
Abstract
Tau aggregation is one of the major features in Alzheimer's disease and in several other tauopathies, including frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), and progressive supranuclear palsy (PSP). More than 35 mutations in the tau gene have been identified from FTDP-17 patients. A group of these mutations alters splicing of exon 10, resulting in an increase in exon 10 inclusion into tau mRNA. Abnormal splicing with inclusion of exon 10 into tau mRNA has also been observed in PSP and AD patients. These results indicate that abnormal splicing of exon 10, leading to the production of tau with exon 10, is probably one of the mechanisms by which tau accumulates and aggregates in tauopathic brains. Therefore, modulation of exon 10 splicing in the tau gene could potentially be targeted to prevent tauopathies. To identify small molecules or compounds that could potentially be developed into drugs to treat tauopathies, we established a cell-based high-throughput screening assay. In this review, we will discuss how realistic, specific biological molecules can be found to regulate exon 10 splicing in the tau gene for potential treatment of tauopathies.
Similar content being viewed by others

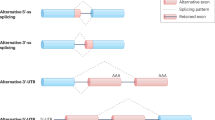

Alternative splicing of exon 10 in the taugene
Splicing of pre-mRNA is a complicated process that is regulated by intron and exon sequences, SR (serine/arginine-rich) proteins, and other non-SR splicing factors, for example, heterogeneous nuclear ribonucleoproteins (hnRNPs). Alternative splicing occurs when the introns of a certain pre-mRNA are excised in more than one way, producing structurally and functionally different proteins from the same gene in different cells, or different developmental stages. It is estimated that more than 60% of human genes undergo alternative splicing, making the cellular process an essential mechanism for generating protein diversity [1–4]. Both constitutive and alternative splicing play important roles in the regulation of gene expression in eukaryotes [5]. A growing number of genetic diseases have been found to be caused by alternative or aberrant splicing events [6–9]. Mutations in cis-elements for splicing are primarily responsible for these diseases. However, tissue-specific splicing factors, such as those identified in amyotrophic lateral sclerosis (ALS), are also suggested to cause the aberrant pathogenic processing of pre-mRNA [10–13]. In addition, polymorphisms and genetic background contribute to variation in alternative splicing in specific genes, leading to modification of phenotypes in diseases (reviewed by Wang and Cooper [14]).
A typical exon is defined by its sequence, which is usually less than 300 base pairs long and, together with its surrounding introns, contains at least three core cis-elements: a 5' splicing site, a 3' splicing site, and a branch point [5, 15]. The splicing events begin with recognition of these elements by assembly of the spliceosome, a complex consisting of different small nuclear ribonuclear particles (snRNPs; U1, U2AF65, U4/U6, and U5), SR proteins (SRP30C, SRP20, SRP40, SRP55, SC35, SF2, hTra2b1), and other factors, including SF1 [5] (reviewed by Kramer [15]). The 5' site is initially paired with U1 snRNP and then replaced by U6 snRNP. The large subunit of splicing factor U2AF binds to the polypyrimidine tract while SF1 interacts with the branch site [16]. SR proteins, on the other hand, function as trans-splicing factors that facilitate the interaction of U1 snRNP with the 5' splice site, and the assembly of spliceosomes. through interactions with U2AF [17], SF1, and other splicing factors. After a series of conformational changes, introns are enzymatically removed and exons are ligated at 5' and 3' splicing sites. Exons, as well as introns, contain cis-regulatory splicing elements that activate or inhibit the splicing of a specific exon. These elements include splicing enhancers and splicing silencers [18–21]. Due to the involvement of many factors in the process of alternative splicing, one would expect that any change of these elements in the overall equation would tip the balance of exon inclusion/exclusion.
The human tau gene is more than 100 kilobases long and has been mapped to chromosome 17q21. It contains 16 exons, with the first exon (-1) as a part of the promoter (Figure 1). Exons 4A, 6, and 8 are not present in the tau mRNAs in human brain [22]. A full-length tau protein is encoded by exons 1–13 and consists of 441 amino acids. Three exons (exons 2, 3, and 10) undergo alternative splicing, giving rise to six isoforms of tau (Figure 1). The fetal tau isoform excludes exons 2, 3, and 10, while adults express all six tau isoforms, which range from 352–441 amino acids in length, depending on inclusion or exclusion of exons 2, 3, and 10 [23]. One of the major characteristics of the tau protein is the presence of four carboxy-terminal tandem repeat sequences (of 31 or 32 amino acids), each of which is encoded by exons 9, 10, 11, and 12 [24–27]. Since exon 10, which encodes the second repeat, is alternatively spliced., isoforms of tau are classified as either 3R taus (three repeats) or 4R taus (four repeats) (Figure 1) [28].

The tau genomic structure and splicing products. The tau gene has 16 exons. Exons 4A, 6, and 8 are not expressed in the human brain. Exons 2, 3, and 10 are alternatively spliced, producing six different mRNAs. The mRNA with or without exon 10 encodes a tau protein with four repeats (4R) or three repeats (3R), respectively. Adapted from Lee et al. [28].
Alternative splicing of exon 10 is developmentally regulated. It has been shown that >95% of tau mRNA in fetal brain excludes exon 10, while 40–50% of tau mRNAs in adult central nervous system includes this exon [29]. Exon 10 is relatively small at 93 base pairs. Exonic splicing enhancers, as well as an exonic splicing silencer, have been identified within this exon [29–31]. By analyzing mini-genes constructed from exon 9, intron 9, exon 10, intron 10 and exon 11 of the tau gene, we and others have also shown that additional cis-elements, including exon 9, exon 11, and trans-factors – for example, SRp20, SRp55, SRp75, SRp30c, ASF, SC35 and hTra2β1 – regulate exon 10 splicing [29, 32–34]. Studies of tau pre-mRNA identified a stem-loop structure at the exon 10-intron 10 boundary, the disruption of which alters exon 10 splicing [30, 35, 36]. Several splicing factors, including hTra2β1, SF2/ASF, SRp55, and SRp30C, affect exon 10 splicing by directly binding to exonic enhancers [37, 38]. However, it is unclear how other cis-elements and trans-factors are involved in exon 10 splicing.
Tau is abundantly expressed in the central nervous system, predominantly located in axons, and expression is also detectable in the axons of peripheral neurons. Only low levels of expression are observed in central nervous system astrocytes and oligodendrocytes [39–41]. The main function of tau is to bind and stabilize microtubules. The binding of tau to microtubules promotes their polymerization [39, 42]. Analysis of the tau sequence revealed that tau has four microtubule-binding motifs that are located in four repeat regions at the carboxyl terminus of the protein within conserved 18 residue long binding elements separated by less conserved spacers of 13–14 amino acids [28]. 4R-tau isoforms, which contain an additional binding element, have a greater affinity for microtubules and are more efficient in promoting their polymerization than 3R-tau isoforms [43, 44]. It is interesting that in adult human brain, the ratio of 3R-tau to 4R-tau isoforms is about 1 [43]. Therefore, one would expect that in a normal individual the ratio of 4R-tau/3R-tau proteins is balanced.
Tauopathies and mutations in Tau
Tauopathies comprise a group of neurodegenerative disorders that share a common pathological feature: the formation of insoluble intraneuronal aggregates composed of filamentous hyperphosphorylated tau proteins [28, 45]. Paired helical filaments and the hyperphosphorylation of tau are two of the most recognized molecular characteristics in Alzheimer's disease (AD) and several other neurodegenerative tauopathies, for example, frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) and progressive supranuclear palsy (PSP) [28]. The genetic relevance between tau and tauopathies came from linkage analysis of FTDP-17 and other neurodegenerative diseases, many of which were mapped to the region on chromosome 17q21–22 where the tau gene is located [46–50]. Subsequently, mutations were identified in the tau gene from FTDP-17 patients [48, 51–53]. To date, at least 35 distinct pathogenic mutations in the tau gene that lead to the formation of filaments made of hyperphosphorylated tau [54] have been described in a large number of families with FTDP-17 (Table 1; http://www.molgen.ua.ac.be/ftdmutations) [22, 55]. Several other mutations have also been described in PSP, Pick's disease, and Parkinson's disease (Table 2; http://www.molgen.ua.ac.be/ftdmutations) [56–60]. It has also been demonstrated that the majority of missense mutations that reside in coding regions of tau exons 9, 10, 12, or 13 alter the ability of tau to interact with microtubules and to promote microtubule assembly [51, 52, 59, 61–63]. On the other hand, additional mutations (for example, three silent mutations in exon 10, a deletion mutation, seven substitutions of the intron following exon 10, and one mutation in intron 9) disrupt exon 10 splicing cis-elements (Table 1). This mostly results in the increased production of tau mRNA with exon 10 inclusion, shifting the 3R-tau/4R-tau ratio in favor of more 4R-tau [51, 64–66]. It has been also demonstrated that the exonic mutations S305N, N279K, Δ280K, and N296H may not only alter tau functions, but also affect splicing enhancers, or destabilize the stem loop structure of tau pre-mRNA, causing abnormal exon 10 inclusion/exclusion [30, 67]. All these results suggest that aberrant exon 10 splicing is one of the most important mechanisms in the pathogenesis of tauopathies.
In addition to the mutations that have been identified in FTDP-17 patients, it has been suggested that additional pathogenic mutations or polymorphisms in introns that affect exon 10 splicing may be present in tauopathies [28]. Analysis of tau protein and mRNA in PSP indicates that more 4R-tau is produced in these patients [68]. However, unlike other tauopathies, PSP has usually been considered to be sporadic in nature. Conrad et al. [69], however, demonstrated an association between a polymorphic dinucleotide repeat marker, (GT)n, found in intron 9 of the tau gene and PSP. These results were subsequently confirmed by several other groups [70, 71]. Further studies indicate that a series of polymorphisms within the tau gene (for example, eight single nucleotide polymorphisms, one deletion, and the (TG)n repeat) are inherited in completed linkage disequilibrium, defining the tau gene as two extended haplotypes, H1 and H2 [72] (reviewed by Schraen-Maschke et al. [22]). Importantly, it was found that the most common allele (H1 haplotype) and genotype (H1/H1) were over-represented in PSP patients compared with normal controls. However, although the dinucleotide polymorphism is associated with extended H1/H2 haplotypes in disequilibrium, it has been considered a variation that may not have biological significance in the disease process. Taken together with the discoveries of the over-production of 4R-tau, and disequilibrium linkage to the tau gene in PSP patients, it is reasonable to presume that variations in intron 9 or intron 10 could be critical to influencing exon 10 inclusion/exclusion in these patients [71, 73–75].
In contrast, dysregulation of tau pre-mRNA splicing contributing to AD pathogenesis is still under debate. Several groups have shown by quantitative RT-PCR that there is no direct correlation between the 4R/3R ratio and tau pathology in AD patients [73–75]. On the other hand, Glatz et al. [76] demonstrated that in the temporal cortex of many AD brains, production of 4R-tau is significantly elevated, suggesting the importance of exon 10 inclusion/exclusion in the pathogenesis of AD. Nevertheless, although mutations have only been identified in the tau gene from FTDP-17 patients, tau polymorphisms, or other factors that may affect tau splicing, appear to be genetic risk factors for neurodegenerative diseases with tauopathy, in particular in PSP and AD patients.
Pharmacological intervention of tausplicing in tauopathy
Tau is hyperphosphorylated and forms aggregates in brains of tauopathic patients. One would expect that small molecules and drugs that prevent these processes could be identified and used as therapeutic treatments. Not surprisingly, extensive efforts have been made to identify these molecules to inhibit formation of tau filaments or tau hyperphosphorylation as the mainstream objective in the search for drugs for treatment of tauopathies [55, 77]. However, the overlooked exon 10 splicing of the tau gene has been gaining ground during the past few years as an area of study for potential treatment of tauopathies.
Several well-documented technologies that target aberrant splicing of specific genes in the treatment of diseases have been discussed in a recent review by Gallo et al. [55]. Among them, antisense oligoribonucleotides have been most commonly cited. For instance, Kalbfuss et al. [78] demonstrated that oligoribonucleotides binding to E10 splicing junctions could suppress the predominant inclusion of exon 10 in tau mRNA in rat PC12 cells. Other examples of modulating alternative splicing with antisense oligoribonucleotides include the genes SMN and Bcl-x [21, 79]. Another strategy that is often employed to stimulate the inclusion or exclusion of a specific exon is to use bifunctional oligoribonucleotides. These molecules are made of an antisense sequence designed to bind to an exonic element and a trans-factor sequence or peptide domain mimicking a trans-splicing factor [80–82]. Relative to antisense oligonuribonucleotides or bifunctional oligoribonucleotides, spliceosome-mediated RNA trans-splicing (SMaRT) is new. SMaRT is a technology that was developed based on the natural occurring trans-splicing event between two independently transcribed pre-mRNAs [83]. By taking advantage of this finding, Puttaraju et al. [83] developed a series of RNA pre-trans-splicing molecules capable of base-pairing to and trans-splicing with a conventional target pre-mRNA. They demonstrated that splicing of the conventional pre-mRNA was modified by these RNA pre-trans-splicing molecules both in vitro and in transfected cells. SMaRT has been tested in tau and SMN and shown great promise in correcting abnormal splicing of these two genes [83–85].
However, one specific area Gallo et al. [55] did not discuss in their review is whether small molecules or chemical compounds can potentially be used to treat diseases by targeting splicing defects. To provide proof-of-principle, we used neurodegenerative spinal muscular atrophy (SMA) as a model. SMA is caused by the homozygous loss of the SMN1 gene. SMN2, which differs from SMN1 by a single nucleotide within exon 7 that affects the efficiency of its incorporation into the mRNA transcript, is preserved in SMA patients [86, 87]. Increased exon 7 insertion into SMN2 mRNA has been suggested as a potentially effective approach for SMA treatment [88]. Using a cell based assay we have developed, we identified the phosphatase inhibitor sodium orthovanadate in a small screen of chemicals known to affect signaling pathways [88]. Additional screening of a library of compounds identified aclarubicin, a chemotherapeutic anthracycline drug that is related to adriamycin [89], and indoprofen, a common non-steroidal anti-inflammatory drug (NSAID) [90]. Aclarubicin, but not adriamycin, increased the amount of full-length transcript produced by the SMN2 gene, which in turn restored SMN protein levels in fibroblasts isolated from a SMA patient. In addition, we showed that aclarubicin has no effects on the splicing of Bcl-x, tau and Myosin V genes [89], indicating a relatively specific effect of aclarubicin on the splicing of the SMN2 gene [89]. We conclude from our studies of SMN that relative specific small molecules able to reverse abnormal splicing may be identified for disease treatment.
Conclusion
Aberrant inclusion of exon 10 into tau mRNA promotes the increased production of 4R-tau and has been increasingly recognized as one of the causes of tauopathies in FTDP-17, PSP, and many AD patients [51, 64–66, 76]. The similarity between exon 7 splicing in the SMN gene and exon 10 splicing in the tau gene prompted us to design a cell-based assay using luciferase as a reporter for the identification of small molecules that can reverse abnormal exon 10 inclusion. The mini-reported genes are displayed in Figure 2[91]. When exon 10 is excluded, the luciferase is expressed, while inclusion of exon 10 shifts luciferase out of the reading frame, resulting in no luciferase activity. Compounds that stimulate luciferase activity would likely act by reducing exon 10 inclusion in the tau mRNA. Based on this assay, we have carried out low- and high-throughput screening. While characterizing more than 500 compounds that stimulated luciferase activity (T Zou et al., unpublished data), we speculate that it is not out of reach that relative specific small molecules will be identified for further development of drugs for treatment of tauopathies.

Validation of a cell-based luciferase system for measurement of exon 10 splicing in the tau gene (adapted from Yu et al. [91]). (A) The luciferase mini-gene construct. The firefly luciferase gene was fused downstream of exon 11 in frame with a SI9/LI10 mini-gene containing the M14 (G to T) mutation. A stop codon was introduced into exon 10 by mutagenizing position 73 in exon 10 (A to T). The nucleotide A of the initiation codon ATG in the luciferase gene was converted to T. (B) A control mini-gene with a fused Renilla luciferase to exon 11. (C) The firefly luciferase mini-gene (A) was co-transfected into SKN-MC cells with constructs of SRp40, SRp20, or SRp55. Relative firefly luciferase activities were normalized using the Renilla luciferase control (B). (D) RT-PCR from cells described in (C) was carried out to quantify exon 10 inclusion/exclusion.
Abbreviations
- AD:
-
Alzheimer's disease
- ALS:
-
amyotrophic lateral sclerosis
- FTDP-17:
-
frontotemporal dementia with Parkinsonism linked to chromosome 17
- PSP:
-
progressive supranuclear palsy
- SMA:
-
spinal muscular atrophy
- SmaRT:
-
spliceosome-mediated RNA trans-splicing
- snRNP:
-
small nuclear ribonuclear particle.
References
Black DL: Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000, 103: 367-370. 10.1016/S0092-8674(00)00128-8.
Graveley BR: Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 2001, 17: 100-107. 10.1016/S0168-9525(00)02176-4.
Goldstrohm AC, Greenleaf AL, Garcia-Blanco MA: Co-transcriptional splicing of pre-messenger RNAs: considerations for the mechanism of alternative splicing. Gene. 2001, 277: 31-47. 10.1016/S0378-1119(01)00695-3.
Caceres JF, Kornblihtt AR: Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002, 18: 186-193. 10.1016/S0168-9525(01)02626-9.
Moore MJ, Query C, Sharp PA: Splicing of precursors to mRNA by the spliceosome. The RNA World. Edited by: Gabreels FJ, Atkins JF. 1993, Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press, 255-303.
Nakai K, Sakamoto H: Construction of a novel database containing aberrant splicing mutations of mammalian genes. Gene. 1994, 141: 171-177. 10.1016/0378-1119(94)90567-3.
Lorson CL, Hahnen E, Androphy EJ, Wirth B: A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999, 96: 6307-6311. 10.1073/pnas.96.11.6307.
Cooper TA, Mattox W: The regulation of splice-site selection, and its role in human disease. Am J Hum Genet. 1997, 61: 259-266. 10.1086/514856.
Bai G, Lipton SA: Aberrant RNA splicing in sporadic amyotrophic lateral sclerosis. Neuron. 1998, 20: 363-366. 10.1016/S0896-6273(00)80979-4.
Grabowski PJ: Splicing regulation in neurons: tinkering with cell-specific control. Cell. 1998, 92: 709-712. 10.1016/S0092-8674(00)81399-9.
Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein JD: Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron. 1998, 20: 589-602. 10.1016/S0896-6273(00)80997-6.
Aoki M, Lin CL, Rothstein JD, Geller BA, Hosler BA, Munsat TL, Horvitz HR, Brown RH: Mutations in the glutamate transporter EAAT2 gene do not cause abnormal EAAT2 transcripts in amyotrophic lateral sclerosis. Ann Neurol. 1998, 43: 645-653. 10.1002/ana.410430514.
Wong PC, Rothstein JD, Price DL: The genetic and molecular mechanisms of motor neuron disease. Curr Opin Neurobiol. 1998, 8: 791-799. 10.1016/S0959-4388(98)80123-2.
Wang GS, Cooper TA: Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007, 8: 749-761. 10.1038/nrg2164.
Kramer A: The structure and function of proteins involved in mammalian pre-mRNA splicing. Annu Rev Biochem. 1996, 65: 367-409. 10.1146/annurev.bi.65.070196.002055.
Berglund JA, Chua K, Abovich N, Reed R, Rosbash M: The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell. 1997, 89: 781-787. 10.1016/S0092-8674(00)80261-5.
Fu XD: The superfamily of arginine/serine-rich splicing factors. Rna. 1995, 1: 663-680.
Inoue K, Hoshijima K, Higuchi I, Sakamoto H, Shimura Y: Binding of the Drosophila transformer and transformer-2 proteins to the regulatory elements of doublesex primary transcript for sex-specific RNA processing. Proc Natl Acad Sci USA. 1992, 89: 8092-8096. 10.1073/pnas.89.17.8092.
Amrein H, Hedley ML, Maniatis T: The role of specific protein-RNA and protein-protein interactions in positive and negative control of pre-mRNA splicing by Transformer 2. Cell. 1994, 76: 735-746. 10.1016/0092-8674(94)90512-6.
Tian M, Maniatis T: A splicing enhancer complex controls alternative splicing of doublesex pre-mRNA. Cell. 1993, 74: 105-114. 10.1016/0092-8674(93)90298-5.
Taylor JK, Zhang QQ, Wyatt JR, Dean NM: Induction of endogenous Bcl-xS through the control of Bcl-x pre-mRNA splicing by antisense oligonucleotides. Nat Biotechnol. 1999, 17: 1097-1100. 10.1038/15079.
Schraen-Maschke S, Dhaenens CM, Delacourte A, Sablonniere B: Microtubule-associated protein tau gene: a risk factor in human neurodegenerative diseases. Neurobiol Dis. 2004, 15: 449-460. 10.1016/j.nbd.2003.12.009.
van Slegtenhorst M, Lewis J, Hutton M: The molecular genetics of the tauopathies. Exp Gerontol. 2000, 35: 461-471. 10.1016/S0531-5565(00)00114-5.
Crowther T, Goedert M, Wischik CM: The repeat region of microtubule-associated protein tau forms part of the core of the paired helical filament of Alzheimer's disease. Ann Med. 1989, 21: 127-132. 10.3109/07853898909149199.
Goedert M, Crowther RA: Amyloid plaques, neurofibrillary tangles and their relevance for the study of Alzheimer's disease. Neurobiol Aging. 1989, 10: 405-406. 10.1016/0197-4580(89)90076-6. discussion 412-404.
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA: Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989, 3: 519-526. 10.1016/0896-6273(89)90210-9.
Andreadis A, Brown WM, Kosik KS: Structure and novel exons of the human tau gene. Biochemistry. 1992, 31: 10626-10633. 10.1021/bi00158a027.
Lee VM, Goedert M, Trojanowski JQ: Neurodegenerative tauopathies. Annu Rev Neurosci. 2001, 24: 1121-1159. 10.1146/annurev.neuro.24.1.1121.
Gao QS, Memmott J, Lafyatis R, Stamm S, Screaton G, Andreadis A: Complex regulation of tau exon 10, whose missplicing causes frontotemporal dementia. J Neurochem. 2000, 74: 490-500. 10.1046/j.1471-4159.2000.740490.x.
D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD: Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA. 1999, 96: 5598-5603. 10.1073/pnas.96.10.5598.
D'Souza I, Schellenberg GD: Determinants of 4-repeat tau expression. Coordination between enhancing and inhibitory splicing sequences for exon 10 inclusion. J Biol Chem. 2000, 275: 17700-17709. 10.1074/jbc.M909470199.
Kondo S, Yamamoto N, Murakami T, Okumura M, Mayeda A, Imaizumi K: Tra2 beta, SF2/ASF and SRp30c modulate the function of an exonic splicing enhancer in exon 10 of tau pre-mRNA. Genes Cells. 2004, 9: 121-130. 10.1111/j.1356-9597.2004.00709.x.
Jiang Z, Tang H, Havlioglu N, Zhang X, Stamm S, Yan R, Wu JY: Mutations in tau gene exon 10 associated with FTDP-17 alter the activity of an exonic splicing enhancer to interact with Tra2 beta. J Biol Chem. 2003, 278: 18997-19007. 10.1074/jbc.M301800200.
Hernandez F, Perez M, Lucas JJ, Mata AM, Bhat R, Avila J: Glycogen synthase kinase-3 plays a crucial role in tau exon 10 splicing and intranuclear distribution of SC35. Implications for Alzheimer's disease. J Biol Chem. 2004, 279: 3801-3806. 10.1074/jbc.M311512200.
Grover A, Houlden H, Baker M, Adamson J, Lewis J, Prihar G, Pickering-Brown S, Duff K, Hutton M: 5' splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem. 1999, 274: 15134-15143. 10.1074/jbc.274.21.15134.
Jiang Z, Cote J, Kwon JM, Goate AM, Wu JY: Aberrant splicing of tau pre-mRNA caused by intronic mutations associated with the inherited dementia frontotemporal dementia with parkinsonism linked to chromosome 17. Mol Cell Biol. 2000, 20: 4036-4048. 10.1128/MCB.20.11.4036-4048.2000.
Wang Y, Wang J, Gao L, Lafyatis R, Stamm S, Andreadis A: Tau exons 2 and 10, which are misregulated in neurodegenerative diseases, are partly regulated by silencers which bind a SRp30c.SRp55 complex that either recruits or antagonizes htra2beta1. J Biol Chem. 2005, 280: 14230-14239. 10.1074/jbc.M413846200.
D'Souza I, Schellenberg GD: Arginine/serine-rich protein interaction domain-dependent modulation of a tau exon 10 splicing enhancer: altered interactions and mechanisms for functionally antagonistic FTDP-17 mutations Delta280K AND N279K. J Biol Chem. 2006, 281: 2460-2469. 10.1074/jbc.M505809200.
Cleveland DW, Hwo SY, Kirschner MW: Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977, 116: 227-247. 10.1016/0022-2836(77)90214-5.
Binder LI, Frankfurter A, Rebhun LI: The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985, 101: 1371-1378. 10.1083/jcb.101.4.1371.
LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI: Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci USA. 1995, 92: 10369-10373. 10.1073/pnas.92.22.10369.
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW: A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975, 72: 1858-1862. 10.1073/pnas.72.5.1858.
Goedert M, Jakes R: Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9: 4225-4230.
Goode BL, Feinstein SC: Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J Cell Biol. 1994, 124: 769-782. 10.1083/jcb.124.5.769.
Delacourte A: The molecular parameters of tau pathology. Tau as a killer and a witness. Adv Exp Med Biol. 2001, 487: 5-19.
Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG: Localization of disinhibition-dementia-parkinsonism-amyotrophy complex to 17q21–22. Am J Hum Genet. 1994, 55: 1159-1165.
Wijker M, Wszolek ZK, Wolters EC, Rooimans MA, Pals G, Pfeiffer RF, Lynch T, Rodnitzky RL, Wilhelmsen KC, Arwert F: Localization of the gene for rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration to chromosome 17q21. Hum Mol Genet. 1996, 5: 151-154. 10.1093/hmg/5.1.151.
Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D'Amato CJ, Gilman S: Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Conference Participants. Ann Neurol. 1997, 41: 706-715. 10.1002/ana.410410606.
Lendon CL, Lynch T, Norton J, McKeel DW, Busfield F, Craddock N, Chakraverty S, Gopalakrishnan G, Shears SD, Grimmett W, et al: Hereditary dysphasic disinhibition dementia: a frontotemporal dementia linked to 17q21–22. Neurology. 1998, 50: 1546-1555.
Bird TD, Wijsman EM, Nochlin D, Leehey M, Sumi SM, Payami H, Poorkaj P, Nemens E, Rafkind M, Schellenberg GD: Chromosome 17 and hereditary dementia: linkage studies in three non- Alzheimer families and kindreds with late-onset FAD. Neurology. 1997, 48: 949-954.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, et al: Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998, 393: 702-705. 10.1038/31508.
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD: Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998, 43: 815-825. 10.1002/ana.410430617.
Spillantini MG, Crowther RA, Kamphorst W, Heutink P, van Swieten JC: Tau pathology in two Dutch families with mutations in the microtubule- binding region of tau. Am J Pathol. 1998, 153: 1359-1363.
Crowther RA, Goedert M: Abnormal tau-containing filaments in neurodegenerative diseases. J Struct Biol. 2000, 130: 271-279. 10.1006/jsbi.2000.4270.
Gallo JM, Noble W, Martin TR: RNA and protein-dependent mechanisms in tauopathies: consequences for therapeutic strategies. Cell Mol Life Sci. 2007, 64: 1701-1714. 10.1007/s00018-007-6513-4.
Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, Koller WC, Bird TD, Trojanowski JQ, Lee VM, et al: An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002, 52: 511-516. 10.1002/ana.10340.
Pastor P, Pastor E, Carnero C, Vela R, Garcia T, Amer G, Tolosa E, Oliva R: Familial atypical progressive supranuclear palsy associated with homozigosity for the delN296 mutation in the tau gene. Ann Neurol. 2001, 49: 263-267. 10.1002/1531-8249(20010201)49:2<263::AID-ANA50>3.0.CO;2-K.
Rizzini C, Goedert M, Hodges JR, Smith MJ, Jakes R, Hills R, Xuereb JH, Crowther RA, Spillantini MG: Tau gene mutation K257T causes a tauopathy similar to Pick's disease. J Neuropathol Exp Neurol. 2000, 59: 990-1001.
Pickering-Brown S, Baker M, Yen SH, Liu WK, Hasegawa M, Cairns N, Lantos PL, Rossor M, Iwatsubo T, Davies Y, et al: Pick's disease is associated with mutations in the tau gene. Ann Neurol. 2000, 48: 859-867. 10.1002/1531-8249(200012)48:6<859::AID-ANA6>3.0.CO;2-1.
Murrell JR, Koller D, Foroud T, Goedert M, Spillantini MG, Edenberg HJ, Farlow MR, Ghetti B: Familial multiple-system tauopathy with presenile dementia is localized to chromosome 17. Am J Hum Genet. 1997, 61: 1131-1138. 10.1086/301594.
Bugiani O: FTDP-17: phenotypical heterogeneity within P301S. Ann Neurol. 2000, 48: 126-10.1002/1531-8249(200007)48:1<126::AID-ANA21>3.0.CO;2-N.
Sperfeld AD, Collatz MB, Baier H, Palmbach M, Storch A, Schwarz J, Tatsch K, Reske S, Joosse M, Heutink P, et al: FTDP-17: an early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann Neurol. 1999, 46: 708-715. 10.1002/1531-8249(199911)46:5<708::AID-ANA5>3.0.CO;2-K.
Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, Redi F, Crowther RA, Pietrini P, Ghetti B, et al: Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol. 1999, 58: 1207-1226. 10.1097/00005072-199912000-00002.
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B: Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998, 95: 7737-7741. 10.1073/pnas.95.13.7737.
Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H, Morris JG, Fulham MJ, Schofield PR: Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain. 2000, 123: 880-893. 10.1093/brain/123.5.880.
Yasuda M, Takamatsu J, D'Souza I, Crowther RA, Kawamata T, Hasegawa M, Hasegawa H, Spillantini MG, Tanimukai S, Poorkaj P, et al: A novel mutation at position +12 in the intron following exon 10 of the tau gene in familial frontotemporal dementia (FTD-Kumamoto). Ann Neurol. 2000, 47: 422-429. 10.1002/1531-8249(200004)47:4<422::AID-ANA4>3.0.CO;2-G.
Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M: FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett. 1999, 443: 93-96. 10.1016/S0014-5793(98)01696-2.
Sergeant N, Wattez A, Delacourte A: Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively "exon 10" isoforms. J Neurochem. 1999, 72: 1243-1249. 10.1046/j.1471-4159.1999.0721243.x.
Conrad C, Andreadis A, Trojanowski JQ, Dickson DW, Kang D, Chen X, Wiederholt W, Hansen L, Masliah E, Thal LJ, et al: Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann Neurol. 1997, 41: 277-281. 10.1002/ana.410410222.
Rojo A, Pernaute RS, Fontan A, Ruiz PG, Honnorat J, Lynch T, Chin S, Gonzalo I, Rabano A, Martinez A, et al: Clinical genetics of familial progressive supranuclear palsy. Brain. 1999, 122: 1233-1245. 10.1093/brain/122.7.1233.
Pittman AM, Myers AJ, Duckworth J, Bryden L, Hanson M, Abou-Sleiman P, Wood NW, Hardy J, Lees A, de Silva R: The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum Mol Genet. 2004, 13: 1267-1274. 10.1093/hmg/ddh138.
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M: Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999, 8: 711-715. 10.1093/hmg/8.4.711.
Connell JW, Rodriguez-Martin T, Gibb GM, Kahn NM, Grierson AJ, Hanger DP, Revesz T, Lantos PL, Anderton BH, Gallo JM: Quantitative analysis of tau isoform transcripts in sporadic tauopathies. Brain Res Mol Brain Res. 2005, 137: 104-109. 10.1016/j.molbrainres.2005.02.014.
Boutajangout A, Boom A, Leroy K, Brion JP: Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer's disease. FEBS Lett. 2004, 576: 183-189. 10.1016/j.febslet.2004.09.011.
Ingelsson M, Ramasamy K, Cantuti-Castelvetri I, Skoglund L, Matsui T, Orne J, Kowa H, Raju S, Vanderburg CR, Augustinack JC, et al: No alteration in tau exon 10 alternative splicing in tangle-bearing neurons of the Alzheimer's disease brain. Acta Neuropathol (Berl). 2006, 112: 439-449. 10.1007/s00401-006-0095-3.
Glatz DC, Rujescu D, Tang Y, Berendt FJ, Hartmann AM, Faltraco F, Rosenberg C, Hulette C, Jellinger K, Hampel H, et al: The alternative splicing of tau exon 10 and its regulatory proteins CLK2 and TRA2-BETA1 changes in sporadic Alzheimer's disease. J Neurochem. 2006, 96: 635-644. 10.1111/j.1471-4159.2005.03552.x.
Chun W, Johnson GV: The role of tau phosphorylation and cleavage in neuronal cell death. Front Biosci. 2007, 12: 733-756. 10.2741/2097.
Kalbfuss B, Mabon SA, Misteli T: Correction of alternative splicing of tau in frontotemporal dementia and parkinsonism linked to chromosome 17. J Biol Chem. 2001, 276: 42986-42993. 10.1074/jbc.M105113200.
Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR: Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007, 5: e73-10.1371/journal.pbio.0050073.
Cartegni L, Krainer AR: Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003, 10: 120-125. 10.1038/nsb887.
Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F: Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci USA. 2003, 100: 4114-4119. 10.1073/pnas.0633863100.
Villemaire J, Dion I, Elela SA, Chabot B: Reprogramming alternative pre-messenger RNA splicing through the use of protein-binding antisense oligonucleotides. J Biol Chem. 2003, 278: 50031-50039. 10.1074/jbc.M308897200.
Puttaraju M, Jamison SF, Mansfield SG, Garcia-Blanco MA, Mitchell LG: Spliceosome-mediated RNA trans-splicing as a tool for gene therapy. Nat Biotechnol. 1999, 17: 246-252. 10.1038/6986.
Rodriguez-Martin T, Garcia-Blanco MA, Mansfield SG, Grover AC, Hutton M, Yu Q, Zhou J, Anderton BH, Gallo JM: Reprogramming of tau alternative splicing by spliceosome-mediated RNA trans-splicing: implications for tauopathies. Proc Natl Acad Sci USA. 2005, 102: 15659-15664. 10.1073/pnas.0503150102.
Coady TH, Shababi M, Tullis GE, Lorson CL: Restoration of SMN function: delivery of a trans-splicing RNA re-directs SMN2 pre-mRNA splicing. Mol Ther. 2007, 15: 1471-1478. 10.1038/sj.mt.6300222.
Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH: The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997, 6: 1205-1214. 10.1093/hmg/6.8.1205.
Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al: Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995, 80: 155-165. 10.1016/0092-8674(95)90460-3.
Zhang M, Lorson C, Androphy E, Zhou J: An in vivo cellular system to identify compounds that stimulate inclusion of exon 7 in SMN2 mRNA. Gene Therapy. 2001, 8: 1532-1538. 10.1038/sj.gt.3301550.
Andreassi C, Zhou J, Coovert DD, Zhang M, Monani UR, Whitney M, Androphy E, Burghes AHM: Identification of a compound that alters splicing of the SMN2 gene to increases full length SMN mRNA and restores gems to cells derived from type I SMA patients. Hum Mole Gene. 2001, 2841-2849. 10.1093/hmg/10.24.2841.
Lunn MR, Root DE, Martino AM, Flaherty SP, Kelley BP, Coovert DD, Burghes AH, Man NT, Morris GE, Zhou J, et al: Indoprofen upregulates the survival motor neuron protein through a cyclooxygenase-independent mechanism. Chem Biol. 2004, 11: 1489-1493. 10.1016/j.chembiol.2004.08.024.
Yu Q, Guo J, Zhou J: A minimal length between tau exon 10 and 11 is required for correct splicing of exon 10. J Neurochem. 2004, 90: 164-172. 10.1111/j.1471-4159.2004.02477.x.
Acknowledgements
This article has been published as part of BMC Neuroscience Volume 9 Supplement 2: 2008 Proceedings of the 8th International Conference on Alzheimer's Disease Drug Discovery The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2202/9?issue=S2.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhou, J., Yu, Q. & Zou, T. Alternative splicing of exon 10 in the taugene as a target for treatment of tauopathies. BMC Neurosci 9 (Suppl 2), S10 (2008). https://doi.org/10.1186/1471-2202-9-S2-S10
Published:
DOI: https://doi.org/10.1186/1471-2202-9-S2-S10