27-10.1017/S1470903106005451. 10.1017/S1470903106005451
Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, Rinehart C, Seidel B, Yee D, Arteaga CL, Engelman JA: Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008, 118: 2609-2619.
Sawabu T, Seno H, Kawashima T, Fukuda A, Uenoyama Y, Kawada M, Kanda N, Sekikawa A, Fukui H, Yanagita M: Growth arrest-specific gene 6 and Axl signaling enhances gastric cancer cell survival via Akt pathway. Mol Carcinog. 2007, 46: 155-164. 10.1002/mc.20211
Ito T, Tanaka H, Kimura A: Establishment and characterization of a novel imatinib-sensitive chronic myeloid leukemia cell line MYL, and an imatinib-resistant subline MYL-R showing overexpression of Lyn. Eur J Haematol. 2007, 78: 417-431. 10.1111/j.1600-0609.2007.00835.x
Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian CN, Kahnoski R, Futreal PA, Furge KA, Teh BT: Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010, 70: 1063-1071. 10.1158/0008-5472.CAN-09-3965
Mizukami Y, Jo WS, Duerr EM, Gala M, Li J, Zhang X, Zimmer MA, Iliopoulos O, Zukerberg LR, Kohgo Y: Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat Med. 2005, 11: 992-997.
Morse MA, Wei J, Hartman Z, Xia W, Ren XR, Lei G, Barry WT, Osada T, Hobeika AC, Peplinski S: Synergism from combined immunologic and pharmacologic inhibition of HER2 in vivo. Int J Cancer. 2009.
Adida C, Berrebi D, Peuchmaur M, Reyes-Mugica M, Altieri DC: Anti-apoptosis gene, survivin, and prognosis of neuroblastoma. Lancet. 1998, 351: 882-883. 10.1016/S0140-6736(05)70294-4
Saitoh Y, Yaginuma Y, Ishikawa M: Analysis of Bcl-2, Bax and Survivin genes in uterine cancer. Int J Oncol. 1999, 15: 137-141.
Ambrosini G, Adida C, Sirugo G, Altieri DC: Induction of apoptosis and inhibition of cell proliferation by survivin gene targeting. J Biol Chem. 1998, 273: 11177-11182. 10.1074/jbc.273.18.11177
Lu CD, Altieri DC, Tanigawa N: Expression of a novel antiapoptosis gene, survivin, correlated with tumor cell apoptosis and p53 accumulation in gastric carcinomas. Cancer Res. 1998, 58: 1808-1812.
Konno R, Yamakawa H, Utsunomiya H, Ito K, Sato S, Yajima A: Expression of survivin and Bcl-2 in the normal human endometrium. Mol Hum Reprod. 2000, 6: 529-534. 10.1093/molehr/6.6.529
Endoh A, Yagihashi A, Asanuma K, Moriai R, Izawa A, Koyanagi Y, Sato T, Kobayashi D, Watanabe N: Hematopoietic progenitor cell counts performed by the Sysmex SE-9000 analyzer can guide timing of peripheral blood stem cell harvest. Anticancer Res. 2001, 21: 601-604.
Gimenez-Bonafe P, Tortosa A, Perez-Tomas R: Overcoming drug resistance by enhancing apoptosis of tumor cells. Curr Cancer Drug Targets. 2009, 9: 320-340. 10.2174/156800909788166600
Gillet JP, Gottesman MM: Mechanisms of multidrug resistance in cancer. Methods Mol Biol. 2010, 596: 47-76. full_text
Tao LY, Liang YJ, Wang F, Chen LM, Yan YY, Dai CL, Fu LW: Cediranib (recentin, AZD2171) reverses ABCB1- and ABCC1-mediated multidrug resistance by inhibition of their transport function. Cancer Chemother Pharmacol. 2009, 64 (5): 961-9. 10.1007/s00280-009-0949-1
Dai CL, Liang YJ, Wang YS, Tiwari AK, Yan YY, Wang F, Chen ZS, Tong XZ, Fu LW: Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett. 2009, 279 (1): 74-83. 10.1016/j.canlet.2009.01.027
Turk D, Szakacs G: Relevance of multidrug resistance in the age of targeted therapy. Curr Opin Drug Discov Devel. 2009, 12: 246-252.
Mahon FX, Belloc F, Lagarde V, Chollet C, Moreau-Gaudry F, Reiffers J, Goldman JM, Melo JV: MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood. 2003, 101: 2368-2373. 10.1182/blood.V101.6.2368
Che XF, Nakajima Y, Sumizawa T, Ikeda R, Ren XQ, Zheng CL, Mukai M, Furukawa T, Haraguchi M, Gao H: Reversal of P-glycoprotein mediated multidrug resistance by a newly synthesized 1, 4-benzothiazipine derivative, JTV-519. Cancer Lett. 2002, 187: 111-119. 10.1016/S0304-3835(02)00359-2
Kotaki M, Motoji T, Takanashi M, Wang YH, Mizoguchi H: Anti-proliferative effect of the abl tyrosine kinase inhibitor STI571 on the P-glycoprotein positive K562/ADM cell line. Cancer Lett. 2003, 199: 61-68. 10.1016/S0304-3835(03)00338-0
Rumpold H, Wolf AM, Gruenewald K, Gastl G, Gunsilius E, Wolf D: RNAi-mediated knockdown of P-glycoprotein using a transposon-based vector system durably restores imatinib sensitivity in imatinib-resistant CML cell lines. Exp Hematol. 2005, 33: 767-775. 10.1016/j.exphem.2005.03.014
Widmer N, Rumpold H, Untergasser G, Fayet A, Buclin T, Decosterd LA: Resistance reversal by RNAi silencing of MDR1 in CML cells associated with increase in imatinib intracellular levels. Leukemia. 2007, 21: 1561-1562. author reply 1562-1564., 10.1038/sj.leu.2404671
Hiwase DK, Saunders V, Hewett D, Frede A, Zrim S, Dang P, Eadie L, To LB, Melo J, Kumar S: Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clin Cancer Res. 2008, 14: 3881-3888. 10.1158/1078-0432.CCR-07-5095
Shukla S, Robey RW, Bates SE, Ambudkar SV: Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos. 2009, 37: 359-365. 10.1124/dmd.108.024612
Elkind NB, Szentpetery Z, Apati A, Ozvegy-Laczka C, Varady G, Ujhelly O, Szabo K, Homolya L, Varadi A, Buday L: Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD Gefitinib). Cancer Res. 1839, 65: 1770-1777. 10.1158/0008-5472.CAN-04-3303.
Thomas J, Wang L, Clark RE, Pirmohamed M: Active transport of imatinib into and out of cells: implications for drug resistance. Blood. 2004, 104: 3739-3745. 10.1182/blood-2003-12-4276
White DL, Saunders VA, Dang P, Engler J, Zannettino AC, Cambareri AC, Quinn SR, Manley PW, Hughes TP: OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood. 2006, 108: 697-704. 10.1182/blood-2005-11-4687
Wang L, Giannoudis A, Lane S, Williamson P, Pirmohamed M, Clark RE: Expression of the uptake drug transporter hOCT1 is an important clinical determinant of the response to imatinib in chronic myeloid leukemia. Clin Pharmacol Ther. 2008, 83: 258-264. 10.1038/sj.clpt.6100268
Gambacorti-Passerini C, Barni R, le Coutre P, Zucchetti M, Cabrita G, Cleris L, Rossi F, Gianazza E, Brueggen J, Cozens R: Role of alpha1 acid glycoprotein in the in vivo resistance of human BCR-ABL(+) leukemic cells to the abl inhibitor STI571. J Natl Cancer Inst. 2000, 92: 1641-1650. 10.1093/jnci/92.20.1641
Gambacorti-Passerini C, Zucchetti M, Russo D, Frapolli R, Verga M, Bungaro S, Tornaghi L, Rossi F, Pioltelli P, Pogliani E: Alpha1 acid glycoprotein binds to imatinib (STI571) and substantially alters its pharmacokinetics in chronic myeloid leukemia patients. Clin Cancer Res. 2003, 9: 625-632.
le Coutre P, Kreuzer KA, Na IK, Lupberger J, Holdhoff M, Appelt C, Schwarz M, Muller C, Gambacorti-Passerini C, Platzbecker U: Determination of alpha-1 acid glycoprotein in patients with Ph+ chronic myeloid leukemia during the first 13 weeks of therapy with STI571. Blood Cells Mol Dis. 2002, 28: 75-85. 10.1006/bcmd.2002.0493
Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL: American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009, 27: 2091-2096. 10.1200/JCO.2009.21.9170
Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M: KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66: 3992-3995. 10.1158/0008-5472.CAN-06-0191
De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y: KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008, 19: 508-515. 10.1093/annonc/mdm496
Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboue R, Tuech JJ: Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer. 2007, 96: 1166-1169. 10.1038/sj.bjc.6603685
Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A: Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008, 26: 5705-5712. 10.1200/JCO.2008.18.0786
Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M: A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007, 12: 395-402. 10.1016/j.ccr.2007.08.030
Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L: PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer. 2007, 97: 1139-1145. 10.1038/sj.bjc.6604009
Perrone F, Lampis A, Orsenigo M, Di Bartolomeo M, Gevorgyan A, Losa M, Frattini M, Riva C, Andreola S, Bajetta E: PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol. 2009, 20: 84-90. 10.1093/annonc/mdn541
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT: PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004, 6: 117-127. 10.1016/j.ccr.2004.06.022
Fujita T, Doihara H, Kawasaki K, Takabatake D, Takahashi H, Washio K, Tsukuda K, Ogasawara Y, Shimizu N: PTEN activity could be a predictive marker of trastuzumab efficacy in the treatment of ErbB2-overexpressing breast cancer. Br J Cancer. 2006, 94: 247-252. 10.1038/sj.bjc.6602926
Bardelli A, Siena S: Molecular Mechanisms of Resistance to Cetuximab and Panitumumab in Colorectal Cancer. J Clin Oncol. 2010, 28 (7): 1254-61. 10.1200/JCO.2009.24.6116
Lu Y, Zi X, Pollak M: Molecular mechanisms underlying IGF-I-induced attenuation of the growth-inhibitory activity of trastuzumab (Herceptin) on SKBR3 breast cancer cells. Int J Cancer. 2004, 108: 334-341. 10.1002/ijc.11445
Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ: Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005, 65: 11118-11128. 10.1158/0008-5472.CAN-04-3841
Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M: Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst. 2001, 93: 1852-1857. 10.1093/jnci/93.24.1852
Shattuck DL, Miller JK, Carraway KL, Sweeney C: Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008, 68: 1471-1477. 10.1158/0008-5472.CAN-07-5962
Sok JC, Coppelli FM, Thomas SM, Lango MN, Xi S, Hunt JL, Freilino ML, Graner MW, Wikstrand CJ, Bigner DD: Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res. 2006, 12: 5064-5073. 10.1158/1078-0432.CCR-06-0913
Lu Y, Li X, Liang K, Luwor R, Siddik ZH, Mills GB, Mendelsohn J, Fan Z: Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007, 67: 8240-8247. 10.1158/0008-5472.CAN-07-0589
Puri N, Salgia R: Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J Carcinog. 2008, 7: 9- 10.4103/1477-3163.44372
Wheeler DL, Iida M, Kruser TJ, Nechrebecki MM, Dunn EF, Armstrong EA, Huang S, Harari PM: Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther. 2009, 8: 696-703.
Valabrega G, Montemurro F, Sarotto I, Petrelli A, Rubini P, Tacchetti C, Aglietta M, Comoglio PM, Giordano S: TGFalpha expression impairs Trastuzumab-induced HER2 downregulation. Oncogene. 2005, 24: 3002-3010. 10.1038/sj.onc.1208478
Casanovas O, Hicklin DJ, Bergers G, Hanahan D: Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005, 8: 299-309. 10.1016/j.ccr.2005.09.005
Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, Chorianopoulos E, Liesenborghs L, Koch M, De Mol M: Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007, 131: 463-475. 10.1016/j.cell.2007.08.038
Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I: Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006, 444: 1083-1087. 10.1038/nature05313
Kopetz S, Hoff PM, Morris JS, Wolff RA, Eng C, Glover KY, Adinin R, Overman MJ, Valero V, Wen S: Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol. 2010, 28: 453-459. 10.1200/JCO.2009.24.8252
Loges S, Schmidt T, Carmeliet P: Mechanisms of Resistance to Anti-Angiogenic Therapy and Development of Third-Generation Anti-Angiogenic Drug Candidates. Genes & Cancer. 2010, 1: 12-25. 10.1177/1947601909356574.
Price-Schiavi SA, Jepson S, Li P, Arango M, Rudland PS, Yee L, Carraway KL: Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer. 2002, 99: 783-791. 10.1002/ijc.10410
Nagy P, Friedlander E, Tanner M, Kapanen AI, Carraway KL, Isola J, Jovin TM: Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer Res. 2005, 65: 473-482.
Haura EB, Tanvetyanon T, Chiappori A, Williams C, Simon G, Antonia S, Gray J, Litschauer S, Tetteh L, Neuger A: Phase I/II Study of the Src Inhibitor Dasatinib in Combination With Erlotinib in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2010, 28 (8): 1387-94. 10.1200/JCO.2009.25.4029
Kim HP, Han SW, Kim SH, Im SA, Oh DY, Bang YJ, Kim TY: Combined lapatinib and cetuximab enhance cytotoxicity against gefitinib-resistant lung cancer cells. Mol Cancer Ther. 2008, 7: 607-615. 10.1158/1535-7163.MCT-07-2068
Park SJ, Kim MJ, Kim YK, Kim SM, Park JY, Myoung H: Combined cetuximab and genistein treatment shows additive anti-cancer effect on oral squamous cell carcinoma. Cancer Lett. 2009.
Acknowledgements
We would to thank Shawna Organ for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JRS drafted and wrote the manuscript. VC helped writing the manuscript. SG corrected and finalized the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12943_2009_590_MOESM1_ESM.DOC
Additional file 1: List of some small molecule TKIs approved by the FDA or currently undergoing clinical trials. (DOC 76 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sierra, J.R., Cepero, V. & Giordano, S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol Cancer 9, 75 (2010). https://doi.org/10.1186/1476-4598-9-75
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-9-75