
Abstract
Background
MicroRNAs (miRNAs) are small (~19-24nt) non-coding RNAs that play important roles in various biological processes. To date, the next-generation sequencing (NGS) technology has been widely used to discover miRNAs in plants and animals. Although evolutionary analysis is important to reveal the functional dynamics of miRNAs, few computational tools have been developed to analyze the evolution of miRNA sequence and expression across species, especially the newly emerged ones,
Results
We developed miREvo, an integrated software platform with a graphical user interface (GUI), to process deep-sequencing data of small RNAs and to analyze miRNA sequence and expression evolution based on the multiple-species whole genome alignments (WGAs). Three major features are provided by miREvo: (i) to identify novel miRNAs in both plants and animals, based on a modified miRDeep algorithm, (ii) to detect miRNA homologs and measure their pairwise evolutionary distances among multiple species based on a WGA, and (iii) to profile miRNA expression abundances and analyze expression divergence across multiple species (small RNA libraries). Moreover, we demonstrated the utility of miREvo with Illumina data sets from Drosophila melanogaster and Arabidopsis, respectively.
Conclusion
This work presents an integrated pipline, miREvo, for exploring the expressional and evolutionary dynamics of miRNAs across multiple species. MiREvo is standalone, modular, and freely available at http://evolution.sysu.edu.cn/software/mirevo.htm under the GNU/GPL license.
Similar content being viewed by others


Background
MicroRNAs (miRNAs) are small non-coding RNAs about 19–24 nt in length that regulate gene expression post-transcriptionally [1, 2]. MiRNAs are ubiquitous in eukaryotes and take part in the regulation of various key developmental events[3]. Extensive studies have been done, both experimental and bioinformatics, to unravel the mechanisms and characteristics of miRNA [4, 5]. An important characteristic of miRNA is the high sequence conservation observed within either plant or metazoan kingdom. Some miRNAs are at least 400 million years old and even many miRNA:target interactions are broadly conserved [3, 6]. In fact, the strong phylogenetic conservation has been a wildly-used criterion for the identification of miRNAs, such as in Arabidopsis thaliana [7] and human [8]. The observed conservation of miRNA between different species suggests that strong purifying selection is acting to maintain the miRNA genes. Despite the similarity of high conservation within kingdom, plant and animal miRNAs are different in many ways, including the biogenesis pathway, hairpin structures and the base-pairing model with their target sites [3, 5]. These different properties has resulted that the miRNA analysis pipelines, such as novel miRNA prediction and target prediction, are quite different between plants and animals [9–11].
In the last few years, the next-generation sequencing (NGS), an inexpensive and high-throughput sequencing method with high sensitivity and specificity, has been widely used to measure the abundance of small-RNAs in multiple species [12–15]. Additionally, NGS can also be adopted to discover novel miRNAs [15, 16]. To take the advantage of NGS, numerous applications, either web-servers or standalone, have been developed to analyze the deep sequencing data of small RNAs for miRNA discovery and/or expression profiling [4, 9, 17–22] (Table 1). Among these tools, miRDeep [15, 22] is the most popular one, due to its great accuracy and performance. These new experimental and analysis techniques greatly accelerate the miRNA studies. To date, there are more than 18,200 reported miRNAs from 168 species in the miRBase database (http://www.mirbase.org, Version 18.0).
Besides the conserved miRNAs, NGS has identified many new miRNA genes, which are highly divergent between closely-related species, such as miR310 cluster in Drosophila[23, 24]. New miRNA genes are shown to have a high birth and death rate [25] and may represent the source of functional novelty [26]. Further evolutionary studies are required to illustrate the phylogenetic relationship and potential functions of these new miRNAs. Nevertheless, few analysis tools are designed for miRNA evolutionary study. Most of the available miRNA analysis tools are web-server and BLAST-searching based [20], which have limitations in screening highly divergent orthologs even with a stringent E-value cut-off. Moreover, many tools are specifically designed for animals [4, 21] and thus less applicable for the evolutionary analysis of plant miRNAs, which have underwent extensive gene duplications and are usually dispersed in the genome as divergent family members with various copy number across species.
Meanwhile, whole genome sequencing has been completed for a great number of species. The multiple-species whole genome alignments (WGAs) provides us a luxuriant resource for comparative genomic analysis and evolutionary studies [27, 29, 29], and also facilitates the genome-wide studies on the miRNA evolutionary pattern globally [30]. For example, the WGA-based approach has been successfully used to reveal the highly dynamic evolutionary flux of miRNAs in both Drosophila[25] and Arabidopsis[16]. Such investigations enable us to understand the history of miRNAs in long term evolution, e.g., how a young miRNA is born and eventually integrates into the regulatory network [24, 25], and how selection acts to maintain miRNA conservation or drive miRNA diversification [23].
Here, we reported miREvo, an integrated miRNA evolutionary analysis platform for NGS experiments by using miRDeep2 [22] as core algorithm in miRNA prediction. miREvo is readily to use for both plant and animal miRNA analysis, including miRNA expression evaluation, novel miRNA prediction, miRNA homology search and between-species comparative analysis based on WGA. miREvo is a flexible, GUI supported, standalone package; we believe it will provide a convenient aid to miRNA research community.
Implementation
User interface overview
MiREvo is committed to provide an efficient and easy-to-use solution for miRNA analysis based on NGS experiments. To achieve this goal, we have implemented miREvo with both command-line interface (CLI) and Graphical User Interface (GUI, Figure 1). The GUI is an interactive application used to create miRNA projects, specify project parameters, run the computer algorithms on the project data, and view the output of computations lively. Under the GUI, users can easily view, control and manipulate miREvo with a few mouse clicks at once (Additional file 1: Figure S1). On the other hand, The CLI is used to deal with heavy tasks with multiple small RNA libraries.

Initial interface of miREvo. Once the GUI is launched, users can open an existing project or create a new project in miREvo.
Software architecture
The system flow of implementation used in miREvo is simplified in Figure 2, which accepts NGS data as input. Typically, for a full analysis run, the following first three analyses are carried out to generate the final results. When NGS data from multiple small RNA libraries are provided, miREvo is also capable to compare miRNA expression profiles between libraries, as described in the last section here.

System flow of the construction of miRNA expression profiles in miREvo. miREvo accepts second-generation sequencing data as input and then can generate miRNA expression profiles, predict novel miRNAs and identify homology sequence of miRNAs.
Preparing the deep-sequencing reads
After removing the adapter at the 3’ends of sequencing reads, the identical reads are collapsed into a unique read and counted. Then, all the unique reads are aligned to the corresponding reference sequences by Bowtie [31] in each step. The Bowtie parameters used for reads mapping are almost identical to the settings of miRDeep2. Firstly, reads are aligned (up to two mismatches) to a user defined database that contains annotated rRNA, small cytoplasmic RNA (scRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), and tRNA or protein coding regions. This is done with these options: bowtie -f -v $MIS -a --best --strata. The –v option is used to retain reads that have no more than $MIS mismatch of a hits to the genome, where $MIS is a user-specified integer value. These aligned reads could be discarded from further analysis. Next, the remaining reads are aligned (bowtie -f -v 1 -a --best --strata --norc) to the precursors of known miRNAs if provided, to detect the expression abundance of these miRNAs. Lastly, after excluding the reads mapped to the known miRNAs, the remaining reads are used to predict novel miRNAs (Figure 2).
Predicting the novel miRNAs
miREvo uses miRDeep2 [22], a freely software package developed by Max Delbrück Center, to identify novel miRNAs from deep sequencing data. MirDeep is a probabilistic-model-based method for miRNA discovery in animals, which has been demonstrated to be sensitive and accurate in several studies [32, 34, 34]. For animals, the default parameters of miRDeep2 are used in miREvo. To extend miRDeep2 for plant miRNA prediction, parameters are well-adjusted according to the statistical characteristics of plant miRNAs [5, 9, 11]. For instance, more repeated matching is allowed in reads alignment step since plant miRNAs typically possess larger families than the animals. Other features including minimum free energy, stability of secondary structures, and excision length, etc., are also restricted as described previously [3], these new parameters are listed in Additional file 2. To further refine the prediction of novel miRNAs, miREvo provides a new feature of crosschecking with known miRNAs from species of interests in miRBase. Known miRNAs with identical seed and/or mature sequence more than 80% identity with the predicted new one will be reported.
Identifying the miRNA homologs
One challenge in homology identification is to produce a single best orthologous match for a given miRNA/sequence. To reduce the interference by tandem duplication sequences, WGA is well-adjusted and optimized for orthologous alignment extension and specificity [35]. miREvo uses a reference guide method to locate the homologous sequences among the WGA between relative species. Firstly, miRNA precursors are precisely mapped to the reference using BLAT. Next, the whole homologous region is excised from the Multiple Alignments File (MAF) using mafsInRegion (http://genomewiki.ucsc.edu/index.php/Kent_source_utilities), according the coordinate of the miRNA precursor located in the reference. Compared with the methods that directly align miRNAs against the reference genome using BLASTN, our WGA-based method is more credible in screening the orthologs of highly divergent miRNAs.
To access MAF files, UCSC (http://genome.ucsc.edu) has built multiple genome alignments for most sequenced animals, such as vertebrates or inserts. For the plants, we have constructed two MAF files. One represents the alignment of 11 green plant genomes (Additional file 1: Table S1) using Arabidopsis thaliana genome as the reference. The other is a pairwised alignment between rice (Oryza sativa) genomes of Japonica cv. Nipponbare and Indica cv. 93–11 (Additional file 1: Table S1), using Nipponbare genome as the reference. Multiple alignments with additional genomes will be regularly added in further release versions. These two MAF files were generated by using BLASTZ [35] and other tools from the UCSC/Penn State Bioinformatics comparative genomics alignment pipeline [35, 37, 37]. Briefly, plant genome sequences were downloaded from Phytozome (http://www.phytozome.net/, version 7.0) or NCBI (see Additional file 1: Table S1 for the full list of aligned species). After repeat masking by Tandem Repeat Finder [38], all the genomes were aligned to the reference genome, e,g. Arabidopsis thaliana genome, using BLASTZ; Two matching alignments next to each other were joined into one fragment using axtChain [37]; Blocks of chained alignments were further grouped into longer stretches of synteny using netChain [37]. Finally, the sequences were retrieved from the synteny-files and the alignments were re-created.
Comparing orthologous miRNA expression
Once the orthologs are obtained, a consequent desire would be to compare the expression divergence of miRNAs across different species of interests. To fulfill this need, we further supplied a simple solution to support cross-species comparison of miRNA expression. The orthologous sequences from each species are extracted from the WGA according to their coordinates, and then used as reference for short reads mapping in each species with Bowtie (bowtie -f -v 1 -a --best --strata --norc). The expression of mature miRNAs from 5’ and 3’ arms of the hairpin precursors is estimated by counting the mapped reads and normalized by the total number of mapped reads, respectively. Then, a Poisson regression model [39] and likelihood ratio test are employed to obtain the statistical significance of differential expression between species. The final results are presented as a table of homologous miRNA expression and a scatter plot.
Results
Identification of novel miRNAs
To illustrate the usage of miREvo, we applied miREvo to two small RNA NGS datasets, which were retrieved from Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). One was from Drosophila simulans embryo (GSM343915) and the other was from Arabidopsis thaliana leaves (GSM707679). As a working example, we only presented the results of Drosophila melanogaster here. The example of Arabidopsis thaliana was provided in the additional files. The full demo data can be downloaded from our website (http://evolution.sysu.edu.cn/software/mirevo.htm). After a full run of analysis, we provided the following results (Figure 3):
-
(1)
Reads Statistics of miRNAs: A total of 4,609,104 reads were sequenced, representing 863,547 unique signatures. A table summarized the reads count for each miRNA was provided. Among the 136 characterized miRNA genes of D. simulans (miRbase V18.0), 101 were observed with a total of 1,746,656 reads (11,538 unique signatures) mapped to the corresponding loci. In addition, another 43 putative new miRNAs with 5,957expressed reads were predicted using default criteria of miRDeep2[32]. Among them, 13 have hits in other Drosophila species by crosschecking with miRBase. For example, the novel miRNA dsi_22_3.9 has been annotated as miR-1009 in D. melanogaster (Figure 3). Full reports, including Html format report generated by miRDeep2, were retained under the user specified project folder.
-
(2)
FASTA sequences and multiple alignments: The orthologous sequences of the predicted new miRNAs in the other 11 Drosophila species were parsed out from the 12 Drosophila species WGA produced by UCSC (http://hgdownload.cse.ucsc.edu/goldenPath/dm3/multiz15way/). A multiple alignment of these orthologous sequences were constructed. To evaluate the RNA secondary structure conservation, the alignments of minimum free energy (mfe) structure were also displayed in bracket notation. Finally, the pairwise genetic distances (Kmir, based on the Kimura two-parameter model [23]) and sequence identity of miRNA homologs were calculated for precursor, mature and seed regions, separately.
-
(3)
The minimum energy and the miRNA hairpin folding. The hairpin folding of individual pre-miRNA was graphed with detailed information of folding free energy. The pre-miRNA sequence was underlined as color-coded according to the reads’ coverage pre base of the hairpin.

Overview of the result viewing interface of miREvo. The miREvo result presenting interface contains four panels: A, miRNA list table; B, result navigator; C, hairpin structure viewing; and D, miRNA expression table.
Identification of homologous miRNAs
Another application of miREvo is to identify the homologous sequences of a given miRNA. To do so, MiREvo requires input files including 1) the precursor miRNA sequence, 2) the mature miRNA sequence, 3) the reference genome, and 4) the MAF files containing the WGA of species of interests; the output file presents the best-fitted results. Taking the advantage of synteny information of WGA, miREvo is particularly useful in identifying orthologs of highly divergent miRNAs, or paralogous members of old miRNA families. To demonstrate that the WGA-based method has better performance than BLASTN, we conducted a genome-wide screening for all the orthologs of the 240 known miRNA of D.melanogaster (miRBase V18) in genome of D.pseudoobscura using both methods. Considering miRNA precursors that have at least 90% identity out of a coverage length greater than 50nt between the two species, miREvo identified 93 (39%) orthologs with at least ten sequencing reads at the embryonic stage in D. pseudoobscura. By contrast, the BLASTN-based search (Options: -e 0.01 -W 7 -b 3 -v 3 -q −1 -F F) only identified 66 (28%) orthologs. Therefore, the WGA-based method is significantly more effective (p value = 0.012, chi-square test).
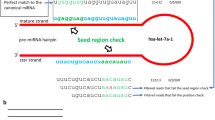
As a case in point, miR-310 is rapidly evolving in Drosophila, which has accumulated a large number of nucleotide substitutions since the split of D. melanogaster and D. pseudoobscura. The orthologous sequence of dme-miR-310 in D. pseudoobscura can be clearly identified with miREvo (Figure 4). By contrast, a BLASTN [40] searching for the same miRNA with word size 7nt generated no hit in D. pseudoobscura. The evolutionary distance (Kmir) of dme-miR-310 between the two species is 0.5, which is half of the averaged genome-wide synonymous divergence [41]. The Kmir/Ks ratio less than 1 suggests that dme-miR-310 is under functional constrain although it might have evolved differentially between species. A more complex case of the homology identification of Arabidopsis ath-miR-169 family was given in Additional file 3. Using miREvo, orthologs for all the members of Arabidopsis ath-miR-169 family can be individually distinguished (Additional file 3: Figure S2).

Othologs of dme-miR-310 across 12 Drosophila genome as identified by miREvo.
Expression profiling of orthologous miRNAs
To demonstrate the utility of miREvo in cross-species comparison of miRNA ortholog expression in an identical cell type, we applied it to two published libraries generated from Drosophila species:D. simulans (GSM343915) and D. pseudoobscura (GSM343916). Using D. melanogaster miRNAs as references, orthologs from the other two species were extracted. The expression of orthologs for each library was analyzed separately, cross-species expression profiles were constructed (Additional file 1: Table S2). Out of 161 orthologous miRNA genes, statistical analysis indentified 47 and 42 mature miRNAs from 5’ and 3’ arms of the hairpin precursors, respectively, that are differentially expressed between D. simulans and D. pseudoobscura (Figure 5 A and B). In summary, miREvo offers the capacity to identify the orthologous miRNAs and also to compare the expression differences among orthologs between species. The demo data and scripts described here can be downloaded at miREvo web site: http://evolution.sysu.edu.cn/software/dro_orth_exp.tar.gz.

Differential expression orthologous miRNAs between D. simulans and D. pseudoobscura. The expression of mature miRNAs from 5’ (A) and 3’ (B) arms of the hairpin precursors were estimated separately. The normorlized expression levels of miRNAs (Log2 TPM) in D. pseudoobscura (y-axes) were plotted against those of their orhologs in D. simulans (x-axes). MiRNAs that differentially expressed between species or not were indicated as solid or open circles, respectively. Dsi, D. simulans; Dps, D. pseudoobscura; TPM, Tags per million mapped reads.
Conclusions
In this report, we introduced miREvo, an integrated platform for miRNA NGS data analysis. miREvo enables batch annotation of the small RNA reads generated by NGS. It also provides flexibility with the tight integration of the miRDeep package, which enables users to predict novel miRNAs for both plants and animals. The main advantage of miREvo is the ability/flexibility to identify miRNA orthologs in closely or distantly related species based on WGA, which is superior to the blast-searching-based tools [4, 20, 26]. To accelerate the evolutionary study of plant miRNAs, we have provided two build-in WGA datasets among multiple plant species. To our knowledge, it is the first available tool for evolutionary analysis of miRNA deep sequencing dataset for both plants and animals. In addition, miREvo is standalone and can be accessed via a GUI or from a CLI. We think miREvo will be a great convenience and facilitate the downstream analysis of miRNA sequencing data.
Availability and requirements
· Project name: miREvo
· Project home page: http://evolution.sysu.edu.cn/software/mirevo.htm
· Operating system(s): Unix/Linux based
· Programming language: Bash, Perl 5 and Perl/TK
· Other requirements: RNAfold, Bowtie, BioPerl, mafsInRegion and BLAT
· License: GNU GPL v3
· Any restrictions to use by non-academics: specified by GNU GPL v3
Before the application can be started, the following open source packages must be installed:
-
1.
Perl 5 – http://www.perl.org/get.html
-
2.
Perl-TK – http://search.cpan.org/~ni-s/Tk-804.027/pod/UserGuide.pod
-
3.
BiolPerl – http://www.bioperl.org/wiki/Getting_BioPerl
-
4.
The following free available packages are also needed by miREvo, which has been deposited into miREvo package, or users can install them independently:
-
(a)
Vienna RNA Package – http://www.tbi.univie.ac.at/~ivo/RNA/
- (b)
-
(c)
masfsInRegion – http://genomewiki.ucsc.edu/index.php/Kent_source_utilities
- (d)
-
e)
ImageMagick (http://www.imagemagick.org/script/index.php)
Such information is given in the user manual accompanied with miREvo. All these packages can also be downloaded from http://evolution.sysu.edu.cn/software/utilities.tar.gz. The documentation also illustrates the usage of each individual functions, including the case-oriented help session that demonstrate the examples described in the paper.
Abbreviations
- miRNA:
-
microRNA
- NGS:
-
Next-generation sequencing
- WGA:
-
whole genome alignment.
References
Bartel DP: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004, 116(2):281–297. 10.1016/S0092-8674(04)00045-5
Voinnet O: Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136(4):669–687. 10.1016/j.cell.2009.01.046
Millar AA, Waterhouse PM: Plant and animal microRNAs: similarities and differences. Funct Integr Genomics 2005, 5(3):129–135. 10.1007/s10142-005-0145-2
Gerlach D, Kriventseva EV, Rahman N, Vejnar CE, Zdobnov EM: miROrtho: computational survey of microRNA genes. Nucleic Acids Res 2009, 37: D111-D117. 10.1093/nar/gkn707
Thakur V, Wanchana S, Xu M, Bruskiewich R, Quick WP, Mosig A, Zhu XG: Characterization of statistical features for plant microRNA prediction. BMC Genomics 2011, 12: 108. 10.1186/1471-2164-12-108
Willmann MR, Poethig RS: Conservation and evolution of miRNA regulatory programs in plant development. Curr Opin Plant Biol 2007, 10(5):503–511. 10.1016/j.pbi.2007.07.004
Adai A, Johnson C, Mlotshwa S, Archer-Evans S, Manocha V, Vance V, Sundaresan V: Computational prediction of miRNAs in Arabidopsis thaliana. Genome Res 2005, 15(1):78–91. 10.1101/gr.2908205
Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E: Phylogenetic shadowing and computational identification of human microRNA genes. Cell 2005, 120(1):21–24. 10.1016/j.cell.2004.12.031
Yang X, Li L: miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 2011, 27: 2614–2615.
Thomas M, Lieberman J, Lal A: Desperately seeking microRNA targets. Nat Struct Mol Biol 2010, 17(10):1169–1174. 10.1038/nsmb.1921
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qi Y, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu JK: Criteria for annotation of plant MicroRNAs. Plant Cell 2008, 20(12):3186–3190. 10.1105/tpc.108.064311
Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung JH, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R, Zhao Y, Zamore PD, Hannon GJ, Marra MA, Weng Z, Perrimon N, Lai EC: Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res 2011, 21(2):203–215. 10.1101/gr.116657.110
Song C, Wang C, Zhang C, Korir NK, Yu H, Ma Z, Fang J: Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics 2010, 11: 431. 10.1186/1471-2164-11-431
Wang J, Czech B, Crunk A, Wallace A, Mitreva M, Hannon GJ, Davis RE: Deep small RNA sequencing from the nematode Ascaris reveals conservation, functional diversification, and novel developmental profiles. Genome Res 2011, 21: 1462–1477. 10.1101/gr.121426.111
Friedlander MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N: Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 2008, 26(4):407–415. 10.1038/nbt1394
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, Carrington JC: High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2007, 2(2):e219. 10.1371/journal.pone.0000219
Wang WC, Lin FM, Chang WC, Lin KY, Huang HD, Lin NS: miRExpress: Analyzing high-throughput sequencing data for profiling microRNA expression. BMC Bioinforma 2009, 10(1):328. 10.1186/1471-2105-10-328
Hackenberg M, Sturm M, Langenberger D, Falcon-Perez JM, Aransay AM: miRanalyzer: a microRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res 2009, 37(Web Server issue):W68-W76.
Moxon S, Schwach F, Dalmay T, Maclean D, Studholme DJ, Moulton V: A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics 2008, 24(19):2252–2253. 10.1093/bioinformatics/btn428
Dezulian T, Remmert M, Palatnik JF, Weigel D, Huson DH: Identification of plant microRNA homologs. Bioinformatics 2006, 22(3):359–360. 10.1093/bioinformatics/bti802
Zhang R, Peng Y, Wang W, Su B: Rapid evolution of an X-linked microRNA cluster in primates. Genome Res 2007, 17(5):612–617. 10.1101/gr.6146507
Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N: miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 2011, 40: 37–52.
Lu J, Fu Y, Kumar S, Shen Y, Zeng K, Xu A, Carthew R, Wu CI: Adaptive evolution of newly emerged micro-RNA genes in Drosophila. Mol Biol Evol 2008, 25(5):929–938. 10.1093/molbev/msn040
Tang TA, Kumar S, Shen Y, Lu JA, Wu ML, Shi SH, Li WH, Wu CI: Adverse interactions between micro-RNAs and target genes from different species. Proc Natl Acad Sci U S A 2010, 107(29):12935–12940. 10.1073/pnas.1007591107
Lu J, Shen Y, Wu Q, Kumar S, He B, Shi S, Carthew RW, Wang SM, Wu CI: The birth and death of microRNA genes in Drosophila. Nat Genet 2008, 40(3):351–355. 10.1038/ng.73
Artzi S, Kiezun A, Shomron N: miRNAminer: a tool for homologous microRNA gene search. BMC Bioinforma 2008, 9: 39. 10.1186/1471-2105-9-39
Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, Ruby JG, Brennecke J, Hodges E, Hinrichs AS, Caspi A, Paten B, Park SW, Han MV, Maeder ML, Polansky BJ, Robson BE, Aerts S, van Helden J, Hassan B, Gilbert DG, Eastman DA, Rice M, Weir M, Hahn MW, Park Y, Dewey CN, Pachter L, Kent WJ, Haussler D, Lai EC, Bartel DP, Hannon GJ, Kaufman TC, Eisen MB, Clark AG, Smith D, Celniker SE, Gelbart WM, Kellis M: Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature 2007, 450(7167):219–232. 10.1038/nature06340
Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, Ward LD, Lowe CB, Holloway AK, Clamp M, Gnerre S, Alfoldi J, Beal K, Chang J, Clawson H, Cuff J, Di Palma F, Fitzgerald S, Flicek P, Guttman M, Hubisz MJ, Jaffe DB, Jungreis I, Kent WJ, Kostka D, Lara M, Martins AL, Massingham T, Moltke I, Raney BJ, Rasmussen MD, Robinson J, Stark A, Vilella AJ, Wen JY, Xie XH, Zody MC, Worley KC, Kovar CL, Muzny DM, Gibbs RA, Warren WC, Mardis ER, Weinstock GM, Wilson RK, Birney E, Margulies EH, Herrero J, Green ED, Haussler D, Siepel A, Goldman N, Pollard KS, Pedersen JS, Lander ES, Kellis M, Inst B, Med BC, Univ W: A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011, 478(7370):476–482. 10.1038/nature10530
Thomas JW, Touchman JW, Blakesley RW, Bouffard GG, Beckstrom-Sternberg SM, Margulies EH, Blanchette M, Siepel AC, Thomas PJ, McDowell JC, Maskeri B, Hansen NF, Schwartz MS, Weber RJ, Kent WJ, Karolchik D, Bruen TC, Bevan R, Cutler DJ, Schwartz S, Elnitski L, Idol JR, Prasad AB, Lee-Lin SQ, Maduro VV, Summers TJ, Portnoy ME, Dietrich NL, Akhter N, Ayele K, Benjamin B, Cariaga K, Brinkley CP, Brooks SY, Granite S, Guan X, Gupta J, Haghighi P, Ho SL, Huang MC, Karlins E, Laric PL, Legaspi R, Lim MJ, Maduro QL, Masiello CA, Mastrian SD, McCloskey JC, Pearson R, Stantripop S, Tiongson EE, Tran JT, Tsurgeon C, Vogt JL, Walker MA, Wetherby KD, Wiggins LS, Young AC, Zhang LH, Osoegawa K, Zhu B, Zhao B, Shu CL, De Jong PJ, Lawrence CE, Smit AF, Chakravarti A, Haussler D, Green P, Miller W, Green ED: Comparative analyses of multi-species sequences from targeted genomic regions. Nature 2003, 424(6950):788–793. 10.1038/nature01858
Wang X, Gu J, Zhang MQ, Li Y: Identification of phylogenetically conserved microRNA cis-regulatory elements across 12 Drosophila species. Bioinformatics 2008, 24(2):165–171. 10.1093/bioinformatics/btm572
Langmead B: Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics 2010. Chapter 11:Unit 11 17 Chapter 11:Unit 11 17
Wei Y, Chen S, Yang P, Ma Z, Kang L: Characterization and comparative profiling of the small RNA transcriptomes in two phases of locust. Genome Biol 2009, 10(1):R6. 10.1186/gb-2009-10-1-r6
Friedlander MR, Adamidi C, Han T, Lebedeva S, Isenbarger TA, Hirst M, Marra M, Nusbaum C, Lee WL, Jenkin JC, Sanchez Alvarado A, Kim JK, Rajewsky N: High-resolution profiling and discovery of planarian small RNAs. Proc Natl Acad Sci U S A 2009, 106(28):11546–11551. 10.1073/pnas.0905222106
Soares AR, Pereira PM, Santos B, Egas C, Gomes AC, Arrais J, Oliveira JL, Moura GR, Santos MA: Parallel DNA pyrosequencing unveils new zebrafish microRNAs. BMC Genomics 2009, 10: 195. 10.1186/1471-2164-10-195
Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W: Human-mouse alignments with BLASTZ. Genome Res 2003, 13(1):103–107. 10.1101/gr.809403
Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, Haussler D, Miller W: Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 2004, 14(4):708–715. 10.1101/gr.1933104
Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D: Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci U S A 2003, 100(20):11484–11489. 10.1073/pnas.1932072100
Benson G: Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 1999, 27(2):573–580. 10.1093/nar/27.2.573
Zuyderduyn S: Statistical analysis and significance testing of serial analysis of gene expression data using a Poisson mixture model. BMC Bioinforma 2007, 8(1):282. 10.1186/1471-2105-8-282
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25(17):3389–3402. 10.1093/nar/25.17.3389
Shen Y, Lv Y, Huang L, Liu W, Wen M, Tang T, Zhang R, Hungate E, Shi S, Wu CI: Testing hypotheses on the rate of molecular evolution in relation to gene expression using microRNAs. Proc Natl Acad Sci U S A 2011, 108(38):15942–15947. 10.1073/pnas.1110098108
Acknowledgements
We thank Yang Lv for useful discussions. This work was supported by grants from National Science Foundation of China (31130069, 31170308, and 41130208), State Key Laboratory of Biocontrol (SKLBC10B01 and SKLBC2011A01), Sun Yat-Sen University (1132985), and Pearl River Science and Technology Foundation of Guangzhou (2012–77).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MW and YS developed miREvo and carried out the analysis. YS and TT conceived and directed the project. MW, YS, SSH and TT wrote the manuscript. All authors have read and approved the final manuscript.
Electronic supplementary material
12859_2012_5270_MOESM2_ESM.doc
Additional file 2: Plant Parameters.doc. The modification of miRDeep2 parameters that are used for the prediction of novel plant miRNAs in miREvo. (DOC 46 KB)
12859_2012_5270_MOESM3_ESM.pdf
Additional file 3: Supplementary Figure S2.pdf. Supplementary Figure S2, which is too large to include in the text. (PDF 22 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wen, M., Shen, Y., Shi, S. et al. miREvo: an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinformatics 13, 140 (2012). https://doi.org/10.1186/1471-2105-13-140
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2105-13-140