
Abstract
Amorphous LiEuTiO4 nanovesicles were fabricated using in situ formed Eu nanoparticles as sacrificial templates. Samples fabricated with these nanovesicles exhibited a good rate performance together with a large reversible discharge capacity of 285.1 mAh g−1 at 0.2 A g−1 and 185.7 mAh g−1 at 10 A g−1 and had an excellent cycle performance with a huge reversible discharge capacity of 282 mAh g−1 after 1000 cycles at 0.1 A g−1. These advantageous characteristics are attributed its unique structure, which reduces the length of Li+/electron transportation and increases the interface between the electrolyte and the electrode. A solid-electrolyte boundary film was formed on the surface of the LiEuTiO4 anode during cycling. However, the rate and cyclic properties of the sample improved after adding vinylene carbonate. Those results demonstrate that LiEuTiO4 nanovesicles can function as an ideal low operating voltage anode material in high-rate lithium ion batteries intended for electric vehicles or hybrid electric vehicles without a corresponding reduction in the battery voltage or energy density.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Rechargeable lithium ion batteries (LIBs) are extensively used in devices that can be easily carried. However, employing LIBs in electric vehicles (EVs) or hybrid electric vehicles (HEVs) requires more advanced safety and improvements in performance and long cycle lifespans [1, 2]. Unfortunately, it is difficult to meet these requirements using present-day commercial graphite anodes owing to their small lithium intercalation potential of 0.1 V (versus Li+/Li), which can bring about lithium plating that in turn can cause safety issues [3, 4]. Therefore, spinel Li4Ti5O12 has been considered as a safe alternative anode material for use in large-rate LIBs for HEVs or EVs because its high lithium intercalation potential of 1.55 V limits lithium plating [5, 6]. However, this relatively high intercalation potential tends to reduce both the battery voltage and energy density [7].
Various alternative anode materials with operating potentials of approximately 1 V have been developed in an attempt to mitigate this problem with the aim of avoiding lithium plating while preserving the battery voltage [8, 9]. These potential alternative compounds include LiEuTiO4 [10,11,12], BiPO4 [13], Li(V0.5Ti0.5)S2 [14], LiVS2 [15], LiTiS2 [16] and Li2C8H4O4 [17]; of those, LiEuTiO4 has recently received significant attention owing to its good electrochemical performance. However, to allow for practical applications of LiEuTiO4, it would be beneficial for this material to exhibit a higher rate value and longer cyclability.
Recently, hollow structures have been shown to be a promising type of morphology for electrode materials for secondary batteries because the unique characteristics of this morphology can promote mass transport by providing a large surface area and small spread length [18,19,20,21]. For instance, Sun et al. fabricated hollow ultrafine Fe2O3 fibers with excellent lithium storage properties [19], and Zhu et al. [20] suggested that hollow manganese silicate spheres can be used as good anodes for LiBs with excellent rate performances. Meanwhile, Miao et al. [21] found that Li9V3(P2O7)3(PO4)2 nanotubes have a good Li+ storage capability when used as a cathode for LIBs. Those works prompted us to investigate the Li+ storage properties of LiEuTiO4 with a hollow structure morphology.
In this paper, we present the successful fabrication of non-crystalline LiEuTiO4 nanovesicles with the aid of plasma using in-situ formed Eu nanoparticles (NPs) as a hard template. The synthesized LiEuTiO4 nanovesicles exhibited a high reversible discharge capacity of 285.1 mAh g−1 at 0.2 A g−1 and 185.7 mAh g−1 at 10 A g−1 and excellent cycle performance with a large discharge capacity of 282 mAh g−1 after 1000 repeats at 0.1 A g−1.
2 Experimental
2.1 Preparation of non-crystalline LiEuTiO4 nanovesicles
All chemicals were used directly as purchased. First, 0.05 mol tris(acetylacetonato)(1,10-phenanthroline)europium was added to a molten salt of 0.05 mol lithium stearate and 0.05 mol diammineteracholotitanate at 230 °C in a 5% H2/95% Ar mixture. After a 30-min reaction with the assistance of plasma (500 V cm−1) in a self-designed reactor [22,23,24] the samples was cooled to 25 °C, and then cleaned three times with an ionic liquid (3-methylimidazolium chloride) and ethanol. Afterwards, the sample was dehydrated in air at 80 °C for 16 h.
2.2 Structural characterization
X-ray diffraction (XRD) spectra of the samples were obtained using an X’Pert X-ray powder diffractometer (CuKα source, λ = 0.15406 nm). The Li, Eu, and Ti concentrations were determined using inductively coupled plasma atomic emission spectroscopy (Irris, Advantage), while the proportion of O in the samples was confirmed using a Vario Microcube elemental analyzer. The Brunauer–Emmett–Teller specific surface areas of the samples were obtained using the N2 adsorption–desorption technique. Prior to these assessments, each sample was degassed for 3 h at 100 °C. Transmission electron microscopy (TEM) patterns and selected-area electron diffraction data of the samples were collected using a Carl Zeiss Libra 200 microscope. A drop of nanoparticles dispersed in ethanol was dropped onto a non-crystalline carbon-spread Cu grid for TEM characterization. X-ray photoelectron spectroscopy (XPS; Perkin-Elmer PHI 5000C ESCA, Al Kα radiation) was used to analyze the surface electronic states of the samples. In preparation for these analyses, samples were first dried under argon, then transferred to an in situ XPS reactor cell of our own design [23, 24]. The binding energies were corrected by using the C1s peak at 284.6 eV of the surface adventitious carbon as a reference. Time-of-flight secondary ion mass spectrometry (ToF–SIMS) data were acquired using a PHI TRIFT II instrument (USA), which has a pulsed liquid metal ion gun. Spectral data were collected using the positive ion mode over the mass range of 1–275 Da. The primary source was Ga+. The applied voltage was 15 kV, while the aperture current was 600 pA. Meanwhile, the raster size was 100 × 100 mm, and the acquisition time was 10 min. At least three different regions were selected for analysis for every sample and data typical of the sample are given herein. 7Li magic angle spinning nuclear magnetic resonance (MAS NMR) spectra were acquired using a Bruker AM-500 spectrometer operating at 194.4 MHz in conjunction with a lab-made probe head. The AM-500 had a Jakobsen 5 mm MAS unit. Rotors made of Si3N4 or zirconia were employed, and the MAS rate was approximately 13.5 kHz. The external chemical shift reference was 1 M LiCl aqueous solution. The obtained nuclear magnetic resonance spectra were analyzed with the software provided with the instrument. Fourier transform infrared (FTIR) spectra were acquired using a Bruker FTIR spectrometer (EQUINOX55).
2.3 Electrochemical testing
The electrochemical properties of LiEuTiO4 prepared in this work and of commercially-available LiEuTiO4 (Changzhen High Technology Co., Ltd.) were assessed with 2032 coin cells. The working electrode was fabricated from a mix of polyvinylidene fluoride (Sigma Aldrich), Super-P carbon black, and LiEuTiO4 in a mass ratio of 10:20:70, spread onto copper foil (99.6%, Goodfellow). A 1 M LiPF6 solution in a 1:1:1 (by volume) mixture of ethyl methyl carbonate, dimethyl carbonate, and ethylene carbonate served as the electrolyte, while Celgrade polypropylene acted as the separator and lithium foil was used as the counter electrode. Cyclic voltammetry data were acquired using an electrochemical workstation (CHI-660a). The scan rate was 1.0 mV s−1. The potential range was from 3.0 to 0.01 V (vs. Li/Li+). The cycling and rate properties were examined in the same potential range at a range of currents. The test instrument used was an Arbin BT-2000. For the in situ XRD analyses, we used a self-designed H-form electrochemical cell with two quartz windows and a built-in Li electrode. The working electrode was placed in a second tube of the H-form cell. It was designed to have two crystalline diamond windows. After passing through the crystalline diamond window, the X-ray beam could be concentrated onto the electrode. The in situ XRD data were obtained during electrochemical cycling.
3 Results and discussion
3.1 Synthesis of material
3.1.1 Characterization
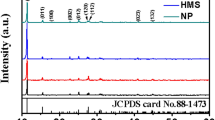
The TEM images (Fig. 1, panels a and b) of the sample show a uniform vesicle-like morphology. Those vesicles have an average size of about 25 nm. Meanwhile, their shells have a thickness of about 3 nm. Furthermore, their specific surface area was 225.4 m2 g−1, which is larger than that of commercial LiEuTiO4 (25.3 m2 g−1). Moreover, the sample was amorphous, as verified by only one halo being visible in the selected electron diffraction patterns (Fig. 1c). The XRD spectra further confirmed that the nanovesicles were neither non-crystalline nor amorphous at room temperature, because no XRD peaks could be observed (Fig. 1d). However, after calcination in argon from 250 to 650 °C for 2 h, crystallized LiEuTiO4 formed gradually (Fig. 1d) [10,11,12]. Although the reported stable phase during calcination was NaEuTiO4 [10, 12], no other phase was formed in our work. The elemental analysis indicate that the sample has composition of Li, Eu, Ti and O with molar ratio of 1:1:1:4. The Eu3d XPS peaks (Fig. 1e) at 1165.0 eV (Eu3d3/2) and 1135.0 eV (Eu3d5/2) indicate that the valence state of Eu in the sample was + 3. Additionally, Ti2p XPS peaks (Fig. 1f) at 465.3 (Ti2p1/2) and 459.6 eV (Ti2p3/2) indicate that the valence state of Ti was + 4. To further confirm the formation of amorphous LiEuTiO4 vesicles, we also conducted a ToF–SIMS analysis (Fig. 1g). The appearance of LiEuTiO4+ (m/z of 267–274), LiEuTiO3+ (m/z of 251–258), LiEuTiO2+ (m/z of 235–242), and LiEuTiO1+ (m/z of 219–226) confirmed that the formed amorphous vesicles were LiEuTiO4. Moreover, no Li–Eu–Ti–O related fragments were observed in the mixture of commercial LiOH, TiO2, and Eu2O3 .

a, b TEM images; c SAED pattern; d XRD patterns; e Eu3d XPS spectra; f Ti2p XPS spectra; g TOF-SIMS spectrum of the sample
3.1.2 Synthetic scheme
To propose a viable vesicle formation scheme, we first observed the fabrication process of LiEuTiO4 nanovesicles (Fig. 2). After 1 min, lots of NPs were formed (Fig. 2a). The XRD pattern (Fig. 2b) and ToF–SIMS spectrum (Fig. 2c) verified that those NPs were composed of metallic Eu (JCPDS-38-0928). This indicated that tris(acetylacetonato)(1,10-phenanthroline)europium could be reduced by H2 in the plasma field as expressed by Eqs. (1) and (2).

a TEM mages; b XRD patterns and c TOF–SIMS spectra during the preparation of the sample, d the possible synthetic scheme
There was no reaction between tris(acetylacetonato)(1,10-phenanthroline)europium and H2 without the plasma. When we extended the reaction time to 2 min, the formation of some LiEuTiO4 was observed (Fig. 2, panels b and c). This suggests that that some Eu NPs may react with TiCl4(NH3)2 and lithium stearate to form LiEuTiO4 according to Eq. (3), namely
and that some of that product had vesicle-like shapes (Fig. 2a). As the reaction progressed, more and more LiEuTiO4 vesicles formed, and the Eu NPs were reduced (Fig. 2a). After 10 min, all NPs had been transformed into vesicles (Fig. 1a). When we further increased the time to 30 min, there was no significant impact on the composition, structure, or morphology of the sample (Fig. 2a).
From the above results, we can conclude that the formation of vesicle-like LiEuTiO4 can be described as presented in Fig. 2d. First, tris(acetylacetonato)(1,10-phenanthroline)europium was reduced to form Eu NPs. Subsequently, the in situ formed Eu NPs act as sacrificial templates and react with lithium stearate and diammineteracholotitanate to form LiEuTiO4 vesicles.
3.1.3 Optimized synthetic conditions
The impact of the reaction conditions, that is, time, temperature, electric field, and molar proportion of tris(acetylacetonato)(1,10-phenanthroline)europium/TiCl4(NH3)2/lithium stearate on the phase composition and yield of the nanovesicles are presented in Table 1. It is apparent that high temperatures and high electric fields are not beneficial for nanovesicle formation. The optimum condition for the preparation LiEuTiO4 nanovesicles with a large specific surface area was a temperature of 230 °C applied for 10 min in conjunction with a tris(acetylacetonato)(1,10-phenanthroline)europium/TiCl4(NH3)2/lithium stearate molar ratio of 1:1:1 and an electric field of 500 V cm−1.
3.2 Electrochemical characteristics
3.2.1 Li+ storage mechanism
LiEuTiO4 nanovesicles were studied as an anode material for LIBs using commercial LiEuTiO4 for comparison purposes. Cyclic voltammograms demonstrated that these nanovesicles have better lithium ion storage properties than commercially obtainable LiEuTiO4 (Fig. 3a) [10,11,12]. This new material has a lower Li+ extraction potential and a higher Li+ insertion potential. This indicates that it has more highly reversible Li+ insertion/extraction properties during discharging/charging [25, 26]. Furthermore, the peaks’ intensities and the areas of the nanovesicles are larger than those of commercial LiEuTiO4. This suggests that the nanovesicles have a higher capacity. The voltammetry plots of both materials are broadly similar throughout subsequent cycles, which means that the Li+ insertion/extraction was stable and reversible for both materials (Fig. S1), although the LiEuTiO4 nanovesicles resulted in larger peak areas.

a CV curves comparison; b initial discharge profiles comparison; c typical XRD patterns during discharging; d the 7Li NMR spectra at different discharge capacities (a-10 mAh g−1; b-27 mAh g−1; c-44 mAh g−1; d-61 mAh g−1; e-78 mAh g−1; f-84.5 mAh g−1; g-91.6 mAh g−1; h-98.8 mAh g−1; i-105.9 mAh g−1; j-113 mAh g−1;); e the relationship between 7Li NMR shifts and discharge capacities; f typical XRD patterns during discharging; g the 7Li NMR spectra at different discharge capacities (a-10 mAh g−1; b-35.3 mAh g−1; c-60.6 mAh g−1; d-85.9 mAh g−1; e-111.2 mAh g−1; f-136.5 mAh g−1; g-161.8 mAh g−1; h-187.1 mAh g−1; i-202.7 mAh g−1; j-213.1 mAh g−1; k-223.4 mAh g−1; l-233.8 mAh g−1; m-244.1 mAh g−1; n-254.5 mAh g−1; o-264.8 mAh g−1; p-275.2 mAh g−1); h the relationship between 7Li NMR shifts and discharge capacities
To determine the Li+ storage scheme, the surface states of the nanovesicles after an initial discharge at 0.1 A g−1 from 3 to 0.01 V (vs. Li/Li+) were analyzed through in situ XPS (Fig. 1, panels e and f). It was determined that Eu3+ was gradually reduced to Eu2+ after Li+ insertion because of charge compensation [10,11,12]. Simultaneously, peaks at approximately 1155.0 (Eu3d3/2) and 1125.0 eV (Eu3d5/2) corresponding to Eu2+ appeared [10,11,12]. Additionally, the Ti4+ valence state remained unchanged (Fig. 1f). This behavior differs from that of Li4Ti5O12, for which the Ti valence state transitions from Ti4+ to Ti3+ after Li+ insertion [27]. Li+ insertion was also confirmed based on the ToF–SIMS depth profile plot of the LiEuTiO4 nanovesicles discharged to 0.01 V (Fig. S2). The nanoparticles were sliced layer-by-layer using a Cs+ ion beam and the proportion of Li atoms in the discharged sample was found to be both higher than in the fresh material while also remaining unchanged in the depth direction, which indicates that the inserted Li+ was distributed uniformly throughout the sample. The higher Li content in the discharged nanovesicles demonstrates that the unique morphology promotes the transport of Li+.
Figure 3b shows the first discharge curves of two samples at a current rate of 0.1 A g−1. The profile of the commercial LiEuTiO4 could be separated into three parts, labeled A, B, and C. In region A, the potential decreased monotonically from the initial discharge voltage to 0.80 V. This was caused by lithium ion insertion into the main part of LiEuTiO4, reaching its solid solubility limit in Li1+xEuTiO4. Region B is a plateau located at approximately 0.80 V and indicates the coexistence of Li-rich and Li-poor phases in Li1+xEuTiO4. Region C is associated with the reversible storage of Li+ ions at accessible interstitial sites on particle interfaces at intercalation voltages below 0.80 V. In this region, electrons can be held up in the so-called “second phase”. This region is generally referred to as the carbon supplement or solid-electrolyte boundary (SEI) layer.
The Li+ storage characteristics of the commercial LiEuTiO4 were confirmed via in situ XRD data (Fig. 3c) [28]. Figure S3 shows that the lattice parameter a decreased, while b, c, and V is increased along with Li+ intercalation in the A and B parts [28]. These linear changes in a, b, c, and V result from Li+ storage at available interstitial sites in LiEuTiO4. The lack of any obvious changes in these same parameters throughout region C suggests that Li+ was instead stored at interfacial sites because the Li+ solid-solution limit in Li1+xEuTiO4 was exceeded. This hypothesis is also confirmed by the sudden transformation of the linear dependence between the 7Li MAS NMR peak shifts and the Li+ storage amount when the Li+ storage transitions to the interfacial sites (the C region) (Fig. 3, panels d and e).
However, the discharge curve of the LiEuTiO4 nanovesicles shows a monotonic voltage change without any plateau (Fig. 3b). There is no obvious distinction between the three parts. Additionally, there was no significant change in the XRD signal during discharge (Fig. 3f). Moreover, no abrupt change in the linear relationship between the 7Li MAS NMR peak shifts and the Li+ storage amount was observed (Fig. 3, panels g and h). In other words, the non-crystalline characteristics of the LiEuTiO4 vesicles mean that they are able to ignore the lattice tension and offer sustained Li+ spread approaches and intercalation sites. This substantially increased the Li+ storage capacity and structure constancy. Thus, the discharge capacity of LiEuTiO4 nanovesicles (Fig. 3b) is higher than the calculated theoretical capacity of LiEuTiO4 (98.97 mAh g−1). However, more work will be required in future to completely understand the detailed process by which Li+ intercalation takes place in LiEuTiO4. We have already commenced with this work in our laboratory.
Figure 3b shows that Li+ storage in the commercial LiEuTiO4 occurred mainly via an intercalation reaction (equivalent to regions A and B). The Li+ ions were randomly distributed throughout the available interstitial sites based on the equation xLi+ + LiEuTiO4 + xe− → Li1+xEuTiO4 (Eq. 4) [10,11,12, 29]. The first discharge capacity and the charge capacity of commercial LiEuTiO4 were 113.0 mAh g−1 (equivalent to Li2.142EuTiO4) and 92.1 mAh g−1 (Li1.931EuTiO4; Fig. 4a), respectively. The material’s irreversible capacity loss reached 18.5%. The first discharge capacity of the LiEuTiO4 nanovesicles was 296.2 mAh g−1 (corresponding to Li3.994EuTiO4). This is larger than the previously reported maximum specific capacity of LiEuTiO4 (Li3.399EuTiO4, 237.3 mAh g−1) [11]. Furthermore, the sample’s first charge capacity was 290.1 mAh g−1 (Li3.933EuTiO4). The capacity loss was only 1.91% (Fig. 4b). The specific capacity reproducibility of our LiEuTiO4 nanovesicles was ascertained based on a minimum of ten replicate trials and the variation was found to be within acceptable limits (± 2.5%). The excellent Li+ storage performance of these LiEuTiO4 nanovesicles can be attributed to their unique structure, which improves the insertion process and enhances the interfacial storage sites. It is commonly known that larger surface area will lead to higher irreversible capacity loss. Because the large specific surface area leads to a larger contact area between the electrode material and the electrolyte. As a result, the formed SEI film causes the increase of the irreversible capacity of the material, and reduces the coulomb efficiency. Obviously, in our work, the unique structure of these LiEuTiO4 nanovesicles makes the increase of their reversible capacity much larger than that of the irreversible capacity caused by their large specific surface area. Therefore, they have higher initial coulomb efficiency than that of commercial LiEuTiO4. The voltage discrepancy between the discharge and charge curves of the LiEuTiO4 nanovesicles was also smaller than that for commercial LiEuTiO4 because of the lower polarization of the former material [25, 26].

a, b Charge–discharge curves during cycling at a current rate of 0.1 A g−1; c cycle performance comparison at a current rate of 0.1 A g−1; d rate performance comparison at different current rates of the samples; e rate performance comparison among ((A) LiEuTiO4 nanovesicles in this work; (B) commercial LiEuTiO4; (C) LiEuTiO4 prepared at 250 °C in this work; (D) LiEuTiO4 prepared at 700 V cm−1 in this work; (E) LiEuTiO4 reported in Ref. [10]; (F) LiEuTiO4 reported in Ref. [11]; (G) LiEuTiO4 nanoparticles in Ref. [11].)
3.2.2 Li+ storage performance
The cycling performances of the two samples at a current rate of 0.1 A g−1 is presented in Fig. 4c. After 100 cycles, the commercial LiEuTiO4 sample showed a charge capacity of only 73.4 mAh g−1 (Li1.742EuTiO4). The capacity loss was 35.0%. Conversely, even after 1000 cycles, the LiEuTiO4 nanovesicles exhibited a minimal capacity decay (4.8%) in conjunction with a charge capacity of 282.0 mAh g−1 (corresponding to Li3.851EuTiO4).
The rate performances of both samples are compared in Fig. 4d. The charge capacity of the LiEuTiO4 nanovesicles at 0.2 and 10.0 A g−1 was 285.1 and 185.7 mAh g−1, respectively. The nanovesicles’ Coulombic efficiency reached almost 100%. For commercial LiEuTiO4, the capacity at 0.2 A g−1 was only 98.0 mAh g−1 and that at 5.0 A g−1 was only 10.2 mAh g−1. The enhanced electrochemical performance of the nanovesicles is caused by their unique structure. Their hollow characteristics enable the quick spread of Li+ and electron transport in the main part of LiEuTiO4. The charge capacity was returned to 285.1 mAh g−1 after the rate was tuned back to 0.2 A g−1. Meanwhile, the LiEuTiO4 nanovesicles exhibited a superior rate performance than that reported for LiEuTiO4 and other LiEuTiO4 electrodes fabricated in this work (Fig. 4e).
3.2.3 Electrochemical impedance spectroscopy analysis
An electrochemical impedance spectroscopic analysis was also conducted for both samples after cycling (Fig. 5a–c). All Nyquist curves showed depressed semicircles (high and medium frequencies) and an inclined line (low frequency). To analyze the impedance spectra of the samples before and after cycling, equivalent circuit models were devised, as shown in the inset of Fig. 5, panels a and c. In these models, Re refers to the electrolyte resistance, while Rsei represents the resistance of the film formed on the boundaries between LiEuTiO4 and the copper foil; moreover, Csei is the capacitance of this film. Additionally, Rct is the charge-migration resistance, i.e., the reactivity of Li+ intercalation/de-intercalation in LiEuTiO4. Cic represents the capacitance resulting from the boundary between the electrolytes and the electrodes. Finally, Zw refers to the diffusion impedance of LiEuTiO4 at low frequency. Table 2 presents the values of Re, Rsei, Rct, and Zw obtained from fitting to the data.

Nyquist plots with fitted lines of LiEuTiO4 nanovesicles and commercial LiEuTiO4 after (a, b) and before cycling (c). The inset is the corresponding equivalent circuits. d C1s XPS spectra and e FTIR spectra of LiEuTiO4 nanovesicles cycled between different potential range; f cycle performance of the samples after addition of 2 wt% VC to 1 M (LiPF6/EC + DMC + EMC) electrolyte
The large impedance semicircle that appeared during the first cycle for the commercial LiEuTiO4 suggests that Li+ insertion into LiEuTiO4 is a difficult process [30]. This is likely because the diffusion and infiltration of the electrolyte into the electrode materials during the first cycle do not proceed readily. As such, the diffusion and infiltration of the electrolyte would be the rate determining step in batteries before cycling (Fig. 5c and Table 2). However, after the first cycle, the charge-migration and diffusion steps together control the electrochemical process (Fig. 5, panels a and b). Apparently, the Re, Rct, and Zw values of the LiEuTiO4 nanovesicles are smaller. This is because of their unique structure (Fig. 1a), which increases the speed of charge migration and spread at the boundary of the electrode/electrolyte. The smaller Rsei of those nanovesicles is the result of their larger surface area and the unique structure of this material, which permits rapid Li+ transportation, reduces the occurrence of disadvantageous reactions, and suppresses the growth of an SEI film. For these reasons, the AC impedance of LiEuTiO4 could potentially be optimized through careful design of the particle morphology.
The simulation results in Table 2 indicate that an interfacial SEI film is formed in both samples during cycling. From the C1s XPS results [31, 32] (Fig. 5d) and FTIR spectra [33,34,35] (Fig. 5e) of the LiEuTiO4 nanovesicles’ electrode, it can be seen that the SEI film contains lots of carbonyl compounds and a small amount of Li2CO3 and ROCO2Li [32,33,34]. When the electrolyte solution is reduced to a low operating voltage (< 0.7 V), a SEI film will form on the electrode’s surface after the cycling between 0 and 3 V. To determine if in our work the SEI results from the reduction of the electrolyte or from reactions between LiEuTiO4 and the electrolyte, we analyzed the surface of the LiEuTiO4 electrode using XPS (Fig. 5d) and FTIR (Fig. 5e) during cyclic tests at 0.01 to 1 V and 1 to 3 V, respectively. In Fig. 3, panels b and c demonstrate that the primary species of the SEI film generated by the LiEuTiO4 nanovesicles cycled over the range of 0.01–1 V were Li2CO3 and ROCO2Li. This is the same as the species observed on the surface of graphite anodes [34]. In sharp contrast, the primary species of the SEI film produced by cycling in the range of 1–3 V were carbonyl compounds (Fig. 5, panels d and e). The discrepancy in the components of the SEI films after cycling in the different voltage ranges means that they have different formation schemes [30]. In fact, the electrolyte solution could only be reduced below 1 V. Thus, the SEI film would have been formed during charging/discharging between 0.01 and 3 V. In other words, it is not necessary to charge/discharge between 1 and 3 V. For this reason, the C=O species must result from reactions between the electrolyte and the LiEuTiO4 electrode. Therefore, both the electrolyte reduction and the reactions between LiEuTiO4 and the electrolyte contributed to the formation of the SEI film for LiEuTiO4 during electrochemical cycling. However, the main source of the film is the reactions, as confirmed by the C1s XPS and FTIR spectra of the samples cycled between 0.01 and 3 V.
3.2.4 Vinylene carbonate effect
The reduction potential of vinylene carbonate in the electrolyte solution was 1.5 V. In other words, it can effectively reduce the detrimental reaction between the electrolyte and the electrode to avoid the formation of SEI films [36]. Consequently, it could play the role of a barrier for the SEI film by suppressing the interfacial reactions between the electrode and electrolyte [37]. Consequently, the cyclic performances of both samples were improved when 2 wt% vinylene carbonate was added to the electrolyte solution (Fig. 5f). The commercial LiEuTiO4 delivered a charge capacity of 78.7 mAh g−1 after 100 cycles with only a 20% loss, while our LiEuTiO4 nanovesicles retained a remarkable value of 289.4 mAh g−1 even after 1000 cycles, with only a 2.3% loss in capacity. These data indicate that the irreversible capacity can be improved through using novel electrolyte additives or surface coatings for materials; related work is ongoing in our laboratory.
4 Conclusions
In summary, we successfully synthesized LiEuTiO4 nanovesicles. They have a large specific surface area of 225.4 m2 g−1. Electrochemical analysis showed that these nanovesicles have an outstanding rate performance together with a large discharge capacity of 285.1 mAh g−1 at 0.2 A g−1 and 185.7 mAh g−1 at 10 A g−1 and also have an excellent cycle performance with a large discharge capacity of 282 mAh g−1 after 1000 cycles at 0.1 A g−1. The excellent electrochemical performances for our LiEuTiO4 nanovesicles can be attributed to their unique structure. First, the small particle size significantly improves the electrical contact with copper foil and reduces the lithium ion/electron transport path lengths. This benefits a quick and reversible lithium ion extraction/insertion. Second, the high specific surface area increases the electrode/electrolyte interface region, thus promoting the rapid migration of Li+ from the electrolyte into the nanovesicles. Additionally, the amorphous structure of the nanovesicles can ignore lattice stress and offer sustained Li+ transport approaches and intercalation sites. This substantially increases lithium ion storage, structure stability, and cycle performance. An SEI film could form on the surface of the LiEuTiO4 anode. After the addition of vinylene carbonate, the rate and cyclic performance of LiEuTiO4 improved. These results demonstrate that LiEuTiO4 can function as an ideal low operating voltage anode material in high-rate LIBs intended for EVs and HEVs without a corresponding reduction in battery voltage or energy density.
References
Palacín M, Guibert A (2016) Batteries: why do batteries fail? Science 351:1253292
Wang C-Y, Zhang G, Ge S, Xu T, Ji Y, Yang X-G, Leng Y (2016) Lithium-ion battery structure that self-heats at low temperatures. Nature 529:515–518
Lüders C, Zinth V, Erhard SV, Osswald PJ, Hofmann M, Gilles R, Jossen A (2016) Lithium plating in lithium-ion batteries investigated by voltage relaxation and in situ neutron diffraction. J Power Sources 342:17–23
Chen S, Xin Y, Zhou Y, Ma Y, Zhou H, Qi L (2016) Self-supported Li4Ti5O12 nanosheet arrays for lithium ion batteries with excellent rate capability and ultralong cycle life. Energy Environ Sci 7:1924–1930
Shen L, Uchaker E, Zhang X (2016) Hydrogenated Li4Ti5O12 nanowire arrays for high rate lithium ion batteries. Adv Mater 24:6502–6506
Armand M, Tarascon J-M (2008) Building better batteries. Nature 451:652–657
Erickson EM, Ghanty C, Aurbach D (2014) New horizons for conventional lithium ion battery technology. J Phys Chem Lett 5:3313–3324
Goodenough JB, Kim Y (2008) Challenges for rechargeable Li batteries. Chem Mater 22:587–603
Oumellal Y, Rougier A, Nazri G, Tarascon J, Aymard L (2008) Metal hydrides for lithium-ion batteries. Nat Mater 7:916–921
Huang J, Yang K, Zhang Z, Yang L, Hirano S-I (2017) Layered perovskite LiEuTiO4 as a 0.8 V lithium intercalation electrode. Chem Commun 53:7800–7803
Wei D, Tang QJ, Tong DG (2019) One-pot synthesis of LiEuTiO4 as an anode material for lithium-ion batteries operating at 0.8 V. J Taiwan Inst Chem Engineers 96:223–228
Chen Y, Zhu D, Ji C, Zhu X, Xu Y, Li D (2019) A facile strategy to upgrade electrochemical performances of LiEuTiO4 by surface modification derived from pyrolysis of urea. Ionics 25:3041–3050
Sun C-F, Hu J, Wang P, Cheng X-Y, Lee SB, Wang Y (2016) Li3PO4 matrix enables a long cycle life and high energy efficiency bismuth-based battery. Nano Lett 16:5875–5882
Clark SJ, Wang D, Armstrong AR, Bruce PG (2016) Li(V0.5Ti0.5)S2 as a 1 V lithium intercalation electrode. Nat Commun 7:10898
Gupta A, Mullins CB, Goodenough JB (2012) Electrochemical probings of Li1+xVS2. Electrochim Acta 78:430–433
Kim Y, Park K-S, Song S-H, Han J, Goodenough JB (2009) Access to M3+/M2+ redox couples in layered LiMS2 sulfides (M = Ti, V, Cr) as anodes for li-Ion battery. J Electrochem Soc 156:A703–708
Armand M, Grugeon S, Vezin H, Laruelle S, Ribière P, Poizot P, Tarascon J-M (2009) Conjugated dicarboxylate anodes for Li-ion batteries. Nat Mater 8:120–125
Wang JP, Bai Y, Wu MY, Yin J, Zhang WF (2009) Preparation and electrochemical properties of TiO2 hollow spheres as an anode material for lithium-ion batteries. J Power Sources 191:614–618
Sun J, Lv C, Lv F, Chen S, Li D, Guo Z, Han W, Yang D, Guo S (2017) Tuning the shell number of multishelled metal oxide hollow Fibers for optimized lithium-ion storage. ACS Nano 11:6186–6193
Zhu JX, Tang CJ, Zhuang ZC, Shi CW, Li NR, Zhou L, Mai LQ (2017) Porous and low-crystalline manganese silicate hollow spheres wired by graphene oxide for high-performance lithium and sodium storage. ACS Appl Mater Interfaces 9:24584–24590
Miao X, Li C, Chu W, Wu NP, Tong DG (2015) Li9V3(P2O7)3(PO4)2 Nanotubes fabricated by a simple molten salt approach with excellent cycling stability and enhanced rate capability in lithium-ion batteries. RSC Adv 5:243–247
Tong DG, Zeng XL, Chu W, Wang D, Wu P (2010) Preparation of monodispersed cobalt–boron spherical nanoparticles and their behavior during the catalytic decomposition of hydrous hydrazine. Mater Res Bull 45:442–447
Miao X, Chen MM, Chu W, Wu P, Tong DG (2016) Mesoporous face-centered-cubic In4Ni alloy nanorices: superior catalysts for hydrazine dehydrogenation in aqueous solution. ACS Appl Mater Interfaces 8:25268–25278
Wei D, Xie J, Tong DG (2018) Amorphous europium hexaboride: a potential room temperature formaldehyde sensing material. ACS Appl Mater Interfaces 8:35681–35684
Besenhard JO (2007) Handbook of battery materials. Wiley, Hoboken
Wu YP, Dai XB, Ma JQ, Cheng YJ (2004) Lithium ion batteries: practice and applications. Chemical Industry Press, Beijing
Yang L, Cao W (2018) Robust macroporous materials of chiral polyaniline composites. Chem Mater 2006:297–300
Park K-S, Benayad A, Kang D-J, Doo S-G (2008) Nitridation-driven conductive Li4Ti5O12 for lithium ion batteries. J Am Chem Soc 130:14930–14931
Song S-H, Alonso JA, Cheng J-G, Goodenough JB (2014) Magnetic phase transformation induced by electrochemical lithium intercalation in Li1+ xEuTiO4 and Li2+2xEu2Ti3O10 (0 ≤ x ≤ 1) compounds. J Solid State Electrochem 18:2047–2060
Cha QX (2002) Kinetics of electrode. Science Press, Beijing
Cai Y, Huang Y, Jia W, Wang X, Guo Y, Jia D, Sun Z, Pang W, Guo Z (2016) Super high-rate, long cycle life of europium-modified, carbon-coated, hierarchical mesoporous lithium-titanate anode materials for lithium ion batteries. J Mater Chem A 4:9949–9957
Dedryvere R, Gireaud L, Grugeon S, Laruelle S, Tarascon JM, Gonbeau D (2005) Characterization of lithium alkyl carbonates by X-ray photoelectron spectroscopy: experimental and theoretical study. J Phys Chem B 109:15868–15875
Guo XW, Fang XP, Sun Y, Shen LY, Wang ZX, Chen LQ (2013) Lithium storage in carbon-coated SnO2 by conversion reaction. J Power Sources 226:75–81
Shu J, Shui MA, Huang FT, Xu D, Ren YL, Hou L, Cui J, Xu JJ (2011) Surface behaviors of conductive acetylene black for lithium-ion batteries at extreme working temperatures. J Phys Chem C 115:6954–6960
Jin XY, Chen SL, Yao EN (1992) Application guide of infrared spectra. Science and Technology Press of Tian Jin, Tian Jin
Dedryvère R, Martinez H, Leroy S, Lemordant D, Bonhomme F, Biensan P, Gonbeau D (2007) Surface film formation on electrodes in a LiCoO2/graphite cell: a step by step XPS study. J Power Sources 174:462–468
He Y, Liu M, Huang Z-D, Zhang B, Yu Y, Li B, Kang F, Kim J-K (2013) Effect of solid electrolyte interface (SEI) film on cyclic performance of Li4Ti5O12 anodes for Li ion batteries. J Power Sources 239:269–276
Acknowledgements
This work was financially supported by Natural Science Foundation of China (21376033), the State Key Laboratory of Geohazard Prevention and Geoenvironment Protection Independent Research Project (SKLGP2016Z012, SKLGP2018Z006). We thank Jim Bailey, PhD, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, Q.L., Tong, D.G. Amorphous LiEuTiO4 nanovesicles as a low-operating potential anode for rechargeable lithium ion batteries. SN Appl. Sci. 2, 539 (2020). https://doi.org/10.1007/s42452-020-2352-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-020-2352-9