
Abstract
Melanomas affecting different components of the uvea occur with differing frequencies and clinical presentations. Uveal melanoma is diagnosed via funduscopic exam and ancillary tests. These lesions may present with visual findings or incidental findings on physical exam. Metastasis occurs in approximately half of all patients with primary uveal melanoma. The liver is the most common site of metastasis. Enucleation was at one time considered the definitive local treatment for primary uveal melanoma, but has been largely replaced by other therapeutic procedures that aim to prevent metastasis while preserving vision. Unfortunately, metastasis of uveal melanoma almost always proves to be fatal. The current treatment of metastatic uveal melanoma is limited by the intrinsic resistance of uveal melanoma to conventional systemic therapies. Advancements in molecular biology have resulted in the identification of a number of promising prognostic and therapeutic targets. Early detection and therapy are important factors in disease survival. It is imperative that the treating physician be familiar with the clinical features of uveal melanoma and distinguish it from mimickers in order to ensure effective and timely treatment.
Similar content being viewed by others


Introduction
Uveal melanoma is the most common primary cancer of the eye in adults [1]. Although both uveal and cutaneous melanomas both originate from melanocytes, their underlying pathogenesis and clinical behavior differ significantly [2].
In the past decade, many details surrounding the underlying pathogenesis of uveal melanoma have emerged, revealing a complex and evolving story. Cytogenetic and molecular genetic features of the uveal cells have been demonstrated to have strong prognostication value in uveal melanoma [3].
The natural history of uveal melanoma often involves the development of metastasis, an event associated with poor prognosis [4]. Therefore, it is imperative that the treating physician be familiar with the clinical features of uveal melanoma allowing for early treatment and aversion of potentially life-threatening metastasis [5].
Uveal melanoma restricted to a limited anatomical region may be controlled by local and locoregional treatments. However, the treatment of metastatic uveal melanoma is limited by the lack of effective systemic treatments [1]. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Epidemiology
Uveal melanoma originates from melanocytes residing in the uveal tract [1]. Approximately 90% of uveal melanomas arise in the choroid, 7% in the ciliary body, and the remaining 3% in the iris [6]. Yet, uveal melanoma is a relatively rare condition. A study based on the National Cancer Database of the USA examined cases of cutaneous and non-cutaneous melanomas between 1985 and 1994. Ocular melanomas constituted 5.2% of identified cases, of which uveal melanoma comprised 85% [7]. Similar results have been reported in other countries with predominantly White populations [8,9,10,11].
Numerous epidemiological studies have examined factors that may increase the risk of uveal melanoma. The risk factors that have been identified and extensively studied include age, gender, race and ethnicity, choroidal nevi, and ocular/oculodermal melanocytosis [2]. The average age of initial diagnosis of uveal melanoma is approximately 60 years [7, 12]. Compared with older patients, younger patients are more likely to present with iris melanomas, have associated melanocytosis, and a lower risk of metastatic disease [13]. Pediatric and congenital cases have been reported, but seldom occur [2, 14]. Although it is uncertain whether or not gender-specific differences in uveal melanoma incidence exist, most symptomatic patients are men [2, 15]. With respect to race and gender, uveal melanoma predominantly affects Caucasians. An analysis of the SEER database from 1992 to 2000 reported that the annual age-adjusted incidence per million for uveal melanoma was 0.31 in Blacks, 0.38 in Asian and Pacific Islanders, 1.67 in Hispanics, and 6.02 in non-Hispanic Whites [16]. Intrinsic host factors that predispose Caucasians to uveal melanoma include ancestry from northern latitudes, fair skin color, light eye color, and propensity to sunburn [17, 18].
Up to half of patients with uveal melanoma will develop distant metastasis, with metastatic spread occurring hematogenously [1]. The Collaborative Ocular Melanoma Study (COMS) identified the 5- and 10-year cumulative metastasis rates of 25% and 34%, respectively [19]. The liver is the most common site of metastasis and is involved in 90% of individuals who develop metastatic disease. The median survival of uveal melanoma patients with liver involvement is reported to be 4–5 months, with a 1-year survival of 10–15% [20, 21]. About 50% of these patients with liver metastasis have extrahepatic involvement. The most common extrahepatic metastasis sites are the lungs (30%), bone (23%), and skin (17%) [22].
The most important predictive factors for metastatic disease include basal tumor diameter, the involvement of the ciliary body, extra-scleral extension, epithelioid melanoma cytology, microvascular density, high mitotic rate, chromosome 3 loss, chromosome 1p loss, chromosome 6p gain, and chromosome 8q gain [23,24,25,26].
Pathogenesis
Alterations on the genetic, molecular, and macroscopic level appear to be instrumental in the development and continued evolution of uveal melanoma. There is evidence that alterations in the function and expression of tumor suppressor pathways allow melanocytes in the uveal tract to enter the cell cycle and undergo unregulated proliferation. The earliest disruptions appear to occur in the retinoblastoma (Rb) tumor suppressor pathway. The Rb protein inhibits the progression of the cell cycle at the G1-S phase checkpoint. In uveal melanoma, Rb is inactivated by hyperphosphorylation [3, 27]. In two-thirds of uveal melanomas this occurs directly via cyclin D overexpression, in the other one-third this process is mediated indirectly by methylation and inactivation of the INK4A gene. INK4A encodes a tumor suppressor protein (p16Ink4a) that activates Rb by inhibiting its phosphorylation by cyclin D/CDK4 [3].
In order for a developing neoplasm to persist, it must establish mechanisms to evade host tumor suppression and promote aberrant cell survival. Two such tumor suppression pathways include the p53 pathway and the Bcl-2 pathway. Uveal melanomas overexpress Bcl-2, a molecule that blocks the release of mitochondrial cytochrome C and the activation of pro-apoptotic caspase proteins [27]. The p53 pathway recognizes a wide variety of oncogenic insults and responds by triggering cell senescence or apoptosis [3]. However, in uveal melanomas a p53 inhibitor, HDM2, is overexpressed resulting in uveal melanoma cell survival [3, 27].
The mitogenic activated protein kinase (MAPK) signalling pathway, associated serine/threonine kinases (i.e., RAS/RAF/ERK/MEK), and the phosphatidylinositol-3-kinase (PI3K)/AKT have also been implicated in the development of various cancers, including uveal melanoma [1, 3, 28, 29]. In contrast to cutaneous melanomas, uveal melanoma lacks a mutation in the BRAF, NRAS, or KIT genes that influence MAPK signalling, suggesting that the two forms of melanoma occur via differing pathogenic pathways [1, 29]. Recent studies have demonstrated that mutations of the GNAQ, GNA11, PLCB4, and CYSLTR4 genes that encode members of the heterotrimeric G-protein alpha subunits may result in their constitutive G-protein activation and upregulation of the MAPK pathway in uveal melanoma [1, 30,31,32,33]. There is also evidence that the PI3K/AKT pathway, a commonly altered signalling pathway in human tumors, is also altered in uveal melanoma. The PI3K/AKT pathway is a cell survival mediator, and is negatively regulated by the tumor suppressor PTEN. PTEN is downregulated in uveal melanoma, promoting tumor cell survival [3, 27, 29]. The RAF/ERK/MEK and PI3K/AKT pathways are also activated by the insulin-like growth factors (IGFs) through their interaction with the insulin-like growth factor-1 receptors (IGFR1). IGFR1 is upregulated in many uveal melanomas, resulting in cell survival and unregulated growth [3].
An inactivating somatic mutation of the BRCA-1 associated protein (BAP1) has been implicated in the progression of uveal melanoma, as the mutation is present in 84% of metastasizing tumors. Germline mutations in BAP1 have been observed in 5% of uveal melanomas and have been associated with larger tumor size and ciliary body involvement.
Two recurrent mutations that are associated with a positive prognosis have been identified in patients with primary uveal melanoma. The first is a recurring mutation occurring at codon 625 of the SF3B1 gene, which encodes the splicing factor 3B subunit 1. SF3B1 mutations and BAP1 mutations are nearly mutually exclusive, suggesting that they may represent alternative pathways in tumor progression [34]. The second mutation associated with a positive prognosis is a mutation in eukaryotic translation initiator factor 1A (EIF1AX), which results in an in-frame alteration at the N-terminus of the protein [35].
The molecular changes discussed above are observed in metastatic and non-metastatic uveal melanomas, implying that these changes occur early in the evolution of the primary tumor. Later changes occur nearly mutually exclusively in either non-metastatic tumors (i.e., gain of chromosome 6p) or in metastatic tumors (i.e., monosomy 3) and represent a bifurcation in tumor progression [36,37,38]. Generally, monosomy 3 has been closely associated with metastasis and mortality [39]. The greater metastatic potential of cells bearing the chromosome 3 monosomy may be due to the loss of specific tumor suppressor genes [40, 41]. Karyotype analysis suggests that this chromosome 3 aneuploidy is an early event in metastasis, followed by secondary chromosomal changes, including chromosome 8 gain (40% of uveal melanomas) and the loss of the q arm of chromosome 6 (25% of uveal melanomas) [42,43,44]. Mortality risk is greater when chromosome 3 loss and chromosome 8 gain co-occur, as is usually the case. Conversely, chromosome 6p gain is associated with a more favorable prognosis, apparently delaying or preventing chromosome 3 loss and delaying metastatic death in the presence of concurrent chromosome 3 loss [24,25,26].
These findings derived from primary uveal tumors have allowed us to identify a gene expression profile that is highly accurate for identifying patients at increased risk for metastatic disease [45, 46]. The development of a clinically practical platform for analyzing gene expression profiles would benefit those patients at high risk for metastatic disease by strengthening surveillance efforts, permitting earlier treatment, and facilitating the enrollment of suitable patients into ongoing clinical trials [45]. These genes are analyzed using a robust, but clinically feasible, PCR-based gene assay. Studies have demonstrated that assays may be performed accurately on fine-needle aspirate biopsy, even though the RNA quantity may be below detectable limits. These assays enhance prognostication efforts, while also guiding future patient management [45].
Apart from the molecular changes outlined above, uveal melanomas also experience microscopic and macroscopic changes. Uveal melanomas are capillary-rich tumors that often display a phenomenon called “vascular mimicry” [27, 47]. This process involves intratumoral channels composed of a PAS-positive basement membrane, devoid of endothelial cells. Although the function of these structures is currently unknown, the presence of these intratumoral channels, high vascular density, and the invasion of tumor cells into these blood vessels and sclera are unfavorable prognostic factors [27, 47,48,49]. Metastasis occurs exclusively by hematogenous spread, as there is no lymphatic drainage of the ocular interior. Uveal melanomas often demonstrates a strong hepatic tropism [3]. The uveal melanoma cells can produce a variety of factors that promote angiogenesis, invasion, and metastasis (e.g., metalloproteinases, fibroblast growth factor, adhesion molecules, vascular endothelial growth factor, etc.) [27, 50,51,52].
Clinical Presentation
Intraocular tumors generally present with associated visual symptoms or are detected as an incidental finding on clinical exam.
Most uveal melanomas are of choroidal origin (90%) [6]. Choroidal melanoma appears as a mass deep to the retina, without retinal feeder vessels, and often produces retinal detachment (Fig. 1). Occasionally, vitreous hemorrhage may occur, obscuring visualization of the tumor. In these instances, the tumor is only visible on ocular ultrasonography [5]. Choroidal melanomas may present as pigmented (55%), non-pigmented (15%), or mixed (30%) [13]. Choroidal melanomas appear in one of three configurations: dome (75%), mushroom (20%), which involves the tumor breaking through the Bruch membrane and herniating into the subretinal space, or diffuse (5%), which are flat lesions often mistaken for choroidal nevus. The mean basal dimension of a choroidal melanoma is 11.3 mm and the mean thickness is 5.5 mm [53]. Choroidal melanomas are classified clinically on the basis of thickness: small (0–3.0 mm), medium (3.1–8.0 mm), and large (8.1 mm or greater) [5].

Fundoscopic image of choroidal melanoma
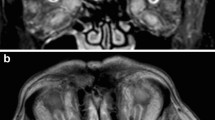
The second most common form of uveal melanoma, ciliary body melanoma (Figs. 2, Fig. 3), is rarely diagnosed as a single entity as it often presents with associated iris or choroidal melanoma due to local extension. Ciliary body melanoma typically presents with prominent episcleral (sentinel) vessels, shallowing of the anterior chamber, unilateral lens changes, unilateral decreased or increased intraocular pressure, a large nodular ciliary body mass, and occasionally, extraocular extension [54]. The ciliary body melanoma can be observed biomicroscopically as a variable pigmented lesion mass with diffuse, nodular, or mixed pattern situated behind the pupil. Macroscopically, choroidal melanomas are classified on the basis of diameter, with larger diameters being associated with a poorer 5-year prognosis: small type (less than 11 mm), medium type (11–15 mm), and large type (greater than 15 mm) [55].

Fundoscopic image of ciliary body melanoma

Gross image of ciliary body melanoma with sentinel vessels
Iris melanoma occurs far less often than uveal melanomas of the posterior segment of the eye (i.e., choroid and ciliary body). Classically, the iris melanoma presents as a gradually expanding pigmented mass (an asymptomatic finding in the large majority), with a consistently demonstrated predilection for the inferior iris (Figs. 4 and 5) [56]. Most iris melanomas have some degree of pigmentation, oftentimes a brown or yellow color. The mean basal dimension of an iris melanoma is 6.0 mm and the mean thickness is 2.0 mm [57]. The majority of melanocytic tumors of the iris are either benign nevi or freckles and tend to remain stable over time. However, tumor growth is not always a sign of malignant degeneration into melanoma [58]. The diagnosis of iris melanoma can only be definitively made by microscopic confirmation. The clinical features that prompt an excisional biopsy include large tumor size, prominent tumor vascularity, tumor seeding, elevated intraocular pressures, and tumor-related ocular complications (e.g., hyphema) [57].

Gross image of iris melanoma

Gross image of iris melanoma
Diagnosis and Prognosis of Uveal Melanoma
Uveal melanoma is diagnosed through funduscopic examination by an experienced clinician, followed by further characterization using a number of ancillary tests [5]. These ancillary tests include ultrasonography, fluorescein angiography, indocyanine green angiography, enhanced depth imaging optical coherence tomography, autofloresence, and fine-needle aspiration biopsy. Cytogenetic analysis of melanoma using DNA or RNA methods can add to the prognostication of uveal melanoma (i.e., mutations in chromosomes 3, 6, and 8; see “Pathogenesis”) [5].
The long-term prognosis of uveal melanoma is poor with death occurring in 50% of cases [59]. Approximately half of patients with uveal melanoma will experience metastasis within 10 years of diagnosis, irrespective of type of treatment received [12, 60,61,62,63,64]. The median survival after metastasis is 6–12 months; however, the median survival of patients receiving treatment is better than those not receiving treatment [59].
The prognosis of uveal melanoma can be estimated by clinical, histopathological, and cytogenetic markers. The most effective measure to minimize poor prognosis is early detection of melanoma when the tumor is small and at lowest risk for metastatic disease [59]. As a result, systemic monitoring is imperative in patients diagnosed with uveal melanoma because of risk of metastasis to the liver, lung, and skin. It is therefore advised that patients undergo physical examination and liver function tests twice yearly and annual chest radiograph and liver imaging (either MRI or ultrasound) [5].
Poor prognosis with uveal melanoma includes several factors, including older patient age, tumor location in the ciliary body, large tumor size, increasing tumor thickness and diameter, presence of subretinal fluid, pigmented melanoma, diffuse (flat) configuration, extraocular extension, epithelioid cell type, increased mitotic activity, infiltrating lymphocytes, tumor vascular networks, chromosomal mutations (e.g., involving chromosomes 3, 6, and 8), and ocular melanocytosis [53, 59, 65, 66].
The 5- and 10-year rates of metastasis, stratified on the basis of type of uveal melanoma (i.e., iris, ciliary body, and choroid), can be found in Table 1. The lower metastasis rate of iris melanoma is related to its lower biologic activity or smaller tumor size [53, 67]. Whereas, the poor prognosis of ciliary body melanoma has been related to larger tumor size, predilection for monosomy 3 and 8q gain, and tumor microvascular patterns [39, 67,68,69].
The American Joint Committee on Cancer Staging Manual (AJCC) 7th edition provides a detailed classification of posterior (ciliary body and choroid) uveal melanoma into defined anatomical and prognostic groups. There are no prognostic stages for iris melanoma in this manual [70].
The AJCC classification for posterior uveal melanoma involves grading tumors on the basis of size via a combination of basal diameter and thickness, labeled as T1, T2, T3, and T4. Tumors in each category can be further subclassified on the basis of the presence or absence of ciliary body involvement and extraocular extension (EOE) [71].
Differential Diagnosis
Lesions that are misdiagnosed as melanomas are referred to pseudomelanomas [72]. Although many of these pseudomelanomas are benign, many of these lesions have the potential to cause a serious threat to vision, undergo malignant transformation, or in some cases may be the harbinger of neoplasm elsewhere. Therefore, it is vital that such lesions be identified appropriately, not only to avoid inappropriate treatment but also to ascertain that these lesions be treated appropriately and in a timely manner [72].
The list of lesions that mimic uveal melanomas is extensive. A study conducted by Shields et al. identified 40 different conditions at final diagnosis in 400 different patients who had been referred with a pseudomelanoma [73]. Some lesions occur more frequently than others, as evidenced by a study that examined the incidence of pseudomelanomas within 1200 patients with a presumed diagnosis of uveal melanoma, during a 25-year period. In 1739 of the patients (14.5%) a final diagnosis different from uveal melanoma was made; the most common pseudomelanomas included choroidal nevus (49%), peripheral exudative hemorrhagic chorioretinopathy (PECHR; 8%), congenital hypertrophy of the retinal pigment epithelium (CHRPE; 6%), idiopathic hemorrhagic detachment of the retina or retinal pigment epithelium (5%), circumscribed choroidal hemangioma (5%), and age-related macular degeneration (4%) [74]. The most common pseudomelanoma identified in the study, choroidal nevus, can have a remarkably similar appearance to choroidal uveal melanoma. Melanoma can be distinguished from a nevus on the basis of risk factors and the fact that melanomas show growth over time, while nevi are stable over time [5].
Pseudomelanomas can be divided into pigmented and non-pigmented lesions (Table 2). Pigmented lesions may be of melanocytic origin or may originate from the iris, ciliary body, or retinal pigmented epithelium. Non-pigmented lesions are more disparate and can be divided into neoplastic, vascular, and inflammatory or reactive [72].
Treatments
Local Tumor Control
Ocular treatment in uveal melanoma is necessary to prevent metastasis of the primary tumor and for the preservation of vision. Treatment strategies depend on the extent of disease, condition of the eye, spread to distant sites of metastasis, and patient’s thoughts and wishes. For asymptomatic patients with smaller tumors (< 2.5 mm height and less than 10 mm in the largest basal dimension), observation for growth before the administration of treatment is recommended [75, 76]. However, there is great contention surrounding this clinical approach. As discussed by Shields et al. [77], of small choroidal melanocytic tumors measuring 3 mm or less in thickness at initial examination, 18% demonstrated growth and 3% metastasized at follow-up. On the basis of their analysis, clinical features of the tumor could be used to predict tumor growth and metastasis, as well as ultimately guide management. Factors predictive of tumor growth include greater tumor thickness (greater relative risk (RR) for initial tumor thickness of 2.1–3.0 mm (RR 5.2) and tumour thickness of 1.1–2.0 mm (RR 4.3) vs. < 1.0 mm in thickness), posterior tumor margin in contact with the optic disc, symptoms of flashes and floaters, blurred vision, orange pigment on tumor surface, and presence of subretinal fluid. Factors predictive of tumor metastasis included posterior tumor margin in contact with optic disc, documented growth, and greater tumor thickness (relative risk for metastasis was greatest for tumor thickness of 1.1–3.0 mm, RR 8.8). The results of this analysis suggest that there may be a valid argument for the active treatment vs. observation of precursor lesions with high-risk clinical features [77].
In those patients with larger tumors and those with symptoms, active treatment is recommended [78]. The major strategies for local tumor control can be subdivided into surgical procedures and radiation therapy (RT).
Surgical options include enucleation (eye removal), orbital exenteration, endoresection, and exoresection [79]. Enucleation is employed in certain circumstances, including in those patients with a clinical diagnosis of uveal melanoma, a tumor that involves more than 40% of the intraocular volume, eyes with neovascular glaucoma, and medium to large uveal melanomas [79, 80]. If the tumor extends into the orbit, orbital exenteration, a more extensive resection that involves the removal of all orbital contents, and adjuvant external beam radiation are commonly employed [79]. In contrast to enucleation and orbital exenteration, endoresection and exoresection are designed to maintain visual functioning of the eye and preserve ocular cosmesis [81]. Endoresection, also called “transretinal resection”, remains controversial because of fears regarding iatrogenic dissemination that may result in local recurrence and metastatic disease [82, 83]. However, long-term follow-up studies have demonstrated that endoresection has local tumor recurrence rates similar to proton beam radiotherapy and plaque brachytherapy (less than 5%) [81, 82]. In initial studies, exoresection (also called eye wall resection or trans-scleral resection) was associated with increased rates of local tumor recurrence when compared to radiotherapy (recurrence rate of 6–57%) [83, 84]; however, with the addition of postoperative ruthenium plaque radiotherapy, local treatment failure rates have fallen considerably (to 5–10%) [81,82,83]. The role of surgical procedures in the treatment of uveal melanoma remains controversial because of fears of tumor cells being disseminated hematologically during surgery and a failure to demonstrate a difference in survival outcomes when compared to RT [82, 85].
RT of uveal melanoma allows for local tumor control, sparing of vision, and conservation of the globe [79]. Radiation is hypothesized to affect the viability of uveal melanoma cells via lethal chromosomal injury, damage to the tumor vasculature, and the induction of reactive oxygen species. RT may be delivered through various modalities, including charged particle therapy (CPT), episcleral radioactive plaque, stereotactic external beam irradiation (SEBI), or transpupillary thermotherapy [86]. CPT involves the use of charged particles (i.e., protons, helium ions, or carbon ions) that are fired with a specific kinetic energy. The particles will enter the tissue and emit the majority of their energy at a fixed depth (i.e., the Bragg peak); little radiation dose is delivered past this point. The depth of the Bragg peak can be altered by changing the initial kinetic energy of the particles. This allows for the delivery of a large dose of radiation to a small volume of tissue [87]. Of all the eye-conserving forms of treatment, CPT is associated with the lowest overall risk of tumor recurrence (local recurrence 3.5% at 5 years, 5% at 10 years) [88, 89]. Apart from CPT, episcleral radioactive plaque, or brachytherapy, is also a commonly used RT. This treatment involves two invasive surgical procedures in which a plaque containing a radioactive material is placed over the region of the tumor (guidelines suggest placing the plaque such that it overlaps the entire tumor margin by at least 2 mm) and later removed after the prescribed dose is delivered [86, 87, 90,91,92]. The radioisotopes that are most commonly used in this procedure are iodine-125, palladium-103, and ruthenium-106 [87]. The COMS reported a 10% recurrence rate at 5 years after iodine-125 plaque brachytherapy [93]. Finally, transpupillary thermotherapy and SEBI are newer treatment modalities that are becoming increasingly accepted as effective methods of achieving local tumor control. Transpupillary thermotherapy involves delivering an infrared beam of light energy into an intraocular neoplasm, resulting in tumor cell necrosis. Despite its advantages (i.e., precision and immediate necrosis of the neoplasm), initial enthusiasm has been dampened by reports of substantial visual loss and local tumor recurrence [94]. This method is a useful primary treatment in a limited group of patients (i.e., small choroidal melanomas near the optic disc or fovea) owing to its precision; however, in most instances this method is best employed as a secondary treatment to plaque radiotherapy [94]. SEBI involves the delivery of a single or several doses of radiation to a well-circumscribed target volume, thus reducing the effects of radiation on the surrounding tissue [95]. This modality of treatment is often utilized in instances where plaque brachytherapy is deemed unsuitable because of large-sized or peripapillary or posteriorly located tumors. The long-term complications associated with SEBI are similar to those observed in CPT and plaque brachytherapy [95]. The choice of RT must optimize dose distribution, while minimizing treatment morbidity, as the radiation dose necessary to control uveal melanoma often exceeds the tolerance of orbital components [79].
Locoregional Treatments of Liver Metastasis
Liver metastasis remains the leading cause of morbidity and mortality in patients with advanced uveal melanoma. Locoregional treatments that aim to control liver metastasis include surgical resection, hepatic intra-arterial chemotherapy (HIA), isolated hepatic perfusion, and percutaneous hepatic perfusion. Most of the evidence for these treatments is based on small, non-comparative, single-institution studies.
There are currently no randomized controlled trials that have compared hepatic resection with best supportive care or chemotherapy. Six of the studies examined included a nonsurgical comparator group. Studies including a nonsurgical comparator group have demonstrated a larger median duration of survival with hepatic resection versus nonsurgical care (i.e., 14–24 vs. 3–12 months, respectively) [96,97,98,99]. Prolonged overall survival times were also demonstrated in non-comparative studies of hepatic resection (range 19–34 months) [100,101,102,103,104,105]. Most studies reported a 5-year overall survival rate in excess of 20% (range 0–42%) [98, 100,101,102,103]. Studies have also consistently demonstrated that negative margin resection, long disease-free survival, low number of lesions, and limited disease distribution are significant positive prognostic indicators [97,98,99,100,101, 104].
Postsurgical recurrence was common, with reported recurrence rates of 72–75% in patients with metastatic melanoma after hepatic resection [102, 103]. Although these findings are suggestive of a survival benefit from surgery, it must be noted that surgical resection is found to be feasible in less than 10% of uveal melanomas metastatic to the liver. Limiting factors include too many metastatic foci, tumors in difficult locations, insufficient hepatic reserve, and tumors invading blood vessels [22].
In HIA, catheters are placed into the hepatic artery either surgically though the gastroduodenal artery or percutaneously through the femoral artery, followed by infusion of chemotherapy (most commonly fotemustine, cisplantin, or melphalan) [106,107,108]. The European Organization for Research and Treatment of Cancer (EORTC) 18021 study, a randomized phase 3 trial, HIA was compared to intravenous delivery of fotemustine in patients with previously untreated, unresectable liver-only metastases from ocular melanoma [109]. Although the study found an improved overall response rate with HIA compared with intravenous fotemustine (11% vs. 2%, respectively), the study failed to demonstrate a difference between the two groups with respect to overall survival. Several non-comparative studies have documented response rates ranging from 16% to 36% and median overall survival durations ranging from 9 to 21 months [107, 108, 110,111,112]. However, it is important to note that several of these studies included patients with extrahepatic disease. HIA is not a treatment free of complications, as the placement of catheters in HIA is associated with a risk of thrombosis, infection, leakage, and displacement [107, 111,112,113].
Hepatic arterial embolization is a technique used to deliver high-dose chemotherapy directly to liver tumor cells, while providing select ischemia. Most studies investigating hepatic arterial embolization utilize the transarterial chemoembolization (TACE) procedure and various chemotherapeutic agents (e.g., fotemustine, cisplantin, and 1,3-bis(2-chloroethyl)-1-nitrosourea) followed by the administration of an embolizing agent [114,115,116,117,118]. In these studies response rates ranged from 0% to 39%, and median overall survival ranged from 5.0 to 8.9 months. Key prognostic factors in TACE treatment include the degree of liver involvement (with more extensive liver involvement being associated with poorer overall median survival durations), baseline lactate dehydrogenase level, and the number of additional visceral sites involved [115,116,117]. In an effort to incite a systemic immune response against tumor cells and improve patient outcomes, Sato et al. performed transaterial embolization with granulocyte macrophage colony-stimulating factor (GM-CSF) in patients with metastatic ocular melanoma limited to the liver [114]. A median overall survival time of 14.4 months was reported. A randomized phase 2 trial comparing immunoembolization with GM-CSF versus bland embolization found that immunoembolization induced more robust inflammatory reactions, which in turn were correlated with delayed progression of extrahepatic systemic metastasis [119].
Isolated hepatic perfusion (IHP) is a surgical procedure that involves isolating the hepatic circulation, and thus allowing the delivery of high doses of chemotherapy directly to the liver [106, 120]. Alkylating agents are favored in this form of therapy, as they are effective over a relatively short exposure time and have steep dose–response curve [121]. The most frequently evaluated agent in patients with hepatic metastatic uveal melanoma is melphalan. Non-comparative studies of IHP treatment of hepatic metastases from uveal melanoma have reported response rates ranging from 33% to 62% and a median overall survival ranging from 10 to 12 months [122,123,124]. Despite these promising results IHP is a complicated procedure with several shortcomings. The procedure is lengthy, non-repeatable, and associated with high morbidity and long hospital stays [122,123,124].
A nonsurgical alternative to IHP has been developed, which is both less complicated and repeatable. The procedure, termed percutaneous hepatic perfusion (PHP), involves inserting a double-balloon catheter in the inferior vena cava to isolate hepatic venous blood. The liver is infused with chemotherapeutic agent, the venous effluent is then filtered extracorporeally before being returned to the systemic circulation via the jugular vein [106]. A recent randomized phase 3 study compared repeated PHP delivery of melphalan (every 4–8 weeks) to best alternative care (BAC) in patients with unresectable hepatic metastases from ocular and cutaneous melanoma. The study found that the median hepatic progression-free survival was significantly prolonged PHP compared to BAC (7.0 vs. 1.6 months, respectively). Although median overall survival did not differ significantly between the PHP and BAC groups (10.6 vs. 10.0 months, respectively), it is likely that this analysis was confounded by patient crossover from the BAC arm to the PHP arm after hepatic progression [125].
Systemic Therapies
Although several novel systemic therapies for patients with metastatic cutaneous melanoma have been shown to be efficacious in randomized trials (i.e., phase 2 and phase 3) and have received US Food and Drug Administration (FDA) approval, the situation for patients with metastatic uveal melanoma is quite different [106]. Several chemotherapeutic and immunomodulatory agents have been examined in patients with uveal melanoma; however, response rates have been low (less than 5%), with median overall survival ranging from 6 to 14 months [126,127,128,129,130,131,132,133,134,135,136,137].
A recent phase 2 clinical trial compared the efficacy of chemotherapy versus selumetinib, a selective, non-ATP competitive inhibitor of MEK1 and MEK2, in patients with advanced uveal melanoma. Selumetinib compared with chemotherapy resulted in a modest improvement in progression free-survival (15.9 vs. 7 weeks, respectively; p < 0.001) and response rate (14% partial response vs. 0% response in accordance with Response Evaluation Criteria in Solid Tumors, respectively). However, no improvement in overall survival was observed [138]. Unfortunately, a phase 3 study of selumetinib in combination with dacarbazine for the treatment of metastatic uveal melanoma did not meet its primary end point of progression-free survival. A full evaluation of the data is ongoing [139].
Another recent study examined the clinical outcomes of patients with stage IV uveal melanoma treated with programmed death receptor 1 (PD-1) and PD-1 ligand (PD-L1) antibodies. The overall response rate was found to be 3.6%. The study concluded that PD-1 and PD-L1 antibodies rarely confer durable remission in patients with metastatic uveal melanoma [140].
Studies examining novel targeted agents are currently underway.
Conclusions and Future Directions
Uveal melanoma is a complex condition that requires a multidisciplinary approach to management and treatment. Early detection and therapy are important factors in disease survival. Therefore, it is imperative that the treating physicians be familiar with the clinical features of uveal melanoma to distinguish it from mimickers and to provide effective and timely treatment.
Current treatment strategies are based on single-center case series examining individual therapies. The rarity of uveal melanoma has made patient enrollment in clinical trials difficult. This is compounded by a lack of uniformity in institutional data collection, which makes comparison across institutions difficult. There is a need for standardization in data collection and collaboration across institutions to better evaluate current and future treatments.
Change history
05 February 2019
The category of this article was incorrectly published as an Original Research Article, when it should have been published as a Review.
References
Spagnolo F, Caltabiano G, Queirolo P. Uveal melanoma. Cancer Treat Rev. 2012;38:549–53.
Yonekawa Y, Kim IK. Epidemiology and management of uveal melanoma. Hematol Oncol Clin N Am. 2012;26:1170–84.
Landerville S, Agapova OA, Harbour JW. Emerging insights into the molecular pathogenesis of uveal melanoma. Future Oncol. 2008;4:629–36.
Nathan P, Cohen V, Coupland S, et al. Uveal melanoma UK national guidelines. Eur J Cancer. 2015. https://doi.org/10.1016/j.ejca.2015.07.013.
Shields CL, Manalac J, Das C, Ferguson K, Shields JA. Choroidal melanoma: clinical features, classification, and top 10 pseudomelanomas. Curr Opin Ophthalmol. 2014;25:177–85.
Damato B. Treatment of primary intraocular melanoma. Expert Rev Anticancer Ther. 2006;6:493–506.
Chang AE, Karnell LH, Menck HR. The national cancer data base report on cutaneous and noncutaneous melanoma: a summary of 84,836 cases from the past decade. Cancer. 1998;83:1664–78.
Virgili G, Gatta G, Ciccolallo L, et al. Incidence of uveal melanoma in Europe. Ophthalmology. 2007;114:2309–15.
Koomen ER, de Vries E, van Kempen LC, et al. Epidemiology of extracutaneous melanoma in the Netherlands. Cancer Epidemiol Biomark Prev. 2010;19:1453–9.
Iscovich J, Ackerman C, Andreev H, Pe’er J, Steinitz R. An epidemiological study of posterior uveal melanoma in Israel, 1961–1989. Int J Cancer. 1995;61:291–5.
Isager P, Østerlind A, Engholm G, et al. Uveal and conjunctival malignant melanoma in Denmark, 1943–97: incidence and validation study. Ophthalmic Epidemiol. 2005;12:223–32.
Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011;118:1881–5.
Shields CL, Kaliki S, Furuta M, Mashayekhi A, Shields JA. Clinical spectrum and prognosis of uveal melanoma based on age at presentation in 8,033 cases. Retina. 2012. https://doi.org/10.1097/iae.0b013e31824d09a8.
Greer CH. Congenital melanoma of the anterior uvea. Arch Ophthalmol. 1966;76:77–8.
Frenkel S, Hendler K, Pe’er J. Uveal melanoma in Israel in the last two decades: characterization, treatment and prognosis. Isr Med Assoc J. 2009;11:280–5.
Hu D-N, Yu G-P, McCormick SA, Schneider S, Finger PT. Population-based incidence of uveal melanoma in various races and enthic groups. Am J Ophthalmol. 2005;140:612–7.
Seddon JM, Gragoudas ES, Glynn RJ, Egan KM, Albert DM, Blitzer PH. Host factors, UV radiation, and risk of uveal melanoma. A case–control study. Arch Ophthalmol. 1990;108:1274–80.
Weis E, Shah CP, Lajous M, Shields JA, Shields CL. The association between host susceptibility factors and uveal melanoma: a meta-analysis. Arch Ophthalmol. 2006;124:54–60.
Diener-West M, Reynolds SM, Agugliaro DJ, et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch Ophthalmol. 2005;123:1639–43.
Gragoudas ES, Egan KM, Seddon JM, et al. Survival of patients with metastases from uveal melanoma. Ophthalmology. 1991;98:383–9 (discussion 390).
Singh AD, Borden EC. Metastatic uveal melanoma. Ophthalmol Clin N Am. 2005;18:143–50.
Bedikian AY. Metastatic uveal melanoma therapy: current options. Int Ophthalmol Clin. 2006;46:151–66.
Singh AD, Shields CL, Shields JA. Prognostic factors in uveal melanoma. Melanoma Res. 2001;11:255–63.
Ewens KG, Kanetsky PA, Richards-Yutz J, et al. Genomic profile of 320 uveal melanoma cases: chromosome 8p-loss and metastatic outcome. Investig Ophthalmol Vis Sci. 2013;54:5721–9.
Damato B, Dopierala JA, Coupland SE. Genotypic profiling of 452 choroidal melanomas with multiplex ligation-dependent probe amplification. Clin Cancer Res. 2010;16:6083–92.
Damato B. Progress in the management of patients with uveal melanoma. The 2012 Ashton Lecture. Eye. 2012;26:1157–72.
Triozzi PL, Eng C, Singh AD. Targeted therapy for uveal melanoma. Cancer Treat Rev. 2008;34:247–58.
Choudhary MM, Triozzi PL, Singh AD. Uveal melanoma: evidence for adjuvant therapy. Int Ophthalmol Clin. 2015;55:45–51.
Zuidervaart W, van Nieuwpoort F, Stark M, et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br J Cancer. 2005;92:2032–8.
Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. English J. 2011;363:2191–9.
Lamba S, Felicioni L, Buttitta F, et al. Mutational profile of GNAQQ209 in human tumors. PLoS One. 2009;4:e6833.
Moore AR, Ceraudo E, Sher JJ, et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet. 2016;48:675–80.
Johansson P, Aoude LG, Wadt K, et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2015;7:4624.
Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45:133–5.
Martin M, Maßhöfer L, Temming P, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45:933–6.
Höglund M, Gisselsson D, Hansen GB, et al. Dissecting karyotypic patterns in malignant melanomas: temporal clustering of losses and gains in melanoma karyotypic evolution. Int J Cancer. 2004;108:57–65.
Tschentscher F, Hüsing J, Hölter T, et al. Tumor classification based on gene expression profiling shows that uveal melanomas with and without monosomy 3 represent two distinct entities. Cancer Res. 2003;63:2578–84.
Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–9.
Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jöckel KH, Becher R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet. 1996;347:1222–5.
Parrella P, Fazio VM, Gallo AP, Sidransky D, Merbs SL. Fine mapping of chromosome 3 in uveal melanoma: identification of a minimal region of deletion on chromosomal arm 3p25.1–p25.2. Cancer Res. 2003;63:8507–10.
Tschentscher F, Prescher G, Horsman DE, et al. Partial deletions of the long and short arm of chromosome 3 point to two tumor suppressor genes in uveal melanoma. Cancer Res. 2001;61:3439–42.
Sisley K, Rennie IG, Parsons MA, et al. Abnormalities of chromosomes 3 and 8 in posterior uveal melanoma correlate with prognosis. Genes Chromosomes Cancer. 1997;19:22–8.
Parrella P, Sidransky D, Merbs SL. Allelotype of posterior uveal melanoma: implications for a bifurcated tumor progression pathway. Cancer Res. 1999;59:3032–7.
Aalto Y, Eriksson L, Seregard S, Larsson O, Knuutila S. Concomitant loss of chromosome 3 and whole arm losses and gains of chromosome 1, 6, or 8 in metastasizing primary uveal melanoma. Investig Ophthalmol Vis Sci. 2001;42:313–7.
Onken MD, Worley LA, Tuscan MD, Harbour JW. An accurate, clinically feasible multi-gene expression assay for predicting metastasis in uveal melanoma. J Mol Diagn. 2010;12:461–8.
Field MG, Decatur CL, Kurtenbach S, et al. PRAME as an independent biomarker for metastasis in uveal melanoma. Clin Cancer Res. 2016;22:1234–42.
Mäkitie T, Summanen P, Tarkkanen A, Kivelä T. Microvascular loops and networks as prognostic indicators in choroidal and ciliary body melanomas. J Natl Cancer Inst. 1999;91:359–67.
Mäkitie T, Summanen P, Tarkkanen A, Kivelä T. Microvascular density in predicting survival of patients with choroidal and ciliary body melanoma. Investig Ophthalmol Vis Sci. 1999;40:2471–80.
Foss AJE, Alexander RA, Jefferies LW, et al. Microvessel count predicts survival in uveal melanoma. Cancer Res. 1996;56:2900–3.
Ten Berge PJM, Danen EHJ, Van Muijen GNP, Jager MJ, Ruiter DJ. Integrin expression in uveal melanoma differs from cutaneous melanoma. Investig Ophthalmol Vis Sci. 1993;34:3635–40.
Natali PG, Hamby CV, Felding-Habermann B, et al. Clinical significance of alpha(v)beta3 integrin and intercellular adhesion molecule-1 expression in cutaneous malignant melanoma lesions. Cancer Res. 1997;57:1554–60.
Väisänen A, Kallioinen M, von Dickhoff K, Laatikainen L, Höyhtyä M, Turpeenniemi-Hujanen T. Matrix metalloproteinase-2 (MMP-2) immunoreactive protein—a new prognostic marker in uveal melanoma? J Pathol. 1999;188:56–62.
Shields CL, Furuta M, Thangappan A, et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch Ophthalmol. 2009;127:989–98.
Demirci H, Shields CL, Shields JA, Honavar SG, Eagle RC. Ring melanoma of the ciliary body: report on twenty-three patients. Retina. 2002;22:698–706.
Costache M, Patrascu OM, Adrian D, et al. Ciliary body melanoma—a particularly rare type of ocular tumor. Case report and general considerations. Maedica (Buchar). 2013;8:360–4.
Kersten R, Tse D, Anderson R. Iris melanoma. Nevus or malignancy. Surv Opthalmol. 1985;29:423–33.
Shields CL, Shields JA, Materin M, Gershenbaum E, Singh AD, Smith A. Iris melanoma: risk factors for metastasis in 169 consecutive patients. Ophthalmology. 2001;108:172–8.
Territo C, Shields CL, Shields JA, Augsburger JJ, Schroeder RP. Natural course of melanocytic tumors of the iris. Ophthalmology. 1988;95:1251–5.
Kaliki S, Shields CL, Shields JA. Uveal melanoma: estimating prognosis. Indian J Ophthalmol. 2015;63:93–102.
Diener-West M, Earle JD, Fine SL, et al. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma, III: initial mortality findings. COMS Report No. 18. Arch Ophthalmol. 2001;119:969–82.
Diener-West M, Hawkins BS, Markowitz JA, Schachat AP. A, review of mortality from choroidal melanoma: II. A meta-analysis of 5-year mortality rates following enucleation, 1966 through 1988. Arch Ophthalmol. 1992;110:245–50.
Kujala E, Mäkitie T, Kivelä T. Very long-term prognosis of patients with malignant uveal melanoma. Investig Ophthalmol Vis Sci. 2003;44:4651–9.
Gamel JW, McLean IW, McCurdy JB. Biologic distinctions between cure and time to death in 2892 patients with intraocular melanoma. Cancer. 1993;71:2299–305.
Virgili G, Gatta G, Ciccolallo L, et al. Survival in patients with uveal melanoma in Europe. Arch Ophthalmol. 2008;126:1413–8.
Shields CL, Kaliki S, Furuta M, Shields JA. Diffuse versus nondiffuse small (≤ 3 mm thickness) choroidal melanoma: comparative analysis in 1,751 cases. The 2012 F. Phinizy Calhoun lecture. Retina. 2013;33:1763–76.
Shields CL, Kaliki S, Livesey M, et al. Association of ocular and oculodermal melanocytosis with the rate of uveal melanoma metastasis: analysis of 7872 consecutive eyes. JAMA Ophthalmol. 2013;131:993–1003.
McLean LCIW, Foster WD, Zimmerman LE. Uveal melanoma. Location, size, cell type, and enucleation as risk factors in metastasis. Hum Pathol. 1982;13:123–32.
Damato B, Coupland SE. A reappraisal of the significance of largest basal diameter of posterior uveal melanoma. Eye (London). 2009;23:2152–60.
Damato B, Duke C, Coupland SE, et al. Cytogenetics of uveal melanoma: a 7-year clinical experience. Ophthalmology. 2007;114:1925–31.
Simpson E, Gallie BL, Saakyan S, et al. International validation of the American Joint Committee on Cancer’s 7th edition classification of uveal melanoma. JAMA Ophthalmol. 2015;133:376–83.
Shields CL, Kaliki S, Furuta M, Fulco E, Alarcon C, Shields JA. American Joint Committee on Cancer classification of posterior uveal melanoma (tumor size category) predicts prognosis in 7731 patients. Ophthalmology. 2013;120:2066–71.
Rennie I. Things that go bump in the light. The differential diagnosis of posterior uveal melanomas. The Duke Elder Lecture 2001. Eye (London). 2002;16:325–46.
Shields J, Augsburger J, Brown G, Rf S. The differential diagnosis of posterior uveal melanoma. Ophthalmology. 1980;87:518–22.
Shields JA, Mashayekhi A, Ra S, Shields CL. Pseudomelanomas of the posterior uveal tract: the 2006 Taylor R. Smith Lecture. Retina J Retina Vitreous Dis. 2005;25:767–71.
Collaborative T, Melanoma O. Factors predictive of growth and treatment of small choroidal melanoma: COMS Report No. 5. The Collaborative Ocular Melanoma Study Group. Arch Ophthalmol. 1997;115:1537–44.
Augsburger JJ. Is observation really appropriate for small choroidal melanomas. Trans Am Ophthalmol Soc. 1993;91:147–75.
Shields CL, Shields JA, Kiratli H, De Potter P, Cater JR. Risk factors for growth and metastasis of small choroidal melanocytic lesions. Trans Am Ophthalmol Soc. 1995;93:259.
McLean MJ, Foster WD, Zimmerman LE. Prognostic factors in small malignant melanomas of choroid and ciliary body. Arch Ophthalmol. 1977;95:48–58.
Nag S, Quivey JM, Earle JD, Followill D, Fontanesi J, Finger PT. The American Brachytherapy Society recommendations for brachytherapy of uveal melanomas. Int J Radiat Oncol Biol Phys. 2003;56:544–55.
Zimmerman LE, McLean I, Foster WD. Statistical analysis of follow-up data concerning uveal melanomas, and the influence of enucleation. Ophthalmology. 1980;87:557–64.
Karkhaneh R, Chams H, Amoli FA, et al. Long-term surgical outcome of posterior choroidal melanoma treated by endoresection. Retina. 2007;27:908–14.
Konstantinidis L, Groenewald C, Coupland SE, Damato B. Long-term outcome of primary endoresection of choroidal melanoma. Br J Ophthalmol. 2014;98:82–5.
Afshar AR, Damato BE. Uveal melanoma: evidence for efficacy of therapy. Int Ophthalmol Clin. 2015;55:23–43.
Damato BE. Local resection of uveal melanoma. Eye (London). 1993;248:388–97.
Hadden PW, Hiscott PS, Damato BE. Histopathology of eyes enucleated after endoresection of choroidal melanoma. Ophthalmology. 2004;111:154–60.
Damato B, Patel I, Campbell IR, Mayles HM, Errington RD. Local tumor control after 106Ru brachytherapy of choroidal melanoma. Int J Radiat Oncol Biol Phys. 2005;63:385–91.
Wang Z, Nabhan M, Schild SE, et al. Charged particle radiation therapy for uveal melanoma: a systematic review and meta-analysis. Int J Radiat Oncol. 2013;86:18–26.
Desjardins L, Lumbroso-Le Rouic L, Levy-Gabriel C, et al. Combined proton beam radiotherapy and transpupillary thermotherapy for large uveal melanomas: a randomized study of 151 patients. Ophthalmic Res. 2006;38:255–60.
Damato B, Kacperek A, Chopra M, Campbell IR, Errington RD. Proton beam radiotherapy of choroidal melanoma: the Liverpool–Clatterbridge experience. Int J Radiat Oncol Biol Phys. 2005;62:1405–11.
Tabandeh H, Chaudhry NA, Murray TG, et al. Intraoperative echographic localization of iodine-125 episcleral plaque for brachytherapy of choroidal melanoma. Am J Ophthalmol. 2000;129:199–204.
Russo A, Laguardia M, Damato B. Eccentric ruthenium plaque radiotherapy of posterior choroidal melanoma. Graefe’s Arch Clin Exp Ophthalmol. 2012;250:1533–40.
Damato B, Patel I, Campbell IR, Mayles HM, Errington RD. Visual acuity after ruthenium106 brachytherapy of choroidal melanomas. Int J Radiat Oncol Biol Phys. 2005;63:392–400.
Jampol LM, Moy CS, Murray TG, et al. The COMS randomized trial of iodine 125 brachytherapy for choroidal melanoma: IV. Local treatment failure and enucleation in the first 5 years after brachytherapy. COMS report no. 19. Ophthalmology. 2002;5:2197.
Shields CL, Shields JA, Perez N, Singh AD, Cater J. Primary transpupillary thermotherapy for small choroidal melanoma in 256 consecutive cases: outcomes and limitations. Ophthalmology. 2002;109:225–34.
Zehetmayer M, Kitz K, Menapace R, et al. Local tumor control and morbidity after one to three fractions of stereotactic external beam irradiation for uveal melanoma. Radiother Oncol. 2000;55:135–44.
Marshall E, Romaniuk C, Ghaneh P, et al. MRI in the detection of hepatic metastases from high-risk uveal melanoma: a prospective study in 188 patients. Br J Ophthalmol. 2012. https://doi.org/10.1136/bjophthalmol-2012-302323.
Rivoire M, Kodjikian L, Baldo S, Kaemmerlen P, Négrier S, Grange JD. Treatment of liver metastases from uveal melanoma. Ann Surg Oncol. 2005;12:422–8.
Mariani P, Piperno-Neumann S, Servois V, et al. Surgical management of liver metastases from uveal melanoma: 16 years’ experience at the Institut Curie. Eur J Surg Oncol. 2009;35:1192–7.
Frenkel S, Nir I, Hendler K, et al. Long-term survival of uveal melanoma patients after surgery for liver metastases. Br J Ophthalmol. 2009;93:1042–6.
Adam R, Chiche L, Aloia T, et al. Hepatic resection for noncolorectal nonendocrine liver metastases: analysis of 1,452 patients and development of a prognostic model. Ann Surg. 2006;244:524–35.
de Ridder J, van Walsum M, Verhoef C, Nagtegaal I, de Wilt J. Hepatic resection for metastatic melanoma in the Netherlands: survival and prognostic factors. Melanoma Res. 2013;23:27–32.
Groeschl RT, Nachmany I, Steel JL, et al. Hepatectomy for noncolorectal non-neuroendocrine metastatic cancer: a multi-institutional analysis. J Am Coll Surg. 2012;214:769–77.
Pawlik TM, Zorzi D, Abdalla EK, et al. Hepatic resection for metastatic melanoma: distinct patterns of recurrence and prognosis for ocular versus cutaneous disease. Ann Surg Oncol. 2006;13:712–20.
Ryu SW, Saw R, Scolyer RA, Crawford M, Thompson JF, Sandroussi C. Liver resection for metastatic melanoma: Equivalent survival for cutaneous and ocular primaries. J Surg Oncol. 2013;108:129–35.
Ripley RT, Davis JL, Klapper JA, et al. Liver resection for metastatic melanoma with postoperative tumor-infiltrating lymphocyte therapy. Ann Surg Oncol. 2010;17:163–70.
Agarwala SS, Eggermont AMM, O’Day S, Zager JS. Metastatic melanoma to the liver. Cancer. 2014;120:781–9.
Peters S, Voelter V, Zografos L, et al. Intra-arterial hepatic fotemustine for the treatment of liver metastases from uveal melanoma: experience in 101 patients. Ann Oncol. 2006;17:578–83.
Siegel R, Hauschild A, Kettelhack C, Kähler KC, Bembenek A, Schlag PM. Hepatic arterial fotemustine chemotherapy in patients with liver metastases from cutaneous melanoma is as effective as in ocular melanoma. Eur J Surg Oncol. 2007;33:627–32.
Leyvraz S, Piperno-Neumann S, Suciu S, et al. Hepatic intra-arterial versus intravenous fotemustine in patients with liver metastases from uveal melanoma (EORTC 18021): a multicentric randomized trial. Ann Oncol. 2014;25:742–6.
Farolfi A, Ridolfi L, Guidoboni M, et al. Liver metastases from melanoma: hepatic intra-arterial chemotherapy. A retrospective study. J Chemother. 2011;23:300–5.
Agarwala SS, Panikkar R, Kirkwood JM. Phase I/II randomized trial of intrahepatic arterial infusion chemotherapy with cisplatin and chemoembolization with cisplatin and polyvinyl sponge in patients with ocular melanoma metastatic to the liver. Melanoma Res. 2004;14:217–22.
Heusner TA, Antoch G, Wittkowski-Sterczewski A, et al. Transarterial hepatic chemoperfusion of uveal melanoma metastases: furvival and response to treatment. RoFo. 2011;183:1151–60.
Becker JC, Terheyden P, Kämpgen E, et al. Treatment of disseminated ocular melanoma with sequential fotemustine, interferon alpha, and interleukin 2. Br J Cancer. 2002;87:840–5.
Sato T, Eschelman DJ, Gonsalves CF, et al. Immunoembolization of malignant liver tumors, including uveal melanoma, using granulocyte-macrophage colony-stimulating factor. J Clin Oncol. 2008;26:5436–42.
Gupta S, Bedikian AY, Ahrar J, et al. Hepatic artery chemoembolization in patients with ocular melanoma metastatic to the liver: response, survival, and prognostic factors. Am J Clin Oncol. 2010;33:474–80.
Patel K, Sullivan K, Berd D, et al. Chemoembolization of the hepatic artery with BCNU for metastatic uveal melanoma: results of a phase II study. Melanoma Res. 2005;15:297–304.
Schuster R, Lindner M, Wacker F, et al. Transarterial chemoembolization of liver metastases from uveal melanoma after failure of systemic therapy: toxicity and outcome. Melanoma Res. 2010;20:191–6.
Sharma KV, Gould JE, Harbour JW, et al. Hepatic arterial chemoembolization for management of metastatic melanoma. AJR Am J Roentgenol. 2008;190:99–104.
Valsecchi ME, Terai M, Eschelman DJ, et al. Double-blinded, randomized phase II study using embolization with or without granulocyte-macrophage colony-stimulating factor in uveal melanoma with hepatic metastases. J Vasc Interv Radiol. 2015;26:523.e2–532.e2.
Alexander HR Jr, Butler CC. Development of isolated hepatic perfusion via the operative and percutaneous techniques for patients with isolated and unresectable liver metastases. Cancer J. 2010;16:132–41.
Vahrmeijer AL, Van De Velde CJH, Hartgrink HH, Tollenaar RAEM. Treatment of melanoma metastases confined to the liver and future perspectives. Dig Surg. 2009;25:467–72.
Alexander HR, Libutti SK, Bartlett DL, Puhlmann M, Fraker DL, Bachenheimer LC. A phase I-II study of isolated hepatic perfusion using melphalan with or without tumor necrosis factor for patients with ocular melanoma metastatic to liver. Clin Cancer Res. 2000;6:3062–70.
Alexander HR Jr, Libutti SK, Pingpank JF, et al. Hyperthermic isolated hepatic perfusion using melphalan for patients with ocular melanoma metastatic to liver. Clin Cancer Res. 2003;9:6343–9.
van Iersel LB, Hoekman EJ, Gelderblom H, et al. Isolated hepatic perfusion with 200 mg melphalan for advanced noncolorectal liver metastases. Ann Surg Oncol. 2008;15:1891–8.
Hughes MS, Zager J, Faries M, et al. Results of a randomized controlled multicenter phase III trial of percutaneous hepatic perfusion compared with best available care for patients with melanoma liver metastases. Ann Surg Oncol. 2015. https://doi.org/10.1245/s10434-015-4968-3.
Bedikian AY, Papadopoulos N, Plager C, Eton O, Ring S. Phase II evaluation of temozolomide in metastatic choroidal melanoma. Melanoma Res. 2003;13:303–6.
Homsi J, Bedikian AY, Papadopoulos NE, et al. Phase 2 open-label study of weekly docosahexaenoic acid-paclitaxel in patients with metastatic uveal melanoma. Melanoma Res. 2010;20:507–10.
Kivelä T, Suciu S, Hansson J, et al. Bleomycin, vincristine, lomustine and dacarbazine (BOLD) in combination with recombinant interferon alpha-2b for metastatic uveal melanoma. Eur J Cancer. 2003;39:1115–20.
O’Neill PA, Butt M, Eswar CV, Gillis P, Marshall E. A prospective single arm phase II study of dacarbazine and treosulfan as first-line therapy in metastatic uveal melanoma. Melanoma Res. 2006;16:245–8.
Pföhler C, Cree IA, Ugurel S, et al. Treosulfan and gemcitabine in metastatic uveal melanoma patients: results of a multicenter feasibility study. Anticancer Drugs. 2003;14:337–40.
Pons F, Plana M, Caminal JM, et al. Metastatic uveal melanoma: is there a role for conventional chemotherapy? A single center study based on 58 patients. Melanoma Res. 2011;21:217–22.
Pyrhönen S, Hahka-Kemppinen M, Muhonen T, et al. Chemoimmunotherapy with bleomycin, vincristine, lomustine, dacarbazine (BOLD), and human leukocyte interferon for metastatic uveal melanoma. Cancer. 2002;95:2366–72.
Schmittel A, Scheulen ME, Bechrakis NE, et al. Phase II trial of cisplatin, gemcitabine and treosulfan in patients with metastatic uveal melanoma. Melanoma Res. 2005;15:205–7.
Schmittel A, Schuster R, Bechrakis NE, et al. A two-cohort phase II clinical trial of gemcitabine plus treosulfan in patients with metastatic uveal melanoma. Melanoma Res. 2005;15:447–51.
Danielli R, Ridolfi R, Chiarion-Sileni V, et al. Ipilimumab in pretreated patients with metastatic uveal melanoma: safety and clinical efficacy. Cancer Immunol Immunother. 2012;61:41–8.
Hofmann UB, Kauczok-Vetter CS, Houben R, Becker JC. Overexpression of the KIT/SCF in uveal melanoma does not translate into clinical efficacy of imatinib mesylate. Clin Cancer Res. 2009;15:324–9.
Mahipal A, Tijani L, Chan K, Laudadio M, Mastrangelo MJ, Sato T. A pilot study of sunitinib malate in patients with metastatic uveal melanoma. Melanoma Res. 2012;22:440–6.
Carvajal RD, Sosman JA, Quevedo JF, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma. JAMA. 2014;311:2397.
AstraZeneca. AstraZeneca provides update on selumetinib in uveal melanoma. (2015). https://www.astrazeneca.com/media-centre/press-releases/2015/astrazeneca-selumetinib-uveal-melanoma-oncology-22072015.html. Accessed 23 Nov 2017.
Algazi AP, Tsai KK, Shoushtari AN, et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer. 2016;122:3344–53.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Manni Singh, Priya Durairaj, and Jensen Yeung have nothing to declare.
Compliance with Ethical Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/CCFCF06073D508CD.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Singh, M., Durairaj, P. & Yeung, J. Uveal Melanoma: A Review of the Literature. Oncol Ther 6, 87–104 (2018). https://doi.org/10.1007/s40487-018-0056-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40487-018-0056-8