
Abstract
Background
MicroRNAs (miRNAs) are a class of noncoding small RNAs (sRNAs) that are 20–24 nucleotides (nt) in length. Extensive studies have indicated that miRNAs play versatile roles in plants, functioning in processes such as growth, development and stress responses. Chilling is a common abiotic stress that seriously affects plants growth and development. Recently, chilling-responsive miRNAs have been detected in several plant species. However, little is known about the miRNAs in the model plant tomato. ‘LA1777’ (Solanum habrochaites) has been shown to survive chilling stress due to its various characteristics.
Results
Here, two small RNA libraries and two degradome libraries were produced from chilling-treated (CT) and non-chilling-treated (NT) leaves of S. habrochaites seedlings. Following high-throughput sequencing and filtering, 161 conserved and 236 novel miRNAs were identified in the two libraries. Of these miRNAs, 192 increased in the response to chilling stress while 205 decreased. Furthermore, the target genes of the miRNAs were predicted using a degradome sequencing approach. It was found that 62 target genes were cleaved by 42 conserved miRNAs, while nine target genes were cleaved by nine novel miRNAs. Additionally, nine miRNAs and six target genes were validated by quantitative real-time PCR (qRT-PCR). Target gene functional analysis showed that most target genes played positive roles in the chilling response, primarily by regulating the expression of anti-stress proteins, antioxidant enzyme and genes involved in cell wall formation.
Conclusions
Tomato is an important model plant for basic biological research. In this study, numerous conserved and novel miRNAs involved in the chilling response were identified using high-throughput sequencing, and the target genes were analyzed by degradome sequencing. The work helps identify chilling-responsive miRNAs in tomato and increases the number of identified miRNAs involved in chilling stress. Furthermore, the work provides a foundation for further study of the regulation of miRNAs in the plant response to chilling stress.
Similar content being viewed by others

Background
Chilling, a type of low temperature stress, is a common abiotic environmental stress that seriously affects the growth and development of plants [1]. Chilling stress has attracted increasing attention. To date, many studies about plant morphological and physiological responses to chilling stress have been reported [2, 3]. In addition, genes related to the chilling response have been identified, such as genes encoding the transcription factors DREB/CBF [4], WRKY [5], MYB [6] and so on.
In recent years, microRNAs (miRNAs) have become a research hotspot [7–9]. MiRNAs are a distinct class of tiny noncoding RNAs approximately 21 nucleotides (nt) in length [10, 11]. The RNAs participate in regulating gene expression at the posttranscriptional level in both plants and animals [12, 13]. Prior to their regulation of gene expression, miRNAs are generated from hairpin precursors by dicer-like (DCL) into miRNA::miRNA* duplexes [14]. The miRNA* strand is then degraded, the mature miRNA joins with argonaute (AGO) and the RNA-induced silencing complex (RISC) is generated. Finally, the silencing complex targets protein-coding mRNAs by cleaving the mRNAs at specific positions or by repressing translation [12, 15].
Since the discovery of miRNA genes in the nematode Caenorhabditis elegans 20 years ago [16], miRNAs have been intensively studied. Notably, in recent years, new generation sequencing technology has led to the identification of numerous miRNAs, especially novel and low copy number miRNAs, in many plant species, such as wild soybean [17], rape [18], radish [19], Medicago truncatula[20] and so on. In addition, the interaction between miRNAs and target genes has received much attention. Degradome sequencing is an emerging technology used to predict and verify target genes of miRNAs. Compared with traditional methods, i.e., bioinformatics prediction and 5’ RACE, degradome sequencing is rapid and effective [17, 21]. With the utilization of the two high-sequencing approaches, numerous miRNAs have been identified, and great progress has been made in elucidating the interaction between miRNAs and target genes, especially chilling stress-related miRNAs. For example, many chilling-responsive miRNAs have been detected in Arabidopsis[22, 23], Brachypodium[24], Prunus persica[25], Populus tomentosa[26], Oryza sativa[27] and Zea may[28]. Studies of the functions of miRNAs have been carried out simultaneously.
Tomato is a globally important vegetable of the Solanum family [29]. Although tomato is widely grown in various temperature zones, its growth and development are rather sensitive to temperature stress, including extreme chilling stress [3]. However, most cultivated and commercial tomato cultivars are considered to be sensitive to chilling stress [3, 30].
With the completion of the tomato genome sequence [31], tomato has become a model system for research on the interaction between miRNAs and their target genes. Although many tomato miRNAs have been identified by traditional Sanger cloning, deep sequencing and bioinformatics approach [29, 32–35], chilling-responsive miRNAs in tomato have not been reported.
‘LA1777’ (S. habrochaites) is one kind of wild tomato. And it exhibits higher chilling-tolerance ability than other cultivars in our previous research, which is in agreement with other studies [36, 37]. In this study, using ‘LA1777’, two small RNA libraries from leaves treated with and without chilling stress (4°C) were constructed for high-throughput Solexa sequencing. Then 4,342,604 and 7,231,609 clean reads were obtained from the two libraries, respectively. The results showed that small RNAs (sRNAs) 21 and 24 nt in length were the most abundant classes. A total of 161 conserved and 236 novel miRNAs were identified in the two libraries. Based on the abundance of miRNAs, the expression amounts between the two libraries were compared, 192 increased and 205 decreased miRNAs were detected in the chilling stress. In addition, degradome sequencing analysis was adopted to identify target genes of the miRNAs. It was found that 62 target genes were cleaved by 42 conserved miRNAs and nine target genes were cleaved by nine novel miRNAs. The verification of nine miRNAs and six target genes by quantitative real-time PCR (qRT-PCR) basically confirmed the sequencing results. Target gene functional analysis showed most target genes were involved in the defense response through regulating the expression of anti-stress proteins, antioxidant enzyme and genes involved in cell wall formation. In addition to identify chilling-responsive conserved and novel miRNAs in tomato, the work lays the foundation for further elucidating the regulation of miRNAs in response to chilling stress.
Results
Analysis of small RNA data from the libraries
To identify miRNAs from tomato that respond to chilling stress, two small RNA libraries from tomato leaves treated with and without chilling stress were constructed and sequenced using an Illumina Genome Analyzer II (LC Sciences, Hangzhou, China). A total of 6,199,678 and 9,919,778 raw reads were generated from the chilling-treated (CT) library and non-chilling-treated (NT) library, respectively (Table 1). After removing 3’ and 5’ adaptors, sequences with <15 nt and >29 nt and junk reads, the remaining reads were searched against the Rfam (http://rfam.xfam.org/), repeat databases (http://www.girinst.org/repbase/update/index.html) and tomato genome sequence (http://solgenomics.net); 13.71% and 12.59% (reads/reads) of total sRNAs in the CT and NT library were filtered out, respectively. Finally, 4,342,604 and 7,231,609 clean reads corresponding to 1,208,259 and 1,795,513 unique reads were obtained from the two libraries, respectively. The number and percentage of various sRNAs were listed in Table 1.
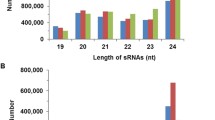
The length distribution of the sRNAs from the two libraries ranged from 15 to 29 nt, as shown in Figure 1. The proportion of 19–25 nt sequences was high in both libraries, comprising over 75% of the sequences; sRNAs 21 nt and 24 nt in length were the two main sRNA classes among the sequences. Among unique sequences, 24 nt sRNA was the most abundant category, which was in agreement with previous reports on cucumber [38], grapevine [39] and rice [40].

Length distribution of sRNAs in two libraries from tomato. a size distribution of total sequences. b size distribution of unique sequences.
Identification of conserved miRNAs from tomato
To detect conserved miRNAs from the two tomato libraries, all clean reads were aligned against mature plant miRNAs and the precursors in miRBase 20.0 (June 2013), allowing for a maximum of one mismatch in the first 16 nt of the miRNA and three mismatches in total between the target miRNAs and the known miRNAs. Finally, 161 conserved miRNAs belonging to 41 miRNA families were identified in the two libraries (Additional file 1: Table S1, Additional file 2: Figure S1). Interestingly, the number of miRNAs varied among families (Figure 2). The largest family was the 156 miRNA family with 21 members, including two members only identified in the NT library and the remaining 19 ones presented in both two libraries, followed by the 171 miRNA family, while the 169 and 384 miRNA families were the smallest, each containing only one miRNA member. In addition, the member of the 169 miRNA family and one member of 408 miRNA family were exclusively detected in the CT library. Among these, 47 miRNAs designated as p3 or p5 were identified for the first time in tomato, which comprised new flanking sequences or other sequences of known miRNAs deposited in miRBase 20.0.

Numbers of conserved miRNAs in different miRNA families in tomato.
In the CT and NT libraries, there were 153 miRNAs in common, while two miRNAs that were sha-miR169c-3p_stu and sha-miR408a-5p_stu were only found in the CT library; six miRNAs that were sha-miR156e-p3_stu, sha-miR156f-p5_stu, sha-miR162-p5_cme, sha-miR166g-p5_nta, sha-miR167-p3 and sha-miR171h-p5_vvi were specific to the NT library (Figure 3, Additional file 1: Table S1). On the other hand, the frequency of miRNAs in the two libraries can be regarded as an index for estimating the expression amounts of miRNAs. Therefore, after sequencing, raw reads obtained by sequencing were normalized for the chi-square test, the fisher-exact test and log2 ratio determination between the CT and NT libraries. The result of this comparison showed that 66 miRNA members increased in response to chilling stress with log2 (CT/NT) > 0, whereas 95 members decreased with log2 (CT/NT) < 0. Only miRNAs with significant expression amounts (chi-square test and fisher-exact test) of both < 0.05 and | log2 (CT/NT) | ≥ 1 were considered to be significantly regulated. Among these miRNAs, 12 miRNAs that were sha-miR166a-p5, sha-miR319, sha-miR397_nta, sha-miR397-p5, sha-miR398a-3p_stu, sha-miR398a-5p_stu, sha-miR398a-p3_cme, sha-miR399-p5, sha-miR408_nta, sha-miR408a-3p_stu, sha-miR408b-5p_stu and sha-miR530a_cme significantly increased in response to chilling stress, while 20 miRNAs significantly decreased (Additional file 1: Table S1). Of the 47 newly identified miRNAs, most had lower expression amounts with norm read <100, possibly because the new flanking sequences or other miRNA sequences were theoretically more volatile than the others [13, 41].

Venn diagram of miRNAs identified in the CT (pink) and NT (green) libraries. a conserved miRNAs. b novel miRNAs.
Identification of novel miRNAs from tomato
To identify potentially novel miRNAs from tomato, after filtering out the conserved miRNAs, sequences ranging from 19 to 24 nt were used to further analysis using strict criteria for miRNA prediction. Second structures of the pre-miRNAs were predicted by UNAfold software (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) [17, 42]; only those with stable hairpin structures were considered. The minimal folding free index (MFEI) was an important feature used to distinguish miRNAs from other noncoding RNAs [43]. In this study, miRNAs with MEFIs exceeding 0.80 were regarded as candidate miRNAs. Ultimately, 236 novel miRNAs corresponding to 224 unique sequences generated from 171 precursors were discovered from tomato in the two libraries (Additional file 3: Table S2, Additional file 4: Figure S2).
As shown in a Venn diagram, 215 novel miRNAs were shared in both libraries; 14 novel miRNAs were unique to the NT library, while seven novel miRNAs were only identified in the CT library (Figure 3). Meanwhile, the expression amounts of the miRNAs between the two libraries were compared. Of the miRNAs identified, 126 miRNAs increased in response to chilling stress, while 110 miRNAs decreased. Moreover, 11 miRNAs significantly increased, while six miRNAs significantly decreased (Additional file 3: Table S2). On the other hand, compared with the conserved miRNAs, most of the novel miRNAs had low expression amounts with norm read <100, which was consistent with previous reports in Arabidopsis[41, 44] and wheat [45]. However, several miRNAs, such as PC-58-3p and PC-58-5p, were present at high amounts with norm read > 1,000, which was different from previous results.
Identification of conserved miRNAs targets in tomato
To understand the function of the miRNAs, high-throughput degradome sequencing, a new technology [21, 46], was utilized to identify the miRNAs targets. In this study, 12,654,699 and 10,844,042 raw reads corresponding to 8,431,161 and 7,202,445 unique reads were initially obtained in the CT and NT library, respectively (Table 2). Following BLAST analysis, 74.14% and 64.13% of the reads could be mapped to mRNA (ftp://ftp.solgenomics.net/est_sequences/species/Tomato), respectively.
For data analysis, CleaveLand3.0 software was used to detect the cleavage products guided by miRNAs. According to the sequencing result, the abundance of sequenced tags was plotted on every transcript, and the cleaved target transcripts were classified into five categories, namely, category 0, category 1, category 2, category 3 and category 4. For the conserved miRNAs, 281 sequences of 62 target genes from 42 conserved miRNAs were identified out of 161 conserved miRNAs (Additional file 5: Table S3). However, the targets of most miRNAs were not identified, perhaps because the abundance of cleavage products was too low to be detected or because the interaction between miRNAs and the targets did not involve cleaving.
Of these sequences, 182 sequences of target genes were shared between the two libraries, including 97 decreased, 83 increased and two constant sequences, while 88 sequences were only identified in the CT library and 11 were only found in the NT library. In the CT library, 270 sequences for conserved miRNAs were identified. The abundance ranged from eight to 3,872 tags per billion. Of these sequences, 146 belonged in category 0, while 13 sequences of conserved miRNAs were in category 1; 96 sequences were in category 2; two were in category 3 and 13 were in category 4. By contrast, 193 sequences of miRNAs were identified in the NT library with abundance ranging from nine to 7,470 tags per billion. Of these, the most abundant category was category 0 including 98 sequences. Additionally, eight, 53 and 34 sequences of conserved miRNAs were grouped into category 1, 2 and 4, respectively (Additional file 5: Table S3). All of the identified target plots (t-plots) were shown in Additional file 6: Figure S3 and additional file 7: Figure S4. Sequenced tags of targets between the two libraries were compared, and the result showed that there were more specific targets detected in the CT library than in the NT library. The result suggested that chilling stress might cause the cleavage of miRNA targets, leading to the accumulation of cleaved fragments.
Among the identified targets, more target genes were predicted to be cleaved by one miRNA. For example, TCP2, TCP4 and TRPB2 were targeted by sha-miR319b_stu. On the other hand, one target was usually cleaved by more than one miRNAs. For example, SPL6 was predicted to be cleaved by sha-miR156a, sha-miR156c, sha-miR156c_nta, sha-miR156d_nta, sha-miR156e_stu, sha-miR156g_stu, sha-miR156h_stu, sha-miR156i_stu and sha-miR156j_stu. By contrast, only one target was found for sha-miR156d_nta and sha-miR393-5p_stu. Interestingly, several targets were cleaved by pairs of miRNAs, such as sha-miR396b_nta and sha-miR396c_nta, both of which targeted CYSPL, RD21A, DRM1, CPR1, CYP40 and DRM2, which suggested that the two miRNAs might cooperate to regulate gene expression (Additional file 5: Table S3).
To understand more about the roles of miRNAs in the response to chilling stress, gene ontology (GO) and kyoto encyclopedia of genes and genomes (KEGG) pathway analysis were performed. GO analysis showed that the targets were associated with various functions (Additional file 8: Figure S5). GO enrichment analysis showed that the target genes of differentially expressed conserved miRNAs were mainly involved in metal ion binding, cytoplasm and superoxide dismutase activity. However, fewer functions were found using KEGG analysis (Additional file 9: Table S4). Gene functional analysis showed that sha-miR398_nta targeted 62 sequences of Cu/Zn superoxide dismutase (EC:1.15.1.1) gene, including 18 found in both libraries. The result suggested that the 18 sequences were not key regulators of the chilling response, while the remaining 44 sequences might play important roles in the stress response. Additionally, transcription factors and stress-responsive proteins were also targeted by miRNAs. For example, scarecrow-like protein, ribosomal protein and TCP family transcription factors targeted by sha-miR171 (sha-miR171a, sha-miR171a_nta, sha-miR171b-3p_stu, sha-miR171c_mtr and sha-miR171d), sha-miR156i-p3_nta and sha-miR319b_stu, respectively, were mainly found in the CT library, which suggested that these miRNAs might play important roles in the chilling stress response, which was in agreement with the results reported in rice [27]. On the other hand, sha-miR396 (sha-miR396a_nta and sha-miR396b_nta) directly targeted low-temperature-induced proteins that might play positive roles in the chilling response, and sha-miR160a and sha-miR167b_nta might participate in the stress response by targeting an auxin response factor gene.
Identification of novel miRNAs targets in tomato
Like the conserved miRNAs, the targets of most novel miRNAs were not identified; out of the 236 novel miRNAs identified, only 34 sequences of nine genes were predicted for nine novel miRNAs (Table 3, Additional file 10: Table S5). Among these target sequences, 20 were identified in both libraries, including 15 decreased and five increased, while 11 were only found in the CT library and three were unique to the NT library. The abundance of the sequenced tags was plotted for every transcript (Additional file 6: Figure S3, Additional file 7: Figure S4, Additional file 10 Table S5); the transcripts were distributed into four categories. Unlike the targets of conserved miRNAs, most tags of novel miRNAs were grouped into category 2. In the CT library, 31 sequences with abundance ranging from eight to 9,509 tags per billion were detected. Of these 31 sequences, 14 were in category 2; 11 were in category 0; five were in category 4 and only one was in category 1. By contrast, 23 sequences with abundance ranging from 18 to 3,381 tags per billion were found in the NT library. There were five, two and 16 sequences in category 0, 1 and 2, respectively.
Unlike the conserved miRNAs, more target tags of the novel miRNAs were detected in the NT library, which suggested that under chilling stress, cleaving might not be the main mode of action between novel miRNAs and target genes. Perhaps eliciting mRNA degradation or arresting mRNA translation played a dominant role in this interaction. GO enrichment analysis showed that integral to membrane, cellulose biosynthetic process and cellular cell wall organization were the main functions of the target genes cleaved by the differentially expressed novel miRNAs (Additional file 8: Figure S5).
Compared with the targets of conserved miRNAs, target gene description showed that the target genes of novel miRNAs were with special functions (Table 3). Receptor-like protein kinase is regarded as the sensor or transducer of stress signals. A receptor-like protein kinase gene targeted by PC-58-5p was identified in the current study. The gene encoding xyloglucan endotransglucosylase/hydrolases (EC:2.4.1.207) targeted by PC-125-3p was detected, and the tag sequences were only detected in the NT library. In addition, PC-102-5p, targeting a cellulose synthase (EC:2.4.1.12) subunit gene, was detected in the two libraries. And the inositol-pentakisphosphate 2-kinase-like (EC:2.7.1.158) gene, which plays a role in the reaction between ATP and ADP and is targeted by PC-146-5p, was also identified.
qRT-PCR verification
To examine the miRNAs expression amounts and verify the sequencing result, nine miRNAs including six conserved miRNAs and three novel miRNAs were selected for further analysis. Following previous studies [39, 47, 48], poly (A)-tailed qRT-PCR was utilized to analyze the temporal and spatial expression patterns of these miRNAs in tomato. For further verification and confirmation, another chilling-tolerant tomato cultivar, ‘Hezuo908’, was subjected to the same treatment as the tomato cultivar ‘LA1777’. Quantitative analysis showed that compared with the expression amounts of miRNAs in the NT libraries, the expression amounts of the nine miRNAs varied in the CT libraries of both cultivars (Figure 4). At 1 h of treatment, the expression amounts of sha-miR156a, sha-miR482b and PC-69-5p were significantly higher in both CT libraries than that in the NT libraries, followed by a marked decrease and then gradually increased. The expression amounts of sha-miR6023-p5 and sha-miR6027 were also significantly higher in both CT libraries at 1 h and then obviously decreased at 12 h. At 4 h, the expression amounts of sha-miR160a, sha-miR398_nta and PC-46-5p were significantly higher in both CT libraries than that in the NT libraries and then decreased, while the expression amounts of PC-170-5p were significantly higher in the CT libraries at 8 h.Meanwhile, the expression amounts of six target genes cleaved by sha-miR160a, sha-miR398_nta, sha-miR482b, sha-miR6027 and PC-46-5p were also verified by qRT-PCR. As shown in Figure 5, the expression amounts of target genes cleaved by sha-miR160a and PC-46-5p were both significantly lower in the CT libraries, followed by an increase, and they were significantly higher in the CT libraries at 48 h. The expression amounts of the target genes cleaved by sha-miR398_nta and sha-miR6027 exhibited the same trend, while they were significantly higher in the CT libraries and peaked at 24 h and 12 h, respectively. At 1 h, the expression amounts of target genes of sha-miR482b were significantly lower in CT libraries, followed by an increase and peaked at 24 h. The results were basically identical to the sequencing data.

qRT-PCR validation of chilling-responsive conserved and novel miRNAs in ‘LA1777’ (a) and ‘Hezuo908’ (b) tomato. The reference gene was 5S rRNA. All reactions were repeated three times. Normalized miRNA expression amount at 1 h without chilling treatment was arbitrarily set to 1. Differences between the NT and CT libraries were tested with a T test. *indicates a significant difference between the two libraries (<0.05).

qRT-PCR validation of target genes in ‘LA1777’ (a) and ‘Hezuo908’ (b) tomato. The reference gene was Actin. All reactions were repeated three times. Normalized target gene expression amount at 1 h without chilling treatment was arbitrarily set to 1. Differences between the NT and CT libraries were tested with a T test. *indicates a significant difference between the two libraries (<0.05).
Network analysis
A total of 71 genes targeted by 51 miRNAs were detected in response to chilling stress. The network between miRNAs and target genes was elucidated according to GO analysis (Figure 6). As shown in the network, the increased sha-miR156 (sha-miR156a, sha-miR156c, sha-miR156c_nta, sha-miR156d_nta, sha-miR156e_stu, sha-miR156g_stu, sha-miR156h_stu, sha-miR156i_stu, sha-miR156j_stu and sha-miR156i-p3_nta), sha-miR171b-3p_stu, sha-miR319b_stu, sha-miR396 (sha-miR396a_nta, sha-miR396b_nta and sha-miR396c_nta), sha-miR398_nta and PC-102-5p played important regulatory roles in the network by regulating the expression of genes encoding anti-stress proteins (SPL, RL, SCL and TCP), antioxidant enzyme (SODC2) and cellulose synthesis (CESA1), while the decreased sha-miR160a, sha-miR167b_nta, sha-miR168 (sha-miR168a-5p and sha-miR168b-5p), sha-miR171a, sha-miR171a_nta, sha-miR171c_mtr, sha-miR171d, PC-58-5p, PC-146-5p and PC-125-3p were involved in signal transduction and cell wall formation, as well as regulation of genes encoding anti-stress proteins (SCL15/27). Of the target genes examined, SPL, SCL, TCP, SODC2, CESA1, auxin response factors genes (ARFV, ARFQ, ARFH and ARFL), IKU2, IPPK and XTH32 might be the most important genes in the entire network.

MicroRNA-gene network analysis. Target genes of microRNAs identified in the CT and NT libraries from tomato were assembled into the network according to GO annotation. The circles represent miRNAs (red indicates upregulated miRNAs and green indicates downregulated miRNAs), and rectangles represent the target genes.
Discussion
MiRNAs, a class of noncoding small RNAs, are important regulators involved in plant growth, development and stress responses that have received increasing amounts of attention [9, 32, 49–51]. In recent years, a new approach for discovering miRNAs, high-throughput sequencing technology, has been widely used to identify conserved and novel miRNAs in plants, which has enlarged the realm of miRNA research and made miRNA a hotspot of epigenetic research. Abiotic stress, a common stress that seriously affects plant production, has also received increasing attention. The miRNA regulation theory and the utilization of high-throughput sequencing have opened up a new area of abiotic stress research. In recent years, great progress has been made in understanding the role of miRNAs in the response to various environmental stresses, such as drought [52, 53], salt [54, 55] and waterlogging stresses [56]. Chilling is a common abiotic stress that seriously affects normal plant growth, development, yield and quality. Plant miRNAs involved in chilling response have been increasingly studied during the past few years. However, to our knowledge, few such studies have been reported in tomato.
Tomato is an important vegetable crop that is grown worldwide [57]. Chilling is a common abiotic stress that affects tomato cultivation, especially in the greenhouse during the cooler seasons [58]. In China, solar greenhouses are the principal systems employed for tomato cultivation in the spring and winter, when chilling stress is common [58]. Since miRNAs are thought to be important for the abiotic stress response and a high-through sequencing approach is available, research on chilling-responsive miRNAs in tomato will help to reveal the response mechanisms of the miRNAs and to further elucidate the mechanism of miRNA regulation in general. More importantly, the study provides a theoretical foundation for breeding chilling-tolerant tomato cultivars and for further research on miRNA regulation of the response to chilling stress.
Conserved and novel miRNAs in tomato
To identify chilling-responsive miRNAs from tomato, two small RNA libraries from tomato seedlings treated with and without chilling stress were constructed. A high-through sequencing approach was utilized to identify conserved and novel miRNAs; 4,342,604 and 7,231,609 clean reads were obtained by high-throughput sequencing of the two libraries, respectively. Following filtering, a total of 161 conserved miRNAs belonging to 41 miRNA families were identified (Additional file 1: Table S1). Unexpectedly, 47 new flanking sequences noted as p3 or p5 were discovered (Additional file 1: Table S1). In miRBase 20.0, there were 46 precursors and 48 mature miRNA sequences from tomato. In this study, 161 conserved miRNA sequences generated from 92 precursors were identified. All conserved miRNAs were divided into 41 miRNA families, and several miRNA families were the first to be detected in tomato, such as miR164, 384, 390, 396, 398, 408, 530, 827, 1886, 3627, 7981, 7982, 7983 and 8007. Although miR395, 1918, 5302, 5303 and 5304 were deposited in miRBase, they were not detected in the current study, perhaps because these miRNAs have specific expression patterns. Additionally, compared with the miRNAs deposited in miRBase, many of the conserved miRNAs identified in the current study were also detected in other members of the Solanaceae family, such as potato and tobacco, which suggested that these miRNAs were conserved in Solanaceae. Therefore, there was more vast development space for tomato miRNAs.
Additionally, 236 novel miRNAs generated from 171 precursors were identified (Additional file 3: Table S2). In contrast to conserved miRNAs, most novel miRNAs exhibited low expression amounts with norm read <100, which was previously reported in Arabidopsis[41, 44, 59], wheat [45] and grapevine [60]. However, several novel miRNAs exhibited moderate or high expression amounts with norm read > 1,000. In the current study, a total of 161 conserved and 236 novel miRNAs were firstly discovered to be associated with the tomato chilling response.
Differentially expressed miRNAs in tomato
The abundance of miRNAs can be regarded as an index for estimating the expression amounts of miRNAs. Therefore, to identify chilling-responsive miRNAs, the expression amounts of miRNAs between the two libraries were compared. In total, 192 increased and 205 decreased miRNAs were differentially expressed between the two libraries. And the miRNAs were expected to play key roles in the chilling response. It is worth to noting that 49 miRNAs were significantly regulated by chilling stress (Additional file 1: Table S1, Additional file 3: Table S2). Of these, 23 miRNAs including 12 conserved and 11 novel miRNAs significantly increased in the response to chilling stress, while 26 miRNAs including 20 conserved and six novel miRNAs significantly decreased. In short, more miRNAs were cold-suppressed miRNAs, which was in agreement with previous research on Brachypodium[24].
Of the miRNAs with significantly altered expression, several chilling-responsive miRNAs were conserved among several plant species. MiR397 was induced by chilling stress in Brachypodium, Populus tomentosa and Arabidopsis[22, 24, 26]. MiR319 was induced by chilling stress in Arabidopsis[22]. In the current study, sha-miR397(sha-miR397_nta and sha-miR397-p5) and sha-miR319 (sha-miR319 and sha-miR319b_stu) were both found to be induced by chilling stress, which indicated that some miRNAs involved in chilling stress showed consistency among several plant species. Additionally, some miRNAs, such as miR169, 172 and 393, increased under the chilling stress in Arabidopsis[22] and Brachypodium[24], but in this study, no obvious change in expression was detected. On the other hand, several previously reported chilling-responsive miRNAs, such as miR402 in Arabidopsis[22], were not detected in the current study, which suggested that these miRNAs might be species-specific and their expression was not specifically altered in tomato, or perhaps the altered expression of these miRNAs did not occur during the duration of the study.To confirm and verify the sequencing result, nine miRNAs, including conserved sha-miR156a, sha-miR160a, sha-miR398_nta, sha-miR482b, sha-miR6023-p5 and sha-miR6027 and the novel PC-46-5p, PC-69-5p and PC-170-5p, were chosen for qRT-PCR verification in the sequenced tomato cultivar ‘LA1777’. As shown in Figure 4, the results of verification were consistent with the sequencing results. For further confirmation, the expression of these miRNAs was also examined in another more chilling-tolerant tomato cultivar, ‘Hezuo908’. The results showed a similar trend in the two tomato cultivars, while the range of altered expression differed among cultivars. The result suggested that there was a consistent relationship between the miRNA sequences of the two tomato cultivars. However, the extent of the responses differed between the cultivars under chilling stress, perhaps due to differences in the levels of chilling tolerance.
Target genes of tomato miRNAs
To study the function of miRNAs in the chilling stress response, the two libraries were also used to degradome sequencing. As expected, many predicted targets for conserved miRNAs were identified, including those that function in various biological processes (Additional file 5: Table S3). For example, SPL, AP2 and MADS-box, which were targeted by sha-miR156 (sha-miR156a, sha-miR156c, sha-miR156c_nta, sha-miR156d_nta, sha-miR156e_stu, sha-miR156g_stu, sha-miR156h_stu, sha-miR156i_stu and sha-miR156j_stu), sha-miR172 (sha-miR172a, sha-miR172b, sha-miR172i_nta and sha-miR172c-3p_aly) and sha-miR396a_nta, respectively, participated in plant development. In addition, genes involved in regulating the plant response to chilling stress were also identified. For example, most genes encoding anti-stress proteins (scarecrow-like protein and ribosomal protein) targeted by sha-miR171a_nta, sha-miR171b-3p_stu, sha-miR171c_mtr and sha-miR156i-p3_nta were only detected in the CT library, which was in agreement with previous results from Arabidopsis[22]. Moreover, the Cu/Zn superoxide dismutase-encoding gene cleaved by increased sha-miR398_nta was also mainly detected in the CT library, which suggested that sha-miR398_nta and the gene played an important role in the chilling response, which was also in agreement with previous studies of Arabidopsis[23]. In the current study, auxin response factors genes were cleaved by decreased sha-miR160a and sha-miR167b_nta and more tag sequences were mainly detected in the NT library, while only miR167-targeted auxin response factors genes were identified in the chilling response in rice [27]. The results suggested that among different plant species, conserved miRNAs might share similar functions by targeting homologous genes in the chilling stress response. Additionally, most of the conserved miRNA targets were classified as category 0 (Additional file 5: Table S3, Additional file 6: Figure S3, Additional file 7: Figure S4), containing a cleavage site in only one position, further confirming the accuracy of the degradome sequencing result.
Targets of novel miRNAs were also examined (Table 3, Additional file 10: Table S5). Compared with conserved miRNAs, only nine target genes of nine novel miRNAs were identified. GO annotation showed that some specific targets cleaved only by novel miRNAs were identified. Receptor-like protein kinases play important roles in defense responses. When plants suffer from abiotic stress, receptor-like kinases may be the first sensor or transducer of stress signals. Moreover, several related genes encoding receptor-like protein induced by chilling stress have been reported in rice [61], maize [28] and Arabidopsis thaliana[62]. In the current study, a receptor-like protein kinase gene targeted by PC-58-5p was also identified, which suggested that PC-58-5p was an important regulator of the chilling response. A cellulose synthase subunit gene targeted by PC-102-5p was detected in the research; this gene played an important role in cellulose synthesis [63] and cell wall formation [64]. In addition, cellulose synthase was involved in the low temperature stress response by affecting fiber quality, cell wall formation and cell wall loosening [65]. Perhaps the cellulose synthase subunit gene played an essential role in the low temperature response. In addition, PC-125-3p targeted a gene encoding xyloglucan endotransglucosylase/hydrolases, which regulated another major component of the plant cell wall. Several related genes in Arabidopsis and rice played a role in the cold stress response [66, 67]. Interestingly, PC-146-5p targeted the inositol-pentakisphosphate 2-kinase-like gene, encoding an enzyme that catalyzed the chemical reaction between the ATP and ADP [68, 69]. Perhaps this enzyme participated in the stress response by controlling the energy supply.
In the CT library, 301 target tags were cleaved by 48 miRNAs with the abundance of the target tags ranging from eight to 9,509 tags per billion, while 216 target tags were cleaved by 48 miRNAs in the NT library with an abundance ranging from nine to 7,470 tags per billion. Of these, 171 and 16 target tags which were cleaved by 33 conserved and four novel miRNAs respectively increased in the stress, while 108 and 18 target tags which were cleaved by 37 conserved and five novel miRNAs respectively decreased. Combined with the miRNA expression analysis, the result showed that the abundance of the target tags was basically consistent with the miRNA expression amount. On the other hand, the expression amounts of six target genes cleaved by sha-miR160a, sha-miR398_nta, sha-miR482b, sha-miR6027 and PC-46-5p were also verified by qRT-PCR. Compared with the expression amounts of these miRNAs, it was found that the decline in expression of target genes was basically accompanied by an increase in miRNAs expression amounts, which suggested that the tomato miRNAs positively regulated their target genes, which were involved in the chilling response.
Surprisingly, several low temperature-induced transcription factor genes, such as genes encoding DREB/CBF, WRKY and MYB were not detected in the current study, perhaps because these genes were not expressed at the time of sampling or in the leaves.
Network analysis
By elucidating the network between miRNAs and their target genes (Figure 6), the increased sha-miR156 (sha-miR156a, sha-miR156c, sha-miR156c_nta, sha-miR156d_nta, sha-miR156e_stu, sha-miR156g_stu, sha-miR156h_stu, sha-miR156i_stu, sha-miR156j_stu and sha-miR156i-p3_nta), sha-miR171b-3p_stu, sha-miR319b_stu, sha-miR396 (sha-miR396a_nta, sha-miR396b_nta and sha-miR396c_nta) and sha-miR398_nta targeted genes with various functions, which suggested that these miRNAs played important regulatory roles in the tomato chilling response network. The corresponding target genes, including SPL, SCL, TCP, DRM1 and SODC2, played important roles in regulating plant development, gene expression and ROS inactivation. The decreased sha-miR160a, sha-miR167b_nta, sha-miR168 (sha-miR168a-5p and sha-miR168b-5p), sha-miR171a, sha-miR171a_nta, sha-miR171c_mtr and sha-miR171d targeted genes that function in signal transduction, regulation of the expression of miRNAs and genes encoding anti-stress proteins. Of the novel miRNAs, PC-102-5p increased in the stress, which played an important role in cellulose synthesis. While PC-58-5p, PC-125-3p and PC-146-5p all decreased in the stress, and their target genes were involved in signal perception, cell wall formation and energy supply. Therefore, these miRNAs might be involved in the chilling response by inhibiting plant development, cell wall formation and genes expression, as well as improving signal perception, energy supply and regulating miRNA expression.
Conclusions
In summary, in the present study, using high-throughput sequencing, hundreds of chilling-responsive miRNAs were identified in tomato. The information fills the gaps in the knowledge of tomato chilling-responsive miRNAs and increases the knowledge of the response of miRNAs to stress. Additionally, many target genes of miRNAs were identified by degradome sequencing. The result showed that the target genes were involved in various functions. Notably, most target genes of differentially expressed miRNAs actively respond to chilling stress by regulating the expression of anti-stress proteins, antioxidant enzyme and genes involved in cell wall formation. The findings lay the foundation for exploring the role of the regulation of miRNAs in the plant response to chilling stress.
Methods
Plant materials and treatments
The wild tomato cultivar ‘LA1777’ (S. habrochaites) with higher chilling-tolerance ability, was obtained from the Tomato Genetic Resource Center, University of California. After soaking and pregermination, the tomato seeds were sown in an aperture disk and cultured in the greenhouse (average day/night temperatures, 25 ± 1°C/20 ± 1°C). Seedlings at the three-leaf stage were transferred into nutrition pots. At the five-leaf stage, healthy tomato seedlings were pretreated for two days under a 14-h light (25°C)/10-h dark (20°C) photoperiod (photo intensity 180 μmol m−2 s−1). The seedlings were then divided into two groups for treatment; one group was exposed to 4°C/4°C for chilling treatment (CT), while the other group was not treated with chilling stress (NT). Both groups were grown under a 14 h light/10 h dark photoperiod at a light intensity of 180 μmol m−2 s−1. The third leaf from the stem apex was harvested at 1, 4, 8, 12, 24 and 48 h after treatment. To reduce the differences between individuals, samples were collected from six tomato seedlings at a single time point. And there were three biological replicates per treatment. Sample from the two groups were immediately frozen in liquid nitrogen and stored at -80°C until use.
Small RNA library construction and deep sequencing
Tomato leaves were harvested at 1, 4, 8, 12, 24 and 48 h after chilling treatment and leaves from seedlings not exposed to chilling treatment were harvested at the same time points. To construct two small RNA libraries, samples from the two treatments harvested at different time points were individually subjected to small RNA extraction using an Illumina Truseq Small RNA Preparation kit (Illumina, San Diego, USA). Equal quantities of small RNA extracted from different time points in one treatment group were pooled as one small RNA library, and the two small RNA libraries were then constructed. Following isolation and ligation, the small RNAs were reverse transcribed to cDNAs. The purified cDNAs from the two RNA libraries were sequenced with an Illumina Genome Analyzer II (LC Sciences, Hangzhou, China) according to the manufacturer’s protocols.
Identification of conserved and novel miRNAs
After sequencing, raw reads were obtained using Illumina sequencing-related software analysis. Next, the ACGT101-miR program (version 4.2; LC Sciences, Hangzhou, China), a proprietary pipeline script, was utilized for further analysis [70]. Raw reads with low quality and low copy numbers were removed. Adapter sequences were removed, and the remaining sequences with lengths between 19 and 24 nt were subjected to analysis with miRBase 20.0 (http://www.mirbase.org/) for conserved and novel miRNAs identification. A maximum of one mismatch in the first 16 nt of the miRNA and three mismatches in total were allowed between the target miRNAs and known miRNAs deposited in the miRBase database [35]. The specific criteria of miRNAs prediction were described by Meyers et al. [71]. Stable hairpin structures were also an important identification index [11, 71]. Therefore, hairpin RNA structures were predicted using UNAfold software (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) [17, 42]. In addition, MFEI was another important feature to be taken into consideration [43]. Only miRNAs with perfect hairpin structures that met the criteria for miRNAs were regarded as novel miRNAs candidates. All of the sequences were aligned to the tomato genome database (http://solgenomics.net) by BLAST. Sequences that matched mRNAs, repeat sequences, tRNAs, sRNAs, snRNAs, snoRNAs, other noncoding RNAs in Rfam (http://www.sanger.ac.uk/software/Rfam) [72] and the noncoding database of GenBank (http://www.ncbi.nlm.nih.gov/) were discarded. Finally, the remaining sequences were grouped into families.
Degradome library construction and data analysis
Two degradome libraries, i.e., the CT library and the NT library, were constructed from tomato leaves following the method reported by Ma et al. [73]. Poly A-enriched RNA was obtained and ligated to oligonucleotide adaptors harboring an MmeI recognition site. Following reverse transcription, first-strand cDNA was generated from the ligated sequence. After a short PCR amplification, additional DNA products were produced. Following purification, digestion, ligation and purification, the cDNA library was subjected to cluster analysis in an Illumina Cluster Station and sequenced with an Illumina Genome Analyzer II (LC Sciences, Hangzhou, China).
After sequencing, a public software package, CleaveLand3.0, was used to analyze the generated sequencing data [46, 74]. MRNA data from the Tomato Genomics Database (http://solgenomics.net) was used as reference data to map the degradome sequencing products.
Alignment between the miRNA and its target was scored by base pairing. Mismatched pairs or single nucleotide bulges were scored as 1, and G:U pairs were scored as 0.5. The score was doubled if the mismatches were located in the core position (2–13) of the miRNA sequence. Based on the abundance of the degradome sequences and cleavage sequences, the targets were grouped into five categories. Category 0 targets were targets in which only one degradome tag was detected, and it was detected at the expected site, which was the most abundant category (>1). Category 1 comprised degradome sequences (>1) whose scores were equal to the maximum, and there were more maximum positions. Category 2 included the sequences (>1) whose abundance fell between the maximum and median values. Category 3 consisted of the sequences (>1) with scores below the median value. The remaining sequences, which included only one raw read, were defined as category 4.
Analysis of miRNAs and target genes by qRT-PCR
Small RNA was extracted from tomato seedlings exposed to chilling treatment or no chilling treatment for 1, 4, 8, 12, 24 and 48 h using RNAiso (Takara, Dalian, China). According to the poly (A) polymerase procedure (Ambion, Austin, TX), small RNA was ligated to poly (A) tails. Using T4 RNA ligase (Invitrogen, Carlsbad, CA), 5’ adaptor (5’-CGACUGGAGCACGAGGACACUGACAUGGACUGAAGGAGUAGAAA-3’) was added to the poly (A)-tailed RNA [47]. Small RNA and RT primer (ATTCTAGAGGCCGAGGCGGCCGACATG-d [T] 30 [A, G or C] [A, G, C or T]) were utilized to amplify the cDNA by reverse transcription [39, 48]. The products were used as templates to analyze the expression of the miRNAs. Meanwhile, total RNA was extracted and utilized to detect the expression amounts of the target genes. qRT-PCR was carried out to detect the expression of the identified miRNAs and target genes. The primers were listed in Additional file 11: Table S6 and Additional file 12: Table S7. The amplification was carried out with SYBR Premix Ex Taq™ (Takara, Dalian, China) in an Eppendorf real-time PCR machine (Mastercycler®ep realplex, Hamburg, Germany) following the manufacturer’s instructions. All reactions were repeated three times, and 5S rRNA and Actin were utilized as the reference genes. The expression amounts of the miRNAs and target genes were calculated using the 2-ΔΔCt method.
For further validation, another chilling tolerant tomato cultivar, ‘Hezuo908’, was subjected to the same treatment as the tomato cultivar ‘LA1777’, and qRT-PCR was performed to examine the expression amounts of miRNAs and target genes using the process described above.
GO, KEGG and network analysis
GO analysis was utilized to study the functions of the target genes of the miRNAs based on the database (http://www.geneontology.org/). KEGG pathway analysis of the miRNAs targets was performed simultaneously based on the KEGG database (http://www.genome.jp/kegg/). Network between miRNAs and their target genes was subsequently assembled according to the GO analysis results.
Statistical analysis
Statistical analysis was performed to compare the miRNAs expression and target genes degradation between the CT and NT libraries. The differences between the two libraries were tested using the chi-square test and the fisher-exact test, and the log2 ratio was regarded as a threshold to detect miRNAs expression and target genes degradation fold changes.
Data access
The sRNA sequence data from this study have been submitted to Gene Expression Omnibus (GEO) under accession number GSE57335 at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57335.
Abbreviations
- MiRNAs:
-
microRNAs
- sRNAs:
-
small RNAs
- nt:
-
nucleotides
- CT:
-
chilling-treated
- NT:
-
non-chilling-treated
- qRT-PCR:
-
quantitative real-time PCR
- DCL:
-
dicer-like
- AGO:
-
argonaute
- RISC:
-
RNA-induced silencing complex
- MFEI:
-
minimal folding free index
- t-plots:
-
target plots
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes.
References
Mahajan S, Tuteja N: Cold, salinity and drought stresses: An overview. Arch Biochem Biophys. 2005, 444 (2): 139-158. 10.1016/j.abb.2005.10.018.
Liu YF, Qi MF, Li TL: Photosynthesis, photoinhibition, and antioxidant system in tomato leaves stressed by low night temperature and their subsequent recovery. Plant Sci. 2012, 196: 8-17.
Liu H, Ouyang B, Zhang JH, Wang TT, Li HX, Zhang YY, Yu CY, Ye ZB: Differential modulation of photosynthesis, signaling, and transcriptional regulation between tolerant and sensitive tomato genotypes under cold stress. PLoS One. 2012, 7 (11): e50785-10.1371/journal.pone.0050785.
Ito Y, Katsura K, Maruyama K, Taji T, Kobayashi M, Seki M, Shinozaki K, Yamaguchi-Shinozaki K: Functional analysis of Rice DREB1/CBF-type transcription factors involved in cold-responsive gene expression in transgenic rice. Plant Cell Physiol. 2006, 47 (1): 141-153.
Zou CS, Jiang WB, Yu DQ: Male gametophyte-specific WRKY34 transcription factor mediates cold sensitivity of mature pollen in Arabidopsis. J Exp Bot. 2010, 61 (14): 3901-3914. 10.1093/jxb/erq204.
Agarwal M, Hao YJ, Kapoor A, Dong CH, Fujii H, Zheng XW, Zhu JK: A R2R3 type MYB transcription factor is involved in the cold regulation of CBF genes and in acquired freezing tolerance. J Biol Chem. 2006, 28 (49): 37636-37645.
Chuck G, Candela H, Hake S: Big impacts by small RNAs in plant development. Curr Opin Plant Biol. 2009, 12 (1): 81-86. 10.1016/j.pbi.2008.09.008.
Dugas DV, Bartel B: MicroRNA regulation of gene expression in plants. Curr Opin Plant Biol. 2004, 7 (5): 512-520. 10.1016/j.pbi.2004.07.011.
Kidner CA, Martienssen RA: The developmental role of microRNA in plants. Curr Opin Plant Biol. 2005, 8 (1): 38-44. 10.1016/j.pbi.2004.11.008.
Ambros V: microRNAs: tiny regulators with great potential. Cell. 2001, 107: 823-826. 10.1016/S0092-8674(01)00616-X.
Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen XM, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, Matzke M, Ruvkun G, Tuschl T: A uniform system for microRNA annotation. RNA. 2003, 9 (3): 277-279. 10.1261/rna.2183803.
Bartel DP: MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004, 116 (2): 281-297. 10.1016/S0092-8674(04)00045-5.
Jones-Rhoades MW, Bartel DP, Bartel B: MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol. 2006, 57: 19-53. 10.1146/annurev.arplant.57.032905.105218.
Kurihara Y, Watanabe Y: Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci U S A. 2004, 101 (34): 12753-12758. 10.1073/pnas.0403115101.
Mallory AC, Elmayan T, Vaucheret H: MicroRNA maturation and action-the expanding roles of ARGONAUTEs. Curr Opin Plant Biol. 2008, 11 (5): 560-566. 10.1016/j.pbi.2008.06.008.
Lee RC, Feinbaum RL, Ambros V: The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993, 75 (5): 843-854. 10.1016/0092-8674(93)90529-Y.
Zeng QY, Yang CY, Ma QB, Li XP, Dong WW, Nian H: Identification of wild soybean miRNAs and their target genes responsive to aluminum stress. BMC Plant Biol. 2012, 12: 182-10.1186/1471-2229-12-182.
Xu MY, Dong Y, Zhang QX, Zhang L, Luo YZ, Sun J, Fan YL, Wang L: Identification of miRNAs and their targets from Brassica napus by high-throughput sequencing and degradome analysis. BMC Genomics. 2012, 13: 421-10.1186/1471-2164-13-421.
Xu L, Wang Y, Xu YY, Wang LJ, Zhai LL, Zhu XW, Gong YQ, Ye S, Liu LW: Identification and characterization of novel and conserved microRNAs in radish (Raphanus sativus L.) using high-throughput sequencing. Plant Sci. 2013, 201–202: 108-114.
Zhou ZS, Zeng HQ, Liu ZP, Yang ZM: Genome-wide identification of Medicago truncatula microRNAs and their targets reveals their differential regulation by heavy metal. Plant Cell Environ. 2012, 35 (1): 86-99. 10.1111/j.1365-3040.2011.02418.x.
German MA, Pillay M, Jeong DH, Hetawal A, Luo SJ, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, De Paoli E, Lu C, Schroth G, Meyers BC, Green PJ: Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol. 2008, 26 (8): 941-946. 10.1038/nbt1417.
Sunkar R, Zhu JK: Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004, 16 (8): 2001-2019. 10.1105/tpc.104.022830.
Sunkar R, Kapoor A, Zhu JK: Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by down-regulation of miR398 and important for oxidative stress tolerance. Plant Cell. 2006, 18 (8): 2051-2065. 10.1105/tpc.106.041673.
Zhang JY, Xu YY, Huan Q, Chong K: Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics. 2009, 10: 449-10.1186/1471-2164-10-449.
Barakat A, Sriram A, Park J, Zhebentyayeva T, Main D, Abbott A: Genome wide identification of chilling responsive microRNAs in Prunus persica. BMC Genomics. 2012, 13: 481-10.1186/1471-2164-13-481.
Chen L, Zhang YY, Ren YY, Xu JC, Zhang ZY, Wang YW: Genome-wide identification of cold-responsive and new microRNAs in Populus tomentosa by high-throughput sequencing. Biochem Biophys Res Commun. 2012, 417 (2): 892-896. 10.1016/j.bbrc.2011.12.070.
Lv DK, Bai X, Li Y, Ding XD, Ge Y, Cai H, Ji W, Wu NLH, Zhu YM: Profiling of cold-stress-responsive miRNAs in rice by microarrays. Gene. 2010, 459 (1–2): 39-47.
Yang G, Zou HD, Wu Y, Liu HK, Yuan YP: Identification and characterization of candidate genes involved in chilling responses in maize (Zea may L.). Plant Cell Tiss Organ Cult. 2011, 106 (1): 127-141. 10.1007/s11240-010-9900-8.
Mueller LA, Solow TH, Taylor N, Skwarecki B, Buels R, Binns J, Lin CW, Wright MH, Ahrens R, Wang Y, Herbst EV, Keyder ER, Menda N, Zamir D, Tanksley SD: The SOL Genomics Network. A comparative resource for Solanaceae biology and beyond. Plant Physiol. 2005, 138 (3): 1310-1317. 10.1104/pp.105.060707.
Venema JH, Linger P, van Heusden AW, van Hasselt PR, Bruggemann W: The inheritance of chilling tolerance in tomato (Lycopersicon spp.). Plant Biol. 2005, 7 (2): 118-130. 10.1055/s-2005-837495.
Sato S, Tabata S, Hirakawa H, Asamizu E, Shirasawa K, Isobe S, Kaneko T, Nakamura Y, Shibata D, Aoki K, Egholm M, Knight J, Bogden R, Li CB, Shuang Y, Xu X, Pan SK, Cheng SF, Liu X, Ren YY, Wang J, Albiero A, Dal Pero F, Todesco S, Van Eck J, Buels RM, Bombarely A, Gosselin JR, Huang MY, Leto JA, et al: The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012, 485 (7400): 635-641. 10.1038/nature11119.
Moxon S, Jing RC, Szittya G, Schwach F, Pilcher RLP, Moulton V, Dalmay T: Deep sequencing of tomato short RNAs identifies microRNAs targeting genes involved in fruit ripening. Genome Res. 2008, 18 (10): 1602-1609. 10.1101/gr.080127.108.
Pilcher RLP, Moxon S, Pakseresht N, Moulton V, Manning K, Seymour G, Dalmay T: Identification of novel small RNAs in tomato (Solanum lycopersicum). Planta. 2007, 226 (3): 709-717. 10.1007/s00425-007-0518-y.
Itaya A, Bundschuh R, Archual AJ, Joung JG, Fei ZJ, Dai XB, Zhao PX, Tang YH, Nelson RS, Ding B: Small RNAs in tomato fruit and leaf development. Biochim Biophys Acta. 2008, 1779 (2): 99-107. 10.1016/j.bbagrm.2007.09.003.
Yin ZJ, Li CH, Han ML, Shen FF: Identification of conserved microRNAs and their target genes in tomato (Lycopersicon esculentum). Gene. 2008, 414 (1–2): 60-66.
Venema JH, Posthumus F, de Vries M, van Hasselt PR: Differential response of domestic and wild Lycopersicon species to chilling under low light: growth, carbohydrate content, photosynthesis and the xanthophyll cycle. Physiol Plant. 1999, 105 (1): 81-88. 10.1034/j.1399-3054.1999.105113.x.
Foolad MR, Lin GY: Relationship between cold tolerance during seed germination and vegetative growth in tomato: germplasm evaluation. J Am Soc Hortic Sci. 2000, 125 (6): 679-683.
Mao WH, Li ZY, Xia XJ, Li YD, Yu JQ: A Combined approach of high-throughput sequencing and degradome analysis reveals tissue specific expression of microRNAs and their targets in cucumber. PLoS One. 2012, 7 (3): e33040-10.1371/journal.pone.0033040.
Wang C, Wang XC, Kibeta NK, Song CN, Zhang CQ, Li XY, Han J, Fang JG: Deep sequencing of grapevine flower and berry short RNA library for discovery of novel microRNAs and validation of precise sequences of grapevine microRNAs deposited in miRBase. Physiol Plant. 2011, 143 (1): 64-81. 10.1111/j.1399-3054.2011.01481.x.
Morin RD, Aksay G, Dolgosheina E, Ebhardt HA, Magrini V, Mardis ER, Sahinalp SC, Unrau PJ: Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res. 2008, 18 (4): 571-584. 10.1101/gr.6897308.
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP: A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20 (24): 3407-3425. 10.1101/gad.1476406.
Zuker M: Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31 (13): 3406-3415. 10.1093/nar/gkg595.
Bonnet E, Wuyts J, Rouze P, Van de Peer Y: Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics. 2004, 20 (17): 2911-2917. 10.1093/bioinformatics/bth374.
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, Carrington JC: High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One. 2007, 2 (2): e219-10.1371/journal.pone.0000219.
Yao YY, Guo GG, Ni ZF, Sunkar R, Du JK, Zhu JK, Sun QX: Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biol. 2007, 8 (6): R96-10.1186/gb-2007-8-6-r96.
Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ: Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol. 2008, 18 (10): 758-762. 10.1016/j.cub.2008.04.042.
Sun X, Korir NK, Han J, Shangguan LF, Kayesh E, Leng XP, Fang JG: Characterization of grapevine microR164 and its target genes. Mol Biol Rep. 2012, 39 (10): 9463-9472. 10.1007/s11033-012-1811-9.
Zhang YP, Yu ML, Yu HP, Han J, Song CN, Ma RJ, Fang JG: Computational identification of microRNAs in peach expressed sequence tags and validation of their precise sequences by miR-RACE. Mol Biol Rep. 2012, 39 (2): 1975-1987. 10.1007/s11033-011-0944-6.
Xu F, Liu Q, Chen LY, Kuang JB, Walk T, Wang JX, Liao H: Genome-wide identification of soybean microRNAs and their targets reveals their organ-specificity and responses to phosphate starvation. BMC Genomics. 2013, 14: 66-10.1186/1471-2164-14-66.
Jiang JX, Lv ML, Liang Y, Ma ZM, Cao JS: Identification of novel and conserved miRNAs involved in pollen development in Brassica campestris ssp. chinensis by high-throughput sequencing and degradome analysis. BMC Genomics. 2014, 15: 146-10.1186/1471-2164-15-146.
Shuai P, Liang D, Zhang ZJ, Yin WL, Xia XL: Identification of drought-responsive and novel Populus trichocarpa microRNAs by highthroughput sequencing and their targets using degradome analysis. BMC Genomics. 2013, 14: 233-10.1186/1471-2164-14-233.
Li BS, Qin YR, Duan H, Yin WL, Xia XL: Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J Exp Bot. 2011, 62 (11): 3765-3779. 10.1093/jxb/err051.
Kantar M, Lucas SJ, Budak H: miRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta. 2011, 233 (3): 471-484. 10.1007/s00425-010-1309-4.
Zhou J, Liu MY, Jiang J, Qiao GR, Lin S, Li HY, Xie LH, Zhuo RY: Expression profile of miRNAs in Populus cathayana L. and Salix matsudana Koidz under salt stress. Mol Biol Rep. 2012, 39 (9): 8645-8654. 10.1007/s11033-012-1719-4.
Zhu JF, Li WF, Yang WH, Qi LW, Han SY: Identification of microRNAs in Caragana intermedia by high-throughput sequencing and expression analysis of 12 microRNAs and their targets under salt stress. Plant Cell Rep. 2013, 32 (9): 1339-1349. 10.1007/s00299-013-1446-x.
Zhai LH, Liu ZJ, Zou XL, Jiang YY, Qiu FZ, Zheng YL, Zhang ZX: Genome-wide identification and analysis of microRNA responding to long-term waterlogging in crown roots of maize seedlings. Physiol Plant. 2013, 147 (2): 181-193. 10.1111/j.1399-3054.2012.01653.x.
Lin T, Zhu GT, Zhang JH, Xu XY, Yu QH, Zheng Z, Zhang ZH, Lun YY, Li S, Wang XX, Huang ZJ, Li JM, Zhang CZ, Wang TT, Zhang YY, Wang AX, Zhang YC, Lin K, Li CY, Xiong GS, Xue YB, Mazzucato A, Causse M, Fei ZJ, Giovannoni JJ, Chetelat RT, Zamir D, Städler T, Li JF, Ye ZB, et al: Genomic analyses provide insights into the history of tomato breeding. Nat Genet. 2014, 46: 1220-1226. 10.1038/ng.3117.
Hu WH, Zhou YH, Du YS, Xia XJ, Yu JQ: Differential response of photosynthesis in greenhouse- and field-ecotypes of tomato to long-term chilling under low light. J Plant Physiol. 2006, 163: 1238-1246. 10.1016/j.jplph.2005.10.006.
Allen E, Xie ZX, Gustafson AM, Sung GH, Spatafora JW, Carrington JC: Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet. 2004, 36 (12): 1282-1290. 10.1038/ng1478.
Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, Burgyan J: Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010, 62 (6): 960-976.
Chen LJ, Wuriyanghan H, Zhang YQ, Duan KX, Chen HW, Li QT, Lu X, He SJ, Ma B, Zhang WK, Lin Q, Chen SY, Zhang JS: An S-domain receptor-like kinase, OsSIK2, confers abiotic stress tolerance and delays dark-induced leaf senescence in rice. Plant Physiol. 2013, 163 (4): 1752-1765. 10.1104/pp.113.224881.
Hong SW, Jon JH, Kwak JM, Nam HG: Identification of a receptor-like protein kinase gene rapidly induced by abscisic acid, dehydration, high salt, and cold treatments in Arabidopsis thaliana. Plant physiol. 1997, 113 (4): 1203-1212. 10.1104/pp.113.4.1203.
Kurek I, Kawagoe Y, Jacob-Wilk D, Doblin M, Delmer D: Dimerization of cotton fiber cellulose synthase catalytic subunits occurs via oxidation of the zinc-binding domains. Proc Natl Acad Sci U S A. 2002, 99 (17): 11109-11114. 10.1073/pnas.162077099.
Desprez T, Juraniec M, Crowell EF, Jouy H, Pochylova Z, Parcy F, Hofte H, Gonneau M, Vernhettes S: Organization of cellulose synthase complexes involved in primary cell wall synthesis in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2007, 104 (39): 15572-15577. 10.1073/pnas.0706569104.
Zheng M, Wang YH, Liu K, Shu HM, Zhou ZG: Protein expression changes during cotton fiber elongation in response to low temperature stress. J Plant Physiol. 2012, 169 (4): 399-409. 10.1016/j.jplph.2011.09.014.
Purugganan MM, Braam J, Fry SC: The Arabidopsis TCH4 xyloglucan endotransglycosylase - Substrate specificity, pH optimum, and cold tolerance. Plant Physiol. 1997, 115 (1): 181-190. 10.1104/pp.115.1.181.
Dong JL, Jiang YY, Chen RJ, Xu ZJ, Gao XL: Isolation of a novel xyloglucan endotransglucosylase (OsXET9) gene from rice and analysis of the response of this gene to abiotic stresses. Afr J Biotechnol. 2011, 10 (76): 17424-17434.
Phillippy BQ, Ullah AHJ, Ehrlich KC: Purification and some properties of inositol 1, 3, 4, 5, 6 -pentakisphosphate 2-kinase from immature soybean seeds. J Biol Chem. 1994, 269 (45): 28393-28399.
Stevenson-Paulik J, Odom AR, York JD: Molecular and biochemical characterization of two plant inositol polyphosphate 6-/3-/5-kinases. J Biol Chem. 2002, 277 (45): 42711-42718. 10.1074/jbc.M209112200.
Li MZ, Xia YL, Gu YR, Zhang K, Lang QL, Chen L, Guan JQ, Luo ZG, Chen HS, Li Y, Li QH, Li X, Jiang AA, Shuai SR, Wang JY, Zhu Q, Zhou XC, Gao XL, Li XW: MicroRNAome of porcine pre-and postnatal development. PLoS One. 2010, 5 (7): e11541-10.1371/journal.pone.0011541.
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen XM, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qi Y, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu JK: Criteria for Annotation of Plant MicroRNAs. Plant Cell. 2008, 20 (12): 3186-3190. 10.1105/tpc.108.064311.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ: MiRBase: tools for microRNA genomics. Nucleic Acids Res. 2008, 36: D154-D158. 10.1093/nar/gkn221.
Ma ZR, Coruh C, Axtell MJ: Arabidopsis lyrata small RNAs: transient MIRNA and small interfering RNA loci within the Arabidopsis genus. Plant Cell. 2010, 22 (4): 1090-1103. 10.1105/tpc.110.073882.
Addo-Quaye C, Miller W, Axtell MJ: CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009, 25 (1): 130-131. 10.1093/bioinformatics/btn604.
Acknowledgments
This work was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Research Innovation Program for College Graduates of Jiangsu Province (CXZZ12-0285).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XC, ZW and FJ designed the experiments. XC and RZ performed the experiments. XC and ZY analyzed the data. XC contributed reagents/materials/analysis tools. XC and ZW wrote the paper. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2014_6877_MOESM1_ESM.xlsx
Additional file 1: Table S1: - Conserved miRNAs from tomato in the CT and NT libraries. asequence in miRbase, “Yes” indicates that the sequence can be found in miRBase; “Different” indicates that the sequence is different from the miRNA sequence deposited in miRBase; “New” indicates that the sequence is newly discovered compared with the miRNA sequences deposited in miRBase; bPre-miRNA sequence in miRBase, “Yes” indicates that the pre-miRNA sequence can be found in miRBase; “Predicted” indicates that the pre-miRNA sequence was not found in miRBase and was predicted by UNAFold software; c*indicates a significant difference between the CT and NT libraries. Chi-square test and fisher-exact test results were both <0.05 and | log2 (CT/NT) | ≥1. (XLSX 34 KB)
12864_2014_6877_MOESM2_ESM.docx
Additional file 2: Figure S1: - Secondary structures of 161 conserved miRNAs identified in the CT and NT libraries. The mature miRNAs sequences are underlined in yellow. (DOCX 13 MB)
12864_2014_6877_MOESM3_ESM.xlsx
Additional file 3: Table S2: - Novel miRNAs identified in the CT and NT libraries in tomato. a“PC” indicates predicted candidate; bdashed boxes indicate two identical sequences; c*indicates significant difference between the CT and NT libraries. Chi-square test and fisher-exact test values were both <0.05 and | log2 (CT/NT) | ≥ 1. (XLSX 38 KB)
12864_2014_6877_MOESM4_ESM.docx
Additional file 4: Figure S2: - Secondary structures of 236 putative novel miRNAs identified in the CT and NT libraries. The mature miRNAs sequences are underlined in yellow. (DOCX 19 MB)
12864_2014_6877_MOESM5_ESM.xlsx
Additional file 5: Table S3: - Targets of conserved miRNAs from tomato in the CT and NT libraries. *indicates significant difference between the CT and NT libraries. Chi-square test and fisher-exact test results were both <0.05 and | log2 (CT/NT) | ≥ 1. (XLSX 37 KB)
12864_2014_6877_MOESM6_ESM.docx
Additional file 6: Figure S3: - T-plots for targets of tomato miRNAs identified in the NT library. According to German et al, t-plots are referred to as “target plots” and the normalized numbers are used to plot the cleavages on target mRNAs. The frequency of degradome tags with 5’ends at the indicated positions is marked in black, and the frequency cleaved at position 10 of the inset miRNA target alignment is highlighted in red. The abscissa number t indicates the cleavage site detected in the target mRNA. (DOCX 14 MB)
12864_2014_6877_MOESM7_ESM.docx
Additional file 7: Figure S4: - T-plots for targets of tomato miRNAs identified in the CT library. According to German et al, t-plots are referred to as “target plots” and the normalized numbers are used to plot the cleavages on target mRNAs. The frequency of degradome tags with 5’ends at the indicated positions is marked in black, and the frequency cleaved at position 10 of the inset miRNA target alignment is highlighted in red. The abscissa number t indicates the cleavage site detected in the target mRNA. (DOCX 13 MB)
12864_2014_6877_MOESM8_ESM.jpeg
Additional file 8: Figure S5: - GO enrichment analysis of the functions of target genes cleaved by conserved and novel miRNAs. (JPEG 728 KB)
12864_2014_6877_MOESM9_ESM.xlsx
Additional file 9: Table S4: - KEGG pathway analysis of target genes of conserved and novel miRNAs in the CT and NT libraries. (XLSX 9 KB)
12864_2014_6877_MOESM10_ESM.xlsx
Additional file 10: Table S5: - Targets of novel miRNAs in the CT and NT libraries. *indicates significant difference between the CT and NT libraries. Chi-square test and fisher-exact test results were both <0.05 and | log2 (CT/NT) | ≥ 1. (XLSX 13 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Cao, X., Wu, Z., Jiang, F. et al. Identification of chilling stress-responsive tomato microRNAs and their target genes by high-throughput sequencing and degradome analysis. BMC Genomics 15, 1130 (2014). https://doi.org/10.1186/1471-2164-15-1130
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-15-1130