
Opinion statement
Allergen immunotherapy (AIT) with whole allergens or allergen extracts has been in use for more than one hundred years. It is clinically efficacious and disease-modifying. However, AIT is also associated with a profile of significant adverse events, including the potential to cause severe, systemic allergic reactions. One alternative to traditional whole-antigen AIT is peptide immunotherapy that uses small synthetic peptide immunoregulatory epitopes (SPIRE) representing T cell epitopes from the allergen of interest. Peptide immunotherapy is being developed for the treatment of allergic and autoimmune diseases where pathogenesis is T cell-dependent. Short, soluble, monomeric peptide fragments avoid the problem of IgE-mediated adverse events (since the peptides will not cross-link allergen-specific IgE on the surface of effector cells, such as mast cells and basophils). However, such peptides retain the ability to induce T cell tolerance and immunoregulation. In early clinical trials, efficacy was demonstrated months to years after the cessation of a short course of treatment, supporting the conclusion that this approach is disease-modifying, changing the natural history of the disease. The improved safety profile of short peptides allows for larger molar-equivalent doses to be administered in shorter time frames than AIT; treatment can be completed with as few as four intradermal injections, while efficacy persists for two years or more.
Similar content being viewed by others
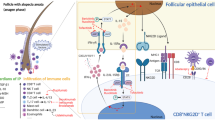


Introduction
Allergen immunotherapy (AIT) is clinically efficacious and disease-modifying. Studies of both subcutaneous immunotherapy (SCIT) and sublingual immunotherapy (SLIT) have demonstrated reductions in symptom scores and medication usage [1, 2]. Clinical benefit persists beyond the treatment period, suggesting a resetting of the allergen-specific adaptive immune response. AIT has been shown to prevent progression of allergic rhinitis to asthma and to reduce the risk of developing new allergic sensitizations. The mechanisms of this approach are not completely understood, but several studies have supported a role for the induction of allergen-specific T cells and B cells with immunoregulatory or suppressive characteristics, such as IL-10-secreting Tr1 T cells and B10 B cells [3–6]. Furthermore, clinical efficacy resulting from treatment with whole allergen molecules, or extracts, is associated with induction of allergen-specific IgG (particularly the IgG4 isotype). While absolute levels of serum allergen-specific IgG4 do not correlate with clinical efficacy, there is increasingly convincing data that high affinity allergen-specific IgG4 may better represent a functional “blocking” antibody that may be capable of sequestering an allergen before it reaches effector cells [7]. However, the use of whole allergen proteins carries a significant risk of adverse events. Allergic adverse events result from conformational epitopes that are able to able to cross-link IgE molecules bound to the surface of effector cells (mast cells and basophils) and can cause both local and systemic allergic reactions. Adverse events lead to dose limitation and a protracted course of treatment that typically lasts about three years and requires monthly injections (SCIT), or daily dosing (SLIT). Lack of compliance is increasingly recognized as a major limiting factor with whole allergen approaches and is related to the onerous nature of the treatment regimen. For example, a recent European study found poor overall real-life compliance to SCIT and SLIT, with only 18 percent of 6,486 patients reaching the minimally required duration of treatment of three years (SCIT, 23 %; SLIT, 7 %) [8••].
Numerous strategies have been developed to overcome adverse allergic events in AIT. These approaches primarily involve altering the structure of the antigen to maintain the ability to induce tolerance, but ameliorate the risk of eliciting allergic reactions. A promising approach is the use of synthetic peptide immunoregulatory epitopes (SPIREs) that are short soluble peptides containing the major T cell epitopes of clinically important allergen proteins. This therapeutic strategy delivers the relevant T cell epitopes required for tolerance induction, but does so using peptide fragments that lack the length and conformational structure that would allow them to cross-link IgE molecules on the surface of effector cells.
It is currently believed that the presentation of peptide epitopes to T cells in the absence of pro-inflammatory signals induces a state of antigen-specific immune tolerance. This manifests itself as inactivation of antigen-specific T cells (anergy), skewing of the T helper cell phenotype from Th2 to Th1, and the induction of regulatory T cells that are further able to down-regulate inflammatory immune responses through production of the tolerogenic cytokines IL-10 and/or TGF-β [3, 4].
Peptide immunotherapy for cat allergies
Allergy to cat dander is relatively common, with 17 % of the population of the United States (US) yielding a positive skin prick test [9]. Almost 30 % of allergic asthma in the US is attributable to cat sensitization [10]. The application of allergen immunotherapy (AIT) has been shown to ameliorate some aspects of a cat allergy [11, 12]. Administration of large doses of cat dander, which contains the primary cat allergen Fel d1, results in significant reductions in skin reactivity and airway hypersensitivity in allergic patients [11, 12]. Despite these positive results, the use of whole allergens in AIT leads to unpredictable anaphylactic reactions in some patients [12]. This is primarily due to the fact that whole Fel d1 molecules are able to cross-link IgE molecules bound to effector cells in sensitized individuals. To circumvent this problem, the regions of Fel d1 recognized by T cells of cat-allergic individuals were identified and formulated into peptide-based therapies.
Based on major histocompatibility complex (MHC) binding studies assessing the binding affinity of candidate T cell epitope peptides (from the sequence of Fel d 1) to the ten most common HLA-DR molecules in the human population, Worm et al. were able to define seven short peptides (13-17 amino acids; ToleroMune Cat) [13•] with moderate to high affinity binding to multiple MHC gene products expressed by more than 95 percent of the population. Cellular reactivity and the potential to induce basophil histamine release among the selected peptides was evaluated in cat-allergic subjects. The selected peptides were formulated into an adjuvant-free therapeutic preparation and evaluated in a Phase 2a clinical trial for safety and surrogate clinical efficacy via two different administration routes. Using an incremental dose cohort design, the authors showed that Cat-PAD was well tolerated in doses from 0.03 nmol to 12 nmol via intradermal administration, or 20 nmol via subcutaneous administration [13•]. Based on the ability of a single administration of Cat-PAD to reduce the magnitude of the late-phase skin reaction (LPSR), 3-nmol (containing approximately 35 μg of Fel d 1 epitopes) and 6-nmol doses and intradermal administration were selected for Phase 2b clinical studies.
The clinical efficacy of the 3-nmol and 6-nmol doses of Cat-PAD was subsequently evaluated in two separate, repeat administration, Phase 2b studies performed in an allergen exposure model [14•, 15••]. Subjects in both studies were exposed to physiologically relevant airborne levels of cat allergen extract (Fel d 1 concentrations of approximately 50 ng/m3) in an environmental exposure chamber (EEC). In the first of these studies, subjects with allergic rhinoconjunctivitis triggered by exposure to cats, attended the EEC for four consecutive days and were exposed to airborne cat allergen extract for three hours on each day. EEC challenges were performed at baseline and 18-22 weeks after the initiation of the study. One hundred and twenty one subjects meeting the inclusion/exclusion criteria were randomized to receive a placebo regimen or one of four Cat-PAD regimens consisting of either 3-nmol or 6-nmol doses administered in either 4 or 8 administrations over 12-14 weeks. Efficacy was assessed in terms of changes to the total rhinoconjuncivitis symptom score (TRSS), which measured eight symptom fields consisting of both nasal (sneezing, itchy nose, runny nose, nasal congestion) and ocular (itchy eyes, watery eyes sore eyes, red eyes) outcomes. Each outcome field had a score of 0-3 points with “0” representing no symptoms and “3” representing severe symptoms. A baseline TRSS score of at least 10 out of 24 (including 6 out of 12 for nasal symptoms) on at least one time point on days 3 and 4 of EEC exposure was a prerequisite for study entry. Treatment with Cat-PAD was safe and well tolerated in this study. The frequency of treatment-emergent adverse events (TEAEs), related and unrelated to treatment, was generally lower in the Cat-PAD-treated arms than in the placebo arms, although in the 6-nmol group the frequency of TEAE was higher. Sub-analysis of respiratory system TEAEs showed low incidence of respiratory symptoms in all groups, with the majority of reported events occurring as a result of the EEC exposure to airborne allergen. Administration of eight intradermal injections of Cat-PAD at the 3-nmol dose at two-week intervals resulted in a greater improvement in symptom scores than the placebo with a mean treatment TRSS difference of 2.9 units between 1-3 hours on Days 2-4 (pre-specified primary outcome) of the EEC challenge [14•].
More recently, Patel and colleagues compared the 3-nmol and 6-nmol dosing schedules in a similar study design utilizing an EEC [15••]. In addition to the post-treatment efficacy outcomes measured at 18-22 weeks, efficacy was also evaluated one year after initiation of the study, nine months after cessation of treatment. Here, as before, treatment with Cat-PAD resulted in improvement in the TRSS at both short-term (18-22 week) outcomes. Two hundred and two subjects were randomised to one of three regimens (8 × 3 nmol two weeks apart; 4 × 6 nmol four weeks apart with infill placebo to maintain blinding; or 8 × placebo). Of these, 174 subjects completed at least one post-treatment EEC visit and these comprised the “intention to treat” (ITT) population. One hundred and seventy subjects completed the first phase of the study, such that they produced EEC data on days 2-4 of the post treatment challenge. At 18-22, weeks the mean change in TRSS in Cat-PAD-treated groups exceeded that of the placebo group (8 × 3 nmol -5.136; 4 × 6 nmol -5.406; placebo -2.786), with both Cat-PAD treatment arms exhibiting similar outcomes.
In a protocol extension, in which investigators and participating subjects remained blinded to treatment allocation, all subjects were invited to participate in a one-year follow-up study. As a result, 89 subjects were re-enrolled in a follow on study and returned to the EEC, at 50-54 weeks, for a repeat of the four-day exposure challenge. The aim of the extension was to assess continuing efficacy following a period of nine months without further treatment. Treatment with the 6-nmol Cat-PAD dose resulted in persistent efficacy. In the non-asthmatic population (pre-defined primary outcome measure), the 6-nmol dose resulted in a decrease in the mean TRSS from a baseline of -6.778 points in comparison with the 3-nmol dose (-3.893) and placebo (-2.908). The change in TRSS observed with the 6-nmol dosage was significantly different to both the 3-nmol dose (p = 0.03) and placebo groups (p = 0.01). Similar results were obtained when a pooled analysis of asthmatic and non-asthmatic subjects was performed. Interestingly, TRSS improvement at weeks 50-54 appeared to be greater for the 6-nmol group than at 18-22 weeks. Analysis of total nasal symptom scores (TNSS) in the non-asthmatic population demonstrated statistically significant decreases in mean change from baseline TNSS for the 6-nmol dose (-3.435) and placebo groups (-1.625; p = 0.02 for 6 nmol vs placebo). Similarly, total ocular symptom scores (TOSS) showed a larger decrease in mean change from baseline TOSS for the 6-nmol dose (-3.343) when compared to both 3-nmol (-1.716; p = 0.03 vs 6 nmol) and placebo groups (-1.283; p = 0.01 vs placebo) (15).
In a further extension of this study, 51 subjects were re-enrolled, with study subject and investigator blinding maintained. Subjects were returned to the EEC for an additional four consecutive days of allergen exposure (102 to 106 weeks after initiation of the study). No further treatment with Cat-PAD or placebo was administered between the initial course of treatment and the two-year EEC outcome. A greater improvement was observed in TRSS in the 6-nmol Cat-PAD group (mean reduction in TRSS from baseline -5.87) compared to the placebo (mean reduction in TRSS from baseline -2.02), with a mean treatment effect on TRSS between the two groups of approximately 5 units at the end of fourth day of the EEC challenge [16].
In a post hoc analysis of peak nasal symptoms (TNSS), nasal symptoms were found to peak between two and three hours on each of the four EEC challenge days. Comparison of the least squares mean TNSS between groups of all four EEC visits demonstrated that symptoms were significantly (p < 0.05) lower in the 6-nmol Cat-PAD group compared to the placebo at the two-year follow up. No significant difference was observed at two years between the 3-nmol treatment group and the placebo group [17].
In order to examine the consistency of the treatment effect of Cat-PAD at an individual study subject level, a post-hoc analysis was performed to evaluate the correlation between the change from baseline in mean TRSS on days 1-4 at the 18-22 week outcome and the change in subsequent (50-54 week and 100-104 week) outcomes. For the 89 subjects who returned at 50-54 weeks, the treatment effect for each individual correlated strongly with their response at 18-22 weeks (regression slope = 0.85; correlation coefficient = 0.74). Similarly, for the 51 subjects who returned for the 100-104 week outcome, the treatment effect correlated strongly (correlation coefficient = 0.89); indeed, the slope of the regression curve (1.36) suggested an increased treatment effect at this follow up [18].
House dust mite (HDM)
Based on in silico and in vitro analysis of the ability of peptide sequences derived from major house dust mite allergens to function as synthetic peptide immune-regulatory epitopes (SPIREs), seven short peptides were selected and formulated for intradermal administration. An escalating dose, Phase 2a study was employed to evaluate “first-into-man” safety/tolerability and to identify active doses of HDM SPIREs to be advanced into Phase 2b studies [19]. Subjects with a history of allergic rhinoconjunctivitis triggered by HDM were recruited to a randomized, double-blind, placebo-controlled trial of HDM SPIREs performed in an EEC challenge model with each challenge period consisting of three consecutive days with four hours of airborne allergen exposure (D. pteronyssinus extract 20-120 ng/m3) [20, 21]. Baseline TRSS was assessed in the EEC prior to treatment with a placebo, or one of three HDM-SPIRE regimens (11 intradermal administrations of 3 nmol, 11 × 6 nmol, or 11 × 12 nmol; all administrations at 4 week intervals). In the initial phase of the study, 20 subjects were recruited per treatment arm. Subjects returned for EEC challenges at weeks 19, 36, and 49-50. At the 19-week time point, following the fourth of eleven administrations (i.e., after 4 × 12-nmol HDM-SPIRE), a pre-specified interim analysis of efficacy was performed in order to identify the most efficacious dose for further study. In the second phase of the study, the 11 × 12-nmol HDM-SPIRE and placebo groups were expanded to 44 subjects. The interim analysis at 19 weeks showed a mean change in TRSS of -3.97 in the 12-nmol HDM-SPIRE group compared to -1.83 in the placebo group. As a result of these positive results, a 4 × 12 nmol regimen was also evaluated in the second phase of the study in an expanded number of subjects (n = 44). At the week 49-50 EEC visit, the 4 × 12-nmol group showed a mean change in TNSS of -5.82 compared to the placebo group at -3.04 (p = < 0.05). A subjective global assessment of symptom improvement revealed that 68 percent of the subjects treated with 4 × 12-nmol HDM-SPIRE scored “any better” compared to 44 percent of the placebo group (p = 0.05). Treatment with the full, 11-injection regimen proved no more effective than 4 administrations [20, 21].
Grass allergy
The efficacy of grass-SPIREs, consisting of seven immunodominant T cell epitopes from several grass species, was evaluated in an environmental exposure unit (EEU) model in which subjects were exposed for four consecutive days (three hours per day) to mean circulating rye grass pollen counts of 3,500 ± 500 grains/m3. EEU visits occurred at baseline (approximately four months prior to the grass pollen season) and approximately 25 weeks after initiation of the grass-SPIRE therapy. TRSS was recorded at 30-minute intervals on a scale of 0-24, encompassing eight nasal and ocular symptom fields. At the end of the final EEU visit, subjects completed a seven-point global assessment of changes in symptomatology comparing baseline to post EEU challenge. Two hundred and eight subjects were randomized to one of three grass-SPIRE dosing regimens. Subjects treated with 8 × 6-nmol grass-SPIRE (two week dosing interval) showed a mean change in TRSS of -5.4 at the protocol-specified primary endpoint (all time points on days 2, 3, and 4 in participants with a mean baseline TRSS > 8 points), in comparison with a mean change of -3.8 in the placebo group. The protocol-specified secondary endpoint (change in mean TRSS at EEU visit 25 weeks after randomization in subjects with baseline TRSS > 12) revealed a greater treatment effect with grass-SPIRE (-5.3 versus placebo -3.4; p = 0.05) [22]. Changes in nasal symptom scores were also apparent in this study. The 8 × 6-nmol group showed a change in mean TNSS of -2.3 versus placebo (-1.6; p < 0.05). Furthermore, evaluation of total non-nasal symptom scores (TNNSS) showed a statistical trend for a greater treatment effect in the 8 × 6-nmol group (-3.1) versus the placebo group (-2.2; p = 0.08). Forty four percent of subjects in the 8 × 6-nmol group considered themselves feeling substantially better after treatment, compared with 22 percent of subjects receiving the placebo (p < 0.01) [23].
Conclusion
Although further well designed clinical trials are required (Phase 3 with Cat-PAD is nearing completion at the time of writing this article), peptide immunotherapy is emerging as a promising treatment option for allergic diseases. By using synthetic peptide immunoregulatory epitopes (SPIREs), functional immune tolerance can be established through a combination of mechanisms, including immune deviation (Th2 to Th1) and the generation of regulatory T cells that appear to exert their suppression of allergen-specific immune responses via the actions of IL-10. Growing evidence that patient compliance with both subcutaneous and sublingual allergen immunotherapy is poor highlights the need for rapid, safe treatment modalities that relieve symptoms for extended periods of time. The studies described here demonstrate that treatment with as few as four intradermal of SPIRE injections can provide sustained symptom relief for two years. Furthermore, clinical studies to date demonstrate a favourable safety profile for this form of therapy. Further studies of peptide immunotherapy may not only produce a new class of specific disease-modifying therapeutics, but also provide fresh insight into the immunological mechanisms underlying allergy and immune tolerance.
References and Recommended Reading
Durham SR, Walker SM, Varga EM, Jacobson MR, O’Brien F, Noble W, et al. Long-term clinical efficacy of grass-pollen immunotherapy. N Engl J Med. 1999;341:468–75.
Durham SR, Emminger W, Kapp A, de Monchy JG, Rak S, Scadding GK, et al. SQ-standardized sublingual grass immunotherapy: confirmation of disease modification 2 years after 3 years of treatment in a randomized trial. J Allergy Clin Immunol. 2012;129(3):717–25.
Larche M. Peptide immunotherapy for allergic diseases. Allergy. 2007;62(3):325–31.
Moldaver D, Larche M. Immunotherapy with peptides. Allergy. 2011;66(6):784–91.
Verhoef A, Alexander C, Kay AB, Larche M. T cell epitope immunotherapy induces a CD4(+) T cell population with regulatory activity. PLoS Med. 2005;2(3):e78.
van de Veen W, Stanic B, Yaman G, Wawrzyniak M, Söllner S, Akdis DG, et al. IgG4 production is confined to human IL-10-producing regulatory B cells that suppress antigen-specific immune responses. J Allergy Clin Immunol. 2013;131(4):1204–12.
Shamji MH, Ljørring C, Francis JN, Calderon MA, Larché M, Kimber I, et al. Functional rather than immunoreactive levels of IgG4 correlate closely with clinical response to grass pollen immunotherapy. Allergy. 2012;67(2):217–26.
Kiel MA, Röder E, Gerth van Wijk RG, Al MJ, Hop WCJ, Rutten-van Mölken MPMH. Real-life compliance and persistence among users of subcutaneous and sublingual allergen immunotherapy. J Allergy Clin Immunol. 2013;132:353–60. A recent study providing real-life compliance data from registry data on over 6,000 allergic individuals prescribed either subcutaneous or sublingual immunotherapy. The article highlights the extremely poor compliance with these forms of therapy.
Calabria CW, Dice JP, Hagan LL. Prevalence of positive skin test responses to 53 allergens in patients with rhinitis symptoms. Allergy Asthma Proc. 2007;28(4):442–8.
Arbes SJ, Gergen PJ, Elliott L, Zeldin DC. Prevalences of positive skin test responses to 10 common allergens in the US population: results from the third National Health and Nutrition Examination Survey. J Allergy Clin Immunol. 2005;116(2):377–83.
Taylor WW, Ohman Jr JL, Lowell FC. Immunotherapy in cat-induced asthma. Double-blind trial with evaluation of bronchial responses to cat allergen and histamine. J Allergy Clin Immunol. 1978;61:283–7.
Ohman JL, Findlay SR, Leitermann KM. Immunotherapy in cat-induced asthma. Double-blind trial with evaluation of in vivo and in vitro responses. J Allergy Clin Immunol. 1984;74(3 Pt 1):230–9.
Worm M, Lee HH, Kleine-Tebbe J, et al. Development and preliminary clinical evaluation of a peptide immunotherapy vaccine for cat allergy. J Allergy Clin Immunol. 2011;127(1):89-97, 97.e1-14. A recent study describing the development of the first SPIRE therapy, Cat-PAD, including the results of the first-into-man Phase 2a clinical trial.
Worm M, Patel D, Creticos PS. Cat peptide antigen desensitisation for treating cat allergic rhinoconjunctivitis. Expert Opin Investig Drugs. 2013;22(10):1347–57. A recent study describing results from the first Phase 2b clinical trial of Cat-PAD.
Patel D, Couroux P, Hickey P, et al. Fel d 1-derived peptide antigen desensitization shows a persistent treatment effect 1 year after the start of dosing: a randomized, placebo-controlled study. J Allergy Clin Immunol. 2013;131(1):103-9.e1-7. A key recent article describing enduring clinical efficacy of a short course of peptide immunotherapy given over a three month period. Despite no further treatment for nine months, a persistent treatment effect was observed one year after initiation of the study.
Couroux P, Patel D, Armstrong K, Larché M, Hafner RP. Fel d 1-derived synthetic peptide immuno-regulatory epitopes show a long-term treatment effect in cat allergic subjects. Clin Exp Allergy. 2015; .doi:10.1111/cea.12488
Hafner RP, Couroux P, Armstrong K, et al. Total nasal symptom scores are reduced in an EEC model of cat allergy two years after administration of four doses of Cat-PAD. Allergy. 2013;68(97):647–8.
Haumann B, Couroux P, Armstrong K, et al. Cat-PAD demonstrates sustained, consistent treatment effect on allergic rhinoconjunctivitis in individual cat allergic patients 1 and 2 years after four intradermal injections. Allergy. 2013;68(97):70.
Larche M, Hickey P, Hebert J, et al. Safety and tolerability of escalating doses of house dust mite- peptide antigen desensitization (HDM-PAD). J Allergy Clin Immunol. 2013;131(2):AB37.
Hafner R, Couroux P, Armstrong K, et al. Persistent treatment effect achieved at one year after four doses of der p derived synthetic peptide immuno-regulatory epitopes in an exposure chamber model of house dust mite allergy. J Allergy Clin Immunol. 2014;133(2):AB289.
Hafner RP, Couroux P, Armstrong K, et al. Four doses of Der p derived synthetic peptide immuno-regulatory epitopes over 3 months results in a persistent treatment effect at 1 year on symptoms of house dust mite allergy. Allergy. 2014;69(99):31–2.
Ellis AK, Frankish CW, Armstrong K, et al. Treatment with synthetic peptide immuno-regulatory epitopes derived from grass allergens leads to a substantial reduction in grass allergy symptoms in the environmental exposure unit. J Allergy Clin Immunol. 2014;133(2):AB290.
Ellis AK, Frankish CW, Armstrong K, et al. A short course of synthetic peptide immuno-regulatory epitopes derived from grass allergens leads to a reduction in grass allergy symptoms. Allergy. 2014;69(99):32.
Compliance with Ethics Guidelines
Conflict of Interest
Mark Larché is a co-founder of Circassia Ltd., and is a current consultant to the company and a shareholder in the company. Mark Larché is a consultant to Adiga Life Sciences. In the last 12 months, Mark Larché has acted as a paid consultant to Brandon Capital Ventures and Touchlight Genetics.
Elizabeth Simms, Ijlal Syed, and Christopher Rudulier declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Specific Immunotherapy
Rights and permissions
About this article

Cite this article
Simms, E., Syed, I., Rudulier, C. et al. Peptide Immunotherapy; Short but Long Lasting?. Curr Treat Options Allergy 2, 64–71 (2015). https://doi.org/10.1007/s40521-014-0043-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40521-014-0043-8