
Abstract
Background
Unbalanced chromosomal translocations may present with a variety of clinical and laboratory findings and provide insight into the functions of genes on the involved chromosomal segments.
Case Presentation
A 9 year-old boy presented to our clinic with Factor VII deficiency, microcephaly, a seizure disorder, multiple midline abnormalities (agenesis of the corpus callosum, imperforate anus, bilateral optic nerve hypoplasia), developmental delay, hypopigmented macules, short 5th fingers, and sleep apnea due to enlarged tonsils. Cytogenetic and fluorescence in situ hybridization analyses revealed an unbalanced translocation involving the segment distal to 16p13 replacing the segment distal to 13q33 [46, XY, der(13)t(13;16)(q33;p13.3)]. Specific BAC-probes were used to confirm the extent of the 13q deletion.
Conclusion
This unique unbalanced chromosomal translocation may provide insights into genes important in midline development and underscores the previously-reported phenotype of Factor VII deficiency in 13q deletions.
Similar content being viewed by others

Background
The identification of chromosomal breakpoints in association with variations in human phenotypes often leads to the discovery of novel genes or the characterization of the clinical importance of known genes. Patients who present with a known Mendelian disorder, but who also exhibit developmental delay or other, seemingly unrelated conditions, may harbor a chromosomal abnormality. We present the case of a child who presented with asymptomatic Factor VII deficiency at age 9 who was found to have an unbalanced 13;16 translocation associated with several other developmental abnormalities.
Case presentation
Clinical presentation
A 9 year-old male presented with Factor VII deficiency and multiple congenital abnormalities. Prior to her pregnancy with this child, his mother (currently 50 years old) had nine in vitro fertilization attempts that ended in miscarriage. One of these miscarriages was documented as trisomy 21 at 18 weeks gestation. This patient was one twin of a fraternal twin pregnancy conceived on the 10th round of in vitro fertilization. His sister was born healthy and her cytogenetic evaluation has not been performed. This twin pregnancy was complicated by premature labor requiring 13 weeks of maternal bed rest prior to delivery via C-section at 36 weeks gestation. The patient's birth weight was 3 lbs, 14.5 oz and his Apgar scores were 9 and 9 at 1 and 5 minutes, respectively.
At birth, the patient was noted to have an imperforate anus that was repaired with a colostomy at 15 days of life. An ultrasound of the brain showed agenesis of the corpus callosum, which was later confirmed on MRI. Both an echocardiogram and a renal ultrasound were normal.
For the first year of life, the patient failed to thrive. He underwent two successful operations for his imperforate anus at ages 9 and 11 months, after which he began to gain weight appropriately. Development was delayed; he sat at 9 months, pulled up at 11 months, walked at 22 months, and spoke single words at 15 months. His pediatricians noted "borderline microcephaly" and low muscle tone. He has been receiving speech therapy and physical therapy since age 1. At age 6 years, he developed grand mal seizures which have been successfully managed with medication. He had repeated upper respiratory infections and was noted to have enlarged tonsils that may have contributed to sleep apnea. As part of his pre-operative work-up for tonsillectomy, he was discovered to have elevated prothrombin and partial thromboplastin times of 21.3 seconds and 36.8 seconds, respectively. Factor VII deficiency (17% of normal) was subsequently diagnosed; Factor X levels were 54% of normal. The patient, however, has never had a severe bleeding disorder. It was the combination of Factor VII deficiency and multiple congenital malformations that prompted the genetics work-up, including karyotype. He had abnormal tooth eruption and required dental surgery and frenulectomy. Neither surgery required coagulation assistance.
The patient's parents are healthy, as is his fraternal twin sister and a younger brother, age 7. Review of systems was notable for chronic constipation requiring daily enemas. He is currently on Depakote 325 mg bid and has no known drug allergies.
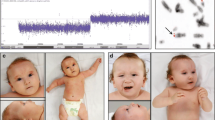
On physical examination (Figure 1), the patient's height is 51 inches (10th centile), his weight is 42 pounds (3rd centile) and his head circumference is 49.5 cm (<3rd centile, the 50th centile for a 2 year-old). His inner and outer canthal distances are at 97th centile. He has thick, curly hair with one hair whorl and a normal posterior hairline. His forehead is prominent and his face is triangular. His palpebral fissures are somewhat almond-shaped and upslanting. He has mild ptosis. He has a high nasal root, hypoplastic alae nasi with prominent columella, a thin upper lip, downturned corners of the mouth, and a small, pointed chin. He has some mild facial asymmetry. His ears are normally placed and rotated without tags or pits; however, there is a slightly flattened appearance to both auricles. His palate is high arched and he has a small notch in his uvula. He has several dental caps and his lower front teeth are abnormally rotated. His chest is normally shaped without pectus deformity. His chest circumference and inter-nipple distance were 50th and 75th centiles, respectively. He has multiple hypopigmented macules on his torso and one café-au-lait spot on his left shoulder measuring approximately 3 cm × 2 cm. He has approximately 10 degrees of scoliosis, mild lordosis and a small sacral dimple. There are surgical scars from his colostomy and anal repair. He has normal male genitalia, Tanner Stage 1. He has decreased elbow and shoulder extension. His distal extensibility is normal. His 5th fingers are slightly short relative to the 4th distal inter-phalangeal crease and show clinodactyly. His palmar creases are normal. Heart, lung, and abdominal examinations were normal. His knees were normal and he has pes planus.

These photographs demonstrate the mild dysmorphic features of the patient, including microcephaly, a triangular and mildly asymmetric face, prominent forehead, slightly downslanting and almond-shaped palpebral fissures, mild ptosis, high nasal bridge and a small chin (panels A and B). He had bilateral 5th finger clinodactyly (panel C) and bilateral optic nerve hypoplasia (panel D).
On ophthalmologic examination, the patients best-corrected visual acuity is 20/50 OU with a +6.25 + 1.25 × 95 OD and +6.25 + 1.75 × 95 OS prescription. He has no measurable stereopsis. Color vision, ocular ductions, ocular alignment, pupillary examination, confrontational visual fields, and slit lamp examination were all normal. Dilated fundoscopic examination showed bilateral, mild optic nerve hypoplasia.
Endocrine studies, including a thyroid panel, a random cortisol, prolactin, and follicle-stimulating were within normal limits. The patient's bone age is at the upper limits of normal by Pyles and Greulich critreria.
Cytogenetic analysis
Routine karyotyping showed a translocation of material of unknown origin onto 13q33 (Figure 2). FISH probes for all telomeres were used to confirm the presence of an unbalanced translocation with distal 16p (16p13.3 → ptelomere) translocated to 13q33. Thus, the patient is monosomic for genes at 13q33 and distal, as well as trisomic for genes at 16p13.3. The nomenclature for this karyotype is 46, XY, der(13)t(13;16)(q33;p13.3). Because the transcription factor gene, ZIC2, lies close to the breakpoint and because mutations in ZIC2 are known to cause holoprosencephaly (a midline defect), we confirmed the breakpoints by performing FISH analysis using ZIC2 specific bacterial artificial chromosome (BAC) probes, RPCI 11 12-G12 selected from NCBI and Ensembl databases (BACPAC Resources, Children's Hospital Oakland Research Institute, Oakland, CA). These FISH studies confirmed that ZIC2 was present on both chromosome 13's and that the breakpoint was distal to 13q32, consistent with our impression that the breakpoint is at 13q33. Cytogenetic analysis of both parents was recommended given the history of previous miscarriages, but has not yet been pursued by the family.

Routine cytogenetics identified a translocation of material of unknown origin onto 13q33 (panel A). Using probes specific for subtelomeric sequences, a translocation was identified from 16p (green) onto 13q (panel B, red probe is for 16q). Similarly, two probes for distal 13q (red and green probes, panel C) were absent from one chromosome 13 identified using an aqua-labeled proximal 13q probe. A FISH BAC probe specific for the ZIC2 gene on 13q (panel D, green probe) confirms that it is not deleted in this patient.
Conclusion
We report on the clinical and cytogenetics findings in a male with a novel, unbalanced 13;16 chromosomal translocation that resulted in monosomy for genes from 13q33 to the q terminus and trisomy for 16p13.3 genes, the most distal band on the short arm of chromosome 16. Examples of genes that map to these segments are given in Figure 3 and 4. Because chromosome 16 may include imprinted loci, it is possible that the phenotype of this patient could be affected by parent-of-origin effects and/or uniparental disomy for 16p13.3. The presence of several midline abnormalities (e.g., absent corpus callosum, optic nerve hypoplasia, small cleft in the uvula, and anal atresia) suggest a midline field abnormality around the time of gastrulation.[1]

Ideogram of chromosomes 16 with the trisomic region highlighted. Examples of genes known to map to these chromosomal segments are noted, according to the information in build 35.1 of the Homo sapiens sequence on the NCBI Map Viewer and Online Mendelian Inheritance in Man. Hypothetical proteins and open reading frames are omitted from the list. In some cases, the exact order of transcripts is ambiguous.
While 13q deletion syndromes are well-recognized,[2] we present a novel translocation associated with 13q deletion and a complex clinical phenotype. The variability in reported phenotypes depends, in part, on the portions of 13q that are deleted and whether other chromosomal abnormalities are involved. Table 1 summarizes published reports of very distal 13q deletions similar to ours. Findings in our patient consistent with 13q deletion include growth and psychomotor retardation, microcephaly, deficiency of coagulation factors VII and X, and mild facial dysmorphisms and asymmetry. His sacral dimple is a finding consistent with the proposal of Luo et al that this region may include genes important in neural tube closure.[3] Similarly, his imperforate anus, muscular hypotonia, microcephaly and dysmorphic features are consistent with the three patients described by Kuhnle et al with sub-telomeric deletions of 13q, although this case is complicated by mosaicism for monosomy 13 and is therefore not included in table 1.[4] Stoll and Alembik describe a patient with 13q33.3→qter with only mild mental retardation, microcephaly, growth delay, a small chin, hypotonia, and a broad forehead[5] and Rivera et al. review six cases with many similar findings, despite a larger 13q deletion.[6] Submicroscopic deletions and/or rearrangements of 13qter have also been reported to lead to "idiopathic" mental retardation.[7]
Less has been reported on trisomy 16p, perhaps because it is less susceptible than other chromosomal regions to translocation[8] and because complete trisomy 16 – the most frequent autosomal anomaly found in miscarriages – is frequently lethal.[9] Again, there is significant phenotypic heterogeneity reported, in part because most patients described have unbalanced translocations and are trisomic for variable portions of 16p. [10–19] Findings consistent with this anomaly in our patient include microcephaly, a broad nasal bridge, developmental delay, and seizures.[16, 20] Cases involving only trisomy of 16p13 are summarized in Table 2. An insertion of 16p13.1→p13.3 into chromosome 1 resulted in mental retardation, short stature, microcephaly, seizures, and multiple dysmorphic features.[21] A 14 year-old male with 46, XY, dup(16)(p13.1→pter) karyotype presented with autism, Tourette's syndrome, short stature, a prominent chin, elongated face, hi-arched palate, small penis/scrotum, poor fine and gross motor movements, and slow basal activity on EEG.[22] Hunter et al. describe a newborn with a 7;16 unbalanced translocation resulting in trisomy of 16p13.1→pter and monosomy of 7p22→pter, chondrodysplasia punctata, absence of the gallbladder, and microcornea.[23] Tschernigg et al. describe a child with "pure" 16pter→p13 due to a tandem duplication of this region, cleft lip/palate, cardiac defects, and club hands/feet. [24]

Ideogram of chromosome 13 with the monosomic region highlighted. Examples of genes known to map to these chromosomal segments are noted, according to the information in build 35.1 of the Homo sapienssequence on the NCBI Map Viewer and Online Mendelian Inheritance in Man. Hypothetical proteins and open reading frames are omitted from the list. In some cases, the exact order of transcripts is ambiguous
Factor VII is a vitamin-K dependent clotting factor in the extrinsic pathway that – when deficient – is inherited as an autosomal recessive trait and produces an elevated partial thromboplastin time (PTT) in the face of a normal activated prothrombin time (PT); hemarthrosis; intracranial hemorrhage; hematuria; spontaneous epistaxis and bruising; and genitourinary and gastrointestinal bleeding. Although levels of activity correlate imprecisely with symptoms, it is likely that levels below 2% of normal are required before major symptoms develop. The Factor VII gene maps to 13q34,[25] is alternatively spliced, and has multiple poly-adenylation signals.[26] Pfeiffer et al. describe two cases of sub-clinical factor VII deficiency associated with a 46, XY, t(13;Y)(q11;q34) translocation and probable deletion of a terminal segment of 13q that manifested as elevated PTT.[27] Hewson and Carter described severe Factor VII deficiency in a case of 13q deletion syndrome;[28] while Fukushima et al. found approximately 50% factor V activity in two of three patients with terminal 13q deletions and normal levels in a patient with trisomy 13.[29] Our patient would be predicted to be monosomic for 13q34. The fact that he exhibits less than 50% activity may be related to a polymorphism on his remaining allele causing slightly reduced activity or reduced levels of protein expression. Factor VII activity was not assayed in his parents.
Bilateral optic nerve hypoplasia is sometimes accompanied by midline anomalies of the central nervous system, such as absent corpus callosum, absent septum pellicidum, and pituitary insufficiency.[30] None of the three genes known to produce this phenotype in animal models – namely netrin (17p13-p12), Hesx1 (3p21),[31] and DCC (18q21.3)[32] – map to either chromosome 13 or 16, suggesting an uncharacterized regulatory gene for midline CNS development in the abnormal regions. Of note, netrin-2 -like gene is located on 16p13.[33] Several other zinc-finger containing proteins of unknown function also map to this region. A three-generation family with dominant congenital cataract and microphthalmia co-segregates with a t(2;16)(p22.3;p13.3) translocation in four balanced carriers and three with monosomy of 16p13.3, suggesting that an important eye developmental gene resides at or near this breakpoint.[34]
Abbreviations
- ASD:
-
atrial-septal defect
- BAC:
-
bacterial artificial chromosome
- CL/P:
-
cleft lip/palate
- EEG:
-
electroencephalogram
- FISH:
-
fluorescent in situ hybridization
- IUGR:
-
intrauterine growth retardation
- MR:
-
mental retardation
- MR:
-
mental retardation
- OD:
-
right eye
- OS:
-
left eye
- OU:
-
both eyes
- PF:
-
palpebral fissures
- PIP:
-
proximal interphalangeal
- PT:
-
activated prothrombin time
- PTT:
-
partial thromboplastin time
- SHH:
-
sonic hedgehog
- SPC:
-
single palmar crease
- SS:
-
short stature
- VSD:
-
ventricular-septal defect.
References
Martinez-Frias ML: Primary midline developmental field. I. Clinical and epidemiological characteristics. Am J Med Genet. 1995, 56 (4): 374-381. 10.1002/ajmg.1320560406.
Jones KL: Deletion 13q Syndrome. Smith's Recognizable Patterns of Human Malformation. Edited by: Jones KL. 1997, Philadelphia , W.B. Saunders Company, 60-61. 5th
Luo J, Balkin N, Stewart JF, Sarwark JF, Charrow J, Nye JS: Neural tube defects and the 13q deletion syndrome: evidence for a critical region in 13q33-34. Am J Med Genet. 2000, 91 (3): 227-230. 10.1002/(SICI)1096-8628(20000320)91:3<227::AID-AJMG14>3.0.CO;2-I.
Kuhnle U, Bartsch O, Werner W, Schuster T: Penoscrotal inversion, hypospadias, imperforate anus, facial anomalies, and developmental delay: definition of a new clinical syndrome. Pediatr Surg Int. 2000, 16 (5-6): 396-399. 10.1007/s003830000379.
Stoll C, Alembik Y: A patient with 13q-syndrome with mild mental retardation and with growth retardation. Ann Genet. 1998, 41 (4): 209-212.
Rivera H, Vasquez AI, Garcia-Cruz D, Crolla JA: Neocentromere at 13q32 in one of two stable markers derived from a 13q21 break. Am J Med Genet. 1999, 85 (4): 385-388. 10.1002/(SICI)1096-8628(19990806)85:4<385::AID-AJMG15>3.0.CO;2-P.
Flint J, Wilkie AO, Buckle VJ, Winter RM, Holland AJ, McDermid HE: The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation. Nat Genet. 1995, 9 (2): 132-140. 10.1038/ng0295-132.
Stoll C: Nonrandom distribution of exchange points in patients with reciprocal translocations. Hum Genet. 1980, 56 (1): 89-93.
Creasy MR, Crolla JA, Alberman ED: A cytogenetic study of human spontaneous abortions using banding techniques. Hum Genet. 1976, 31 (2): 177-196. 10.1007/BF00296145.
Bofinger MK, Opitz JM, Soukup SW, Ekblom LS, Phillips S, Daniel A, Greene EW: A familial MCA/MR syndrome due to translocation t(10;16) (q26;p13.1): report of six cases. Am J Med Genet. 1991, 38 (1): 1-8. 10.1002/ajmg.1320380102.
Brandt CA, Lyngbye T, Pedersen S, Bolund L, Friedrich U: Value of chromosome painting in determining the chromosomal outcome in offspring of a 12;16 translocation carrier. J Med Genet. 1994, 31 (3): 234-237.
Carrasco Juan JL, Cigudosa JC, Otero Gomez A, Acosta Almeida MT, Garcia Miranda JL: De novo trisomy 16p. Am J Med Genet. 1997, 68 (2): 219-221. 10.1002/(SICI)1096-8628(19970120)68:2<219::AID-AJMG19>3.0.CO;2-X.
O'Connor TA, Higgins RR: Trisomy 16p in a liveborn infant and review of trisomy 16p. Am J Med Genet. 1992, 42 (3): 316-319. 10.1002/ajmg.1320420311.
Dallapiccola B, Curatolo P, Balestrazzi P: 'De novo' trisomy 16q11 to pter. Hum Genet. 1979, 49 (1): 1-6.
Mori MA, Gomar JL, Diaz de Bustamante A, Ananias A, Pinel I, Martinez-Frias ML: Partial duplication 16p resulting from a 3:1 segregation of a maternal reciprocal translocation. Am J Med Genet. 1987, 26 (1): 203-206. 10.1002/ajmg.1320260130.
Leonard C, Huret JL, Imbert MC, Lebouc Y, Selva J, Boulley AM: Trisomy 16p in a liveborn offspring due to maternal translocation t(16;21)(q11;p11) and review of the literature. Am J Med Genet. 1992, 43 (3): 621-625. 10.1002/ajmg.1320430324.
McMorrow LE, Bornstein S, Fischer RH, Gluckson MM: Partial trisomy 16p due to maternal balanced translocation. J Med Genet. 1984, 21 (4): 315-316.
Roberts SH, Duckett DP: Trisomy 16p in a liveborn infant and a review of partial and full trisomy 16. J Med Genet. 1978, 15 (5): 375-381.
Leschot NJ, De Nef JJ, Geraedts JP, Becker-Bloemkolk MJ, Talma A, Bijlsma JB, Verjaal M: Five familial cases with a trisomy 16p syndrome due to translocation. Clin Genet. 1979, 16 (3): 205-214.
Jalal SM, Day DW, Garcia M, Benjamin T, Rogers J: Familial transmission of 16p trisomy in an infant. Hum Genet. 1989, 81 (2): 196-198. 10.1007/BF00293904.
Kokalj-Vokac N, Medica I, Zagorac A, Zagradisnik B, Erjavec A, Gregoric A: A case of insertional translocation resulting in partial trisomy 16p. Ann Genet. 2000, 43 (3-4): 131-135.
Hebebrand J, Martin M, Korner J, Roitzheim B, de Braganca K, Werner W, Remschmidt H: Partial trisomy 16p in an adolescent with autistic disorder and Tourette's syndrome. Am J Med Genet. 1994, 54 (3): 268-270. 10.1002/ajmg.1320540316.
Hunter AG, Rimoin DL, Koch UM, MacDonald GJ, Cox DM, Lachman RS, Adomian G: Chondrodysplasia punctata in an infant with duplication 16p due to a 7;16 translocation. Am J Med Genet. 1985, 21 (3): 581-589. 10.1002/ajmg.1320210320.
Tschernigg M, Petek E, Leonhardtsberger A, Wagner K, Kroisel PM: Terminal tandem duplication of 16p: a case with "pure" partial trisomy (16)(pter-->p13). Genet Couns. 2002, 13 (3): 303-307.
Gilgenkrantz S, Briquel ME, Andre E, Alexandre P, Jalbert P, Le Marec B, Pouzol P, Pommereuil M: Structural genes of coagulation factors VII and X located on 13q34. Ann Genet. 1986, 29 (1): 32-35.
O'Hara PJ, Grant FJ, Haldeman BA, Gray CL, Insley MY, Hagen FS, Murray MJ: Nucleotide sequence of the gene coding for human factor VII, a vitamin K-dependent protein participating in blood coagulation. Proc Natl Acad Sci U S A. 1987, 84 (15): 5158-5162.
Pfeiffer RA, Ott R, Gilgenkrantz S, Alexandre P: Deficiency of coagulation factors VII and X associated with deletion of a chromosome 13 (q34). Evidence from two cases with 46,XY,t(13;Y)(q11;q34). Hum Genet. 1982, 62 (4): 358-360. 10.1007/BF00304557.
Hewson MP, Carter JM: Severe congenital Factor VII deficiency associated with the 13q deletion syndrome. Am J Hematol. 2002, 71 (3): 232-233. 10.1002/ajh.10237.
Fukushima Y, Kuroki Y, Iizuka A: Activity and antigen of coagulation factors VII and X in five patients with abnormal chromosome 13. Jinrui Idengaku Zasshi. 1987, 32 (2): 91-96.
Brodsky MC: Congenital Optic Disc Anomalies. Ophthalmology. Edited by: Yanoff M. 2004, St. Louis , Mosby, 1255-1258.
Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC: Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998, 19 (2): 125-133. 10.1038/477.
Deiner MS, Sretavan DW: Altered midline axon pathways and ectopic neurons in the developing hypothalamus of netrin-1- and DCC-deficient mice. J Neurosci. 1999, 19 (22): 9900-9912.
Van Raay TJ, Foskett SM, Connors TD, Klinger KW, Landes GM, Burn TC: The NTN2L gene encoding a novel human netrin maps to the autosomal dominant polycystic kidney disease region on chromosome 16p13.3. Genomics. 1997, 41 (2): 279-282. 10.1006/geno.1997.4659.
Yokoyama Y, Narahara K, Tsuji K, Ninomiya S, Seino Y: Autosomal dominant congenital cataract and microphthalmia associated with a familial t(2;16) translocation. Hum Genet. 1992, 90 (1-2): 177-178. 10.1007/BF00210770.
Luquet I, Favre B, Nadal N, Madinier N, Khau Van Kien P, Huet F, Nivelon-Chevallier A, Mugneret F: Two cases of terminal deletion of chromosome 13: clinical features, conventional and molecular cytogenetic analysis. Ann Genet. 1999, 42 (1): 33-39.
Turleau C, Seger J, de Grouchy J, Dore F, Job JC: [Del (13) (q33). Exclusion of esterase D (ESD) from 13q33 and q34]. Ann Genet. 1978, 21 (3): 189-192.
Emanuel BS, Zackai EH, Moreau L, Coates P, Orrechio E: Interstitial deletion 13q33 resulting from maternal insertional translocation. Clin Genet. 1979, 16 (5): 340-346.
Mucke J, Sandig KR, Trautmann U: [13q syndrome--partial monosomy of the long arm of chromosome 13]. Klin Padiatr. 1983, 195 (5): 361-364.
Bottani A, Xie YG, Binkert F, Schinzel A: A case of Hirschsprung disease with a chromosome 13 microdeletion, del(13)(q32.3q33.2): potential mapping of one disease locus. Hum Genet. 1991, 87 (6): 748-750. 10.1007/BF00201741.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2350/7/2/prepub
Acknowledgements
Written consent was obtained from the patient's mother for the publication of this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
JAT initially saw the patient and requested a karyotype. BPB and JMM performed the initial karyotype and the FISH studies to delineate the translocation. BRH and CB performed the FISH using specific BAC probes for ZIC2. BPB and DB performed a complete physical examination, clinical genetics evaluation, and ophthalmologic examination.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Brooks, B.P., Meck, J.M., Haddad, B.R. et al. Factor VII deficiency and developmental abnormalities in a patient with partial monosomy of 13q and trisomy of 16p: case report and review of the literature. BMC Med Genet 7, 2 (2006). https://doi.org/10.1186/1471-2350-7-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2350-7-2