
Abstract
Neonates, infants, and children differ from adults in many aspects, not just in age, weight, and body composition. Growth, maturation and environmental factors affect drug kinetics, response and dosing in pediatric patients. Almost 80 % of drugs have not been studied in children, and dosing of these drugs is derived from adult doses by adjusting for body weight/size. As developmental and maturational changes are complex processes, such simplified methods may result in subtherapeutic effects or adverse events. Kidney function is impaired during the first 2 years of life as a result of normal growth and development. Reduced kidney function during childhood has an impact not only on renal clearance but also on absorption, distribution, metabolism and nonrenal clearance of drugs. ‘Omics’-based technologies, such as proteomics and metabolomics, can be leveraged to uncover novel markers for kidney function during normal development, acute kidney injury, and chronic diseases. Pharmacometric modeling and simulation can be applied to simplify the design of pediatric investigations, characterize the effects of kidney function on drug exposure and response, and fine-tune dosing in pediatric patients, especially in those with impaired kidney function. One case study of amikacin dosing in neonates with reduced kidney function is presented. Collaborative efforts between clinicians and scientists in academia, industry, and regulatory agencies are required to evaluate new renal biomarkers, collect and share prospective pharmacokinetic, genetic and clinical data, build integrated pharmacometric models for key drugs, optimize and standardize dosing strategies, develop bedside decision tools, and enhance labels of drugs utilized in neonates, infants, and children.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Changes in kidney function during childhood modify not only renal clearance but also absorption, distribution, metabolism and nonrenal clearance of drugs, affecting pharmacokinetics, response, and dosing of drugs. |
New renal biomarkers are needed. ‘Omics’-based technologies, such as proteomics and metabolomics, can be leveraged to uncover novel markers for kidney function during normal development, acute kidney injury, and chronic diseases. |
Pharmacometric modeling and simulation can be applied to simplify design of pediatric investigations, characterize effects of kidney function on drug exposure and response, and fine-tune dosing in pediatric patients, especially in those with impaired kidney function. |
Collaborative efforts are required to evaluate new renal biomarkers, optimize and standardize dosing strategies, develop bedside decision tools, and enhance labels of drugs utilized in neonates, infants, and children. |
1 Introduction
Administering drugs in neonates, infants, and children is challenging. Up to 80 % of drugs prescribed in pediatric patients have not been formally tested in this population, and are therefore not labeled for use in neonates, infants, and children (‘off-label use’) [1, 2]. Clinical research involving children is more difficult than in adults for several reasons [3–6]: (1) a small number of pediatric patients are eligible for clinical trials; (2) the ethical hurdles of conducting studies with placebo/control arms in sick children; (3) cumbersome procedures for obtaining informed consent and, if appropriate, informed assent; (4) lack of suitable infrastructure to conduct pediatric clinical studies; (5) limited market size, and thus economically less attractive for pharmaceutical companies; and (6) technical challenges of collecting laboratory, imaging, or clinical data in pediatric patients.
A majority of drug dosing regimens for use in neonates, infants, and children are derived from adult data by adjusting for body weight/size. Such a simple extrapolation from adults to pediatric individuals may not be appropriate. Why? Children are not ‘small adults’. They differ from adults in many aspects, not just in age and body weight: (1) body composition changes in neonates, infants, and children [7–9]; (2) organ maturation and development can affect pharmacokinetics and response of drugs, especially in children younger than 2 years of age [10, 11]; and (3) therapeutic window (TW), also called therapeutic index (i.e. exposure range with optimal efficacy/safety balance) may change over time in children. As a result of organ maturation and development, kidney function changes during the first 2 years of life, which in turn will affect both drug exposure and response in neonates and infants [12–14].
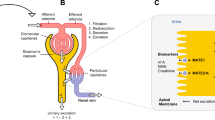
Various factors can affect kidney function in neonates, infants, and children: (1) development and maturation of kidneys as described earlier [10]; (2) underlying kidney diseases and comorbidities [15]; (3) medications and other therapeutic interventions, such as hypothermia in neonates [16–21]; and (4) environmental and genetic factors [22] (Fig. 1).

Factors affecting kidney function in neonates, infants, and children
The focus of this article is to (1) review physiological differences between neonates, infants, children, and adults; (2) review markers for assessing and monitoring kidney function; (3) understand factors that affect kidney function and its impact on drug exposure and response in pediatric patients; (4) introduce quantitative approaches, such as pharmacometric modeling and simulation, to simplify designs of studies in pediatric patients, characterize effects of the kidney on drug exposure and response, and fine-tune dose strategies in pediatric patients, especially in those with impaired kidney function; and (5) outline opportunities to facilitate development and optimize utilization of therapeutics in neonates, infants, and children. The majority of our examples and related discussion will focus on neonates and young infants, who are particularly subject to pharmacokinetic changes due to rapid growth, development, and maturation of organs, including the kidneys. Most studies using quantitative approaches such as pharmacometric modeling and simulation are conducted in this age range.
2 Method
Relevant articles in the PubMed and EMBASE databases were identified using the following keywords: ‘neonates’, ‘infant’, ‘children’, ‘pediatric’, ‘drug development’, ‘pharmacokinetics’, ‘kidney function’, ‘modeling’, ‘simulation’, ‘drug dosing’, and ‘pharmacometrics’. Our search was limited to English-language studies published in peer-reviewed journals. Additional publications were identified from review articles.
3 Physiological Differences Between Adults and Children
Neonates, infants, and children differ from adults in many aspects, not just in age, body weight and composition: capacity of drug-metabolizing enzymes and kidney function change due to organ maturation and development, resulting in altered drug exposure and response, especially in children younger than 2 years of age. Stages of growth are illustrated in Fig. 2 [23].

Stages of growth. Modified from the National Heart, Lung, and Blood Institute [23]
3.1 How Does Body Size and Composition Change in Neonates, Infants, and Children?
Neonates, infants, and children represent a heterogeneous population with nonlinear growth in size and bodyweight from less than 500 g to more than 100 kg. Most changes in body composition take place during the first 2–3 years of life. Body weight doubles within 5 months, and triples within 1 year, while body length increases by 50 % during the first year, and doubles within 4 years. Proportions of body weight contributed by fat, protein, intracellular and extracellular water significantly change during childhood. Total body water (TBW) (i.e. the sum of intracellular and extracellular water) constitutes 80 % of body weight in preterm neonates and 75 % in term neonates. It decreases to adult values at 4 months and remains relatively constant from this age onwards [24] (Fig. 3).

Body composition and growth. Adapted from Bechard et al. [24]
3.2 How Does Development and Maturation of Organs Affect Drugs in Neonates, Infants, and Children?
Changes due to development and maturation of organs can affect various aspects of drug pharmacokinetics, especially in neonates and infants. Potential key changes in absorption, distribution, metabolism, and elimination of drugs during the first years of life are presented in Table 1 [10, 25–27].
Absorption is decreased in neonates and infants, and increases progressively during childhood, mainly due to altered gastric pH, gastric emptying, and intestinal transit time [10, 28]. Colonization of the gastrointestinal tract by bacteria varies with age, route of delivery (vaginal vs. cesarean section), type of feeding, and concurrent drug therapy. This process influences metabolism of bile salts and drugs by intestinal cytochrome P450 (CYP) as well as intestinal motility and absorption [29]. An adult pattern of microbial products is established around 5–12 months of age [30, 31].
Distribution is modified essentially due to changes in body composition and protein-binding capacity. The amount of TBW is higher in neonates and infants [7]. Although the percentage of TBW does not change considerably after 1 year of age, there is continuous decrease in extracellular water from infancy to young adulthood. It should be noted that drug-binding capacity to plasma proteins is decreased in neonates and infants, resulting in an increased volume of distribution of water-soluble drugs [32]. Other factors such as altered regional blood flow and immaturity of the blood-brain barrier can also affect distribution of drugs in neonates, infants, and children.
Metabolism of many drugs is dependent on hepatic blood flow and activity of drug-metabolizing enzymes and transporters. Hepatic blood flow is reduced in neonates, and increases with increasing cardiac output over time. Due to ontogeny, the capacity of drug-metabolizing enzymes and transporters changes in neonates and infants. Activity of phase I enzymes is reduced in neonates [11], increases progressively during the first 6 months of life, may exceed adult rates for a few years, slows during adolescence, and approaches adult rates by late puberty [33]. Similar age-dependent changes are observed for the phase II enzymes, uridine diphosphate glucuronosyltransferase (UGT), sulfotransferase, glutathione-S-transferase (GST), and N-acetyltransferase (NAT) [10]. It is important to realize that different isoenzymes within a family of enzymes can mature at different rates during the first years of life.
Elimination of a majority of drugs from the body occurs primarily via the kidney. Nephrogenesis starts at weeks 5–6 of gestation and is completed around weeks 34–35 of gestation. This process of nephrogenesis is followed by postnatal changes in intrarenal blood flow, but kidney function is still impaired compared with that of adults. This is due to a combination of factors: (1) immature glomerular filtration and tubular function; (2) reduced kidney perfusion pressure; and (3) inadequate osmotic load to produce full counter-current effects. Glomerular filtration rate (GFR) increases rapidly in neonates because of a postnatal drop in kidney vascular resistance and an increase in renal blood flow. GFR continues to increase gradually, approaching adult levels by 12 months of age (Fig. 4), then exceeding adult rates during preschool years to finally reach adult values at prepubertal age [10, 34–39]. A transient increase has been explained by some authors to be based on a larger increase of kidney weight compared with body weight in preschool-age children. This might be explained by an augmented increase of glomerular and tubular cell size and an increased number of capillaries [40, 41].

Typical maturation of GFR as a function of PNA, expressed as a percentage of adult GFR. Adapted from Goyal [41]. GFR glomerular filtration rate, PNA postnatal age
Other factors influencing developmental changes in kidney excretion are prematurity, kidney/urologic fetal malformations, and concomitant medications. Preterm infants are particularly susceptible because of ongoing nephrogenesis [42]. A twofold increase of vancomycin clearance, a drug almost exclusively excreted by the kidney, from week 24 of postmenstrual age (PMA) to week 34 is described [43]. Neonates born small for gestational age (SGA) were found to have a 16 % reduction in drug clearance compared with preterm neonates who are appropriate for gestational age (AGA) [44]. Drugs such as betamethasone or indomethacin have been shown to alter the normal pattern of postnatal kidney maturation in preterm neonates [45].
3.3 What Do We Know About the Therapeutic Window of Drugs in Pediatric Patients?
Developmental changes can modify pharmacokinetic profiles of drugs (e.g. increase high peak-to-trough ratios or variability in exposure), which may impact the efficacy/safety balance [46]. Developmental changes may also directly impact drug response without modifying pharmacokinetic profiles in children [11, 47–50]. Changing expression of receptors during the first years of life can affect efficacy/safety response of drugs in children [51]. A study of sotalol in the treatment of supraventricular tachycardia showed that neonates exhibited a higher sensitivity towards QTc interval prolongation compared with older children (Fig. 5) [52]. Augmented response to warfarin and cyclosporine in children [48, 49], increased sensitivity to d-tubocurarine, an antagonist of nicotinic neuromuscular acetylcholine receptors, in neonates and infants compared with children, adolescents, and adults [53], and different sensitivity to bronchodilators because of a lack of smooth muscles in the airways in neonates [54] are other examples that illustrate that developmental changes can impact the TW of drugs in neonates, infants, and children.

Narrower therapeutic window can alter efficacy/safety balance of drugs in children/neonates (b) compared with that in adults (a). Example of sotalol [52]
3.4 What Do We Know About Pharmacogenetics in Pediatric Patients?
As in the adult population, polymorphism in drug-metabolizing enzymes, drug-transport systems, and drug targets can be associated with clinically relevant differences in drug disposition and/or efficacy/safety profile. Polymorphisms, also known as single nucleotide polymorphisms (SNPs), are defined as genetic variations occurring in at least 1 % of the population. An increasing number of studies are being published that describe differences in drug response as a result of individual genetic background, but most of these reports include only adult individuals [55, 56]. A few studies have shown the impact of pharmacogenomics in pediatric patients and highlighted differences between children and adults [55, 57].
In children with kidney or heart transplants, expression of CYP3A5 affects clearance, dose requirement, and immunosuppressive effects of tacrolimus. Children expressing CYP3A5 (those carrying the A nucleotide, defined as the *1 allele) have a higher dose requirement than non-expressers (those homozygous for the G nucleotide, defined as the *3 allele) [58, 59]. In children with asthma and the CYP3A4*22 allele, fluticasone treatment is associated with better asthma control than in those with the wild-type allele [60]. Pharmacogenetic effects on drug exposure and response can also impact efficacy/safety profiles of drugs in pediatric patients. Cisplatin ototoxicity has been associated with variants in the GST gene family [61, 62]. It is estimated that 10 % of the population have heterozygous mutations in the thiopurine S-methyltransferase (TPMT) gene, leading to decreased levels of the enzyme, while as many as 1 in 300 have homozygous mutations with very low levels of function of the enzyme [63]. SNPs in the TPMT gene are associated with an increased risk of developing severe and life-threatening TPMT-mediated myelotoxicity or hepatotoxicity in children treated with conventional dose of thiopurines [63, 64]. SNPs in the TPMT gene are also associated with a risk of developing severe, potentially life-threatening bone marrow toxicity when treated with conventional doses of azathioprine or mercaptopurine [65]. Studies have found a strong association between HLA-B*1502 and carbamazepine-induced severe cutaneous adverse reactions [66–68]. In such cases, drug labels should include treatment strategies based on pharmacogenetic factors; see example labels for 6-mercaptopurine [69], azathioprine (TPMT variants) [70], and carbamazepine (HLA-B*1502 allele) [71, 72]. Atomoxetine and pimozide are drugs with specific genotype-based dose recommendations for children [56, 73]. Other examples of dose recommendations can be found on the PharmGKB website (http://www.pharmgkb.org).
Pediatric patients present unique pharmacogenetic challenges as neonates, infants, and children have the additional complexity of ontological phenotypes that impact their responses to drugs. Thus, the role and involvement of pharmacogenetics may differ between adult and pediatric patients, and dosing strategies developed in adults may be inaccurate in neonates, infants, and/or children. An example is the role of CYP2C9 and VKORC1 genetic variations in warfarin treatment [74]. In adult patients, such genetic variations are key contributors to intersubject variability in warfarin exposure, whereas in pediatric patients, age and body size have a more pronounced impact on intersubject variability in warfarin exposure than genetic variations [57, 74, 75].
4 Kidney Function and its Impact on Drugs in Pediatric Patients
This section reviews (1) factors that can alter kidney function; (2) effects of impaired kidney function on drug exposure (pharmacokinetics) and response; and (3) measures for assessing and monitoring kidney function and markers for detecting kidney injury/disease in pediatric patients.
4.1 Which Factors Affect Kidney Function in Pediatric Patients?
In neonates, infants, and children, multiple factors affect kidney function: (1) development and maturation of the kidneys as described earlier [10]; (2) acute kidney injury (AKI), underlying kidney diseases and comorbidities [15]; (3) medications, renal replacement therapy (RRT) and other therapeutic interventions, such as hypothermia in neonates [16–19, 76, 77]; and (4) environmental and genetic factors [22].
4.1.1 Chronic Kidney Disease (CKD)
Although CKD is seen less frequently in pediatric patients than in adult patients, it is not a rare disease as the overall prevalence is 75 cases per million children [78]. In adults, diabetic nephropathy and hypertension are the main causes of CKD, whereas in pediatric patients, congenital disease and glomerular disorders are frequent causes of CKD [79–81].
4.1.2 AKI
In ‘developed countries’ the most prevalent causes of an AKI associated with abrupt decrease in kidney function include sepsis, congenital heart disease, and renal ischemia [79, 82, 83]. Prospective studies report AKI incidence rates of 4.5 and 2.5 % in children admitted to intensive care units [82, 83]. In ‘less developed countries’, acute tubular necrosis secondary to gastroenteritis and primary kidney diseases such as hemolytic uremic syndrome and acute glomerulonephritis are more likely involved. Neonates, especially preterm newborns, are susceptible to acquiring a kidney disease due to immature function of their kidneys, rapid hemodynamic changes at birth, and increased risk of hypovolemia as a result of insensible water losses and exposure to nephrotoxic drugs [84].
4.1.3 Medications/Treatments
Nonsteroidal anti-inflammatory drugs (NSAIDs) [85–88], aminoglycosides (gentamicin, amikacin, tobramycin, netilmicin) [89–97] and glycopeptide antibiotics (vancomycin, teicoplanin) [98–101], amphotericin B [102, 103], antiviral agents [104, 105], angiotensin-converting enzyme (ACE) inhibitors [106, 107], calcineurin inhibitors [108], radiocontrast media [109], and cytostatic drugs [110–114] can be nephrotoxic and cause AKI in neonates, infants, and children [17, 115]. Direct pathophysiological mechanisms of nephrotoxicity include constriction of intrarenal vessels, acute tubular necrosis, acute interstitial nephritis, and, more infrequently, tubular obstruction [17]. Drugs without nephrotoxic effect may increase exposure to potentially nephrotoxic agents by exhibiting drug–drug interactions and altering drug-metabolizing enzymes or transporters.
RRT is a particular case of ‘acquired’ changes in the clearance of drugs. The use of RRT associated with AKI and CKD was 15 per million children in the US in 2008 [116]. Drugs can be removed during RRT by diffusion (hemodialysis) or convection (hemofiltration). Molecular weight and size, protein binding, volume of distribution and electrostatic charge are key characteristics affecting drug dialyzability [117–119]. Drugs with high protein binding (>80 %), lipophilic drugs, cationic drugs (retained by anionic protein charges in blood), and drugs with high molecular weight are poorly dialyzable [117, 118, 120–124].
4.1.4 Genetic Factors
The influence of variants in genes encoding for receptors, for example angiotensin II receptor 1 or toll-like receptor 9 (TLR-9), and peptides, for example vasopressin, involved in the pathogenesis of kidney disease have been discussed [22, 125–129]. Polymorphisms of genes encoding for proteins involved in drug elimination could predispose to drug nephrotoxicity. An association between ABCB1 polymorphisms, encoding for the P-glycoprotein (P-gp), and tacrolimus-associated nephrotoxicity in pediatric patients following liver transplant is reported, suggesting that genotyping to find such polymorphisms may have the potential to individualize tacrolimus therapy and enhance drug safety [130]. SNPs in the genes encoding for CYP3A enzymes and the nicotinamide adenine dinucleotide phosphate–CYP oxidoreductase (POR), a protein that functions as an electron donor for CYP monooxygenase enzymes [131], have been shown to influence tacrolimus dose requirements. Individuals carrying the CYP3A4*22 T-variant allele have a lower tacrolimus dose requirement than individuals with the CYP3A4*22 CC genotype, and this effect appears to be independent of CYP3A5 genotype status [132–134]. Individuals carrying the POR*28 T-variant allele have a higher tacrolimus dose requirement than POR*28 CC homozygotes in CYP3A5-expressing individuals [135–137]. Their influence on the risk of nephrotoxicity is still inconsistent but they may also relate to tacrolimus-induced chronic nephrotoxicity [138]. Furthermore, the role of polymorphism in the organic cation transporter 2 (OCT 2) gene to cisplatin-induced nephrotoxicity has been reported [139].
4.2 How Does Impaired Kidney Function Impact Drug Pharmacokinetics and Response in Pediatric Patients?
Impaired kidney function affects not only renal clearance but also absorption, distribution, metabolism, and nonrenal clearance of drugs (Table 2) [122, 140–147].
Modification of distribution due to edema and decreased protein-binding capacity may be especially significant in children treated with highly hydrophilic drugs (such as aminoglycosides). An already large TBW compartment inherent of being a young child combined with a higher TBW due to kidney disease could result in a severe increased volume of distribution. The problem is often underestimated and may therefore go undetected. Even if a decrease in protein capacity induces an increase of the free fraction, the total drug concentration stays within the acceptable therapeutic range.
4.3 How to Assess Kidney Function and Detect Kidney Injury/Disease in Pediatric Patients?
Exact determination of kidney function is problematic in children. Measurement of GFR markers, such as inulin, iohexol, 51Cr-EDTA or 99mTc-diethylenetriaminepentaacetic acid is difficult in pediatric patients due to ethical and practical reasons [158–161].
Equations based on serum creatinine measurements have been developed to estimate GFR. In adults, the most widely used equations for estimating GFR are the Cockroft–Gault formula, the Modification of Diet in Renal Disease (MDRD), and the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI). These equations tend to overestimate GFR in children and should not be used in pediatric populations [162]. Several equations have been developed for pediatric patients: Schwartz, Counahan–Barratt, Leger, the Bedside Chronic Kidney Disease in Children (CkiD), Morris, Shull, Traub, Rudd, Dechaux, Ghazali–Barratt and van den Anker formulas [163–170]. The most commonly used equations in children are the Schwartz formula, Leger, CkiD, and Counahan–Barrat formulas. The Schwartz formula, the most extensively validated formula, is based on serum creatinine and was first validated with data from 186 children, then with data from 2192 children in 14 subsequent validation studies. The Schwartz and Leger equations appear to overestimate GFR in children with decreasing kidney function when compared with measured inulin clearance [171–174]. The CKiD and Counahan–Barrat equations have been developed in children with a median GFR of 40–45 ml/min/1.73 m2, and tend to underestimate kidney function in children with a GFR >60 ml/min [175]. It should be noted that serum creatinine is influenced by muscle mass, gender, diet, and tubular secretion. In children, serum creatinine may also be affected by diseases such as neuromuscular disease and anorexia nervosa [176]. At birth, the serum creatinine values measured in the neonate reflect maternal serum creatinine values because the placenta allows free transfer of creatinine between the mother and her unborn infant. Primarily in preterm infants the serum creatinine values increase during the first days of life, reaching a peak concentration around 5–7 days after birth before a gradual decrease in values are seen [177–179]. The highest values are seen in the most premature infants [180, 181]. As already shown in the rabbit kidney model, this increase in serum creatinine values is caused by an increased tubular reabsorption of creatinine in these preterm infants [182, 183]. Therefore, serum creatinine is a poor marker for GFR in neonates as it does not fulfill the assumptions of a purely filtered substance from which to calculate GFR. This underlines the need for, and evaluation of, newer, earlier markers for kidney function.
Serum creatinine can be measured with different analytical methods: alkaline picrate method, Jaffe method classic and compensated, and an enzymatic method traceable to isotope dilution mass spectrometry (IDMS) [184, 185]. Interlaboratory variation is high with some of these methods [186, 187]; studies have reported median method group variation coefficients of 6.4 % at a concentration of 80 μml/L [188]. Variation in serum creatinine values lead to variation in derived calculations of kidney function. Discrepancies are more pronounced in children aged 1–5 years [189]. Standardization initiatives have been launched to reduce interlaboratory variation in creatinine assay calibration [190].
In order to compensate for inaccuracy of existing equations based on serum creatinine, alternative equations utilizing cystatin C have been developed. Cystatin C is a nonglycosylated protein produced in cells, not influenced by gender, body habitus, or composition (Table 3) [191–195].
Cystatin C does not cross the placental barrier and no correlation was found between maternal and neonatal serum cystatin [196]. Its reference value obtained in children aged 4–19 years is 0.75 ± 0.09 mg/L [197]; cystatin C levels are higher at birth (up to 4.2 mg/L) [191], and decrease in neonates [196] and infants over time [193]. Cystatin C has not been shown to be superior to calculation of GFR through the Schwartz formula in neonates [198]. Data on cystatin C in children receiving RRT are scarce [199–202].
‘Omics’-based technologies, such as proteomics and metabolomics, are uncovering new markers for kidney injury/disease [203–212]. Neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury molecule-1 (KIM-1) are promising candidates to detect kidney injuries in the early stages. These proteins are expressed by renal tubules, and clinical investigations have shown that they are massively expressed in cases of AKI in both adults and children [203, 205, 207, 208, 213–217]. NGAL concentrations seem to correlate with the severity/stage of CKD [204]. The ability to measure these newer markers noninvasively in urine represents an advantage over current serum markers, especially in children. Other new biomarkers are evaluated for (1) AKI: interleukin (IL)-18, liver-type fatty acid-binding protein (L-FABP), urinary insulin-like growth factor-binding protein 7 (IGFBP7), and tissue inhibitor of metalloproteinases-2 (TIMP-2) [206, 218–222]; (2) CKD: β-trace protein (BTP), L-FABP, and asymmetric dimethylarginine (ADMA) [223–227]; and (3) nephrotoxicity: N-acetyl-glucosaminidase (NAG), GST, gamma-glutamyl transpeptidase (GGT), alanine aminopeptidase (AAP), and lactate dehydrogenase (LDH) [228–231].
Animal studies have shown modifications of the metabolome in plasma and kidney tissues in renal ischemia/reperfusion injury. Increase in prostaglandins, higher catabolism of tryptophan and accumulation of citrulline in the kidney could be metabolic signatures of intrarenal inflammation associated with ischemia/reperfusion injury [209]. Clinical studies in neonates, infants, and older children are warranted to (1) evaluate potential measures for kidney function, especially during the first days of life; (2) test new markers such as NGAL and KIM-1 [232]; and (3) explore ‘omics’-based methods in order to improve detection and management of kidney injury/disease in neonates, infants, and children [233].
5 Quantitative Approaches and Opportunities in Pediatric Patients with Impaired Kidney Function
This section introduces quantitative approaches, such as pharmacometric modeling and simulation, to (1) simplify designs of studies in pediatric patients; (2) characterize effects of the kidney on drug exposure/response; (3) fine-tune dosing in pediatric patients with impaired kidney function; and (4) facilitate development and optimize utilization of therapeutics in neonates, infants, and children.
5.1 What is Pharmacometrics?
Pharmacometrics is an emerging science of developing and applying mathematical and statistical methods for characterizing, understanding, and predicting a drug’s pharmacokinetics, and its effects on biomarker and clinical responses over time [234, 235]. With pharmacometric approaches, biological knowledge can be translated into compartmental models with mathematical and statistical components [236]. ‘Population approaches’, introduced by Sheiner in the 1970s, can be utilized to quantify intersubject variability, at the population level, and test covariate effects on model parameters such as impact of body weight or kidney function on drug clearance. Such population models can also be applied to (1) project individual pharmacokinetic, biomarker and clinical responses, e.g. by Bayesian-inference [237–241]; (2) evaluate the impact of alternative doses on pharmacokinetics, biomarker and clinical responses; and (3) provide a scientific rationale for individualized dosing strategies [242–245]. Quantitative approaches such as pharmacometrics have been suggested to quantify the impact of kidney function on drugs and the impact of drugs on kidney function [246–248].
In pediatric patients, body weight reflecting growth, and age describing maturation, are key covariates. As described in Fig. 6, different age descriptors need to be considered in pediatric patients, especially in neonates, including PMA, gestational age (GA) and postnatal age (PNA), also called chronological age [249].

Age terminology during the perinatal period
Pharmacometric approaches include pharmacostatistical, exposure-response, and disease progression models [236, 250, 251]. Systems pharmacology approaches may represent more complex models, such as physiology-based pharmacokinetic models (PBPK) consisting of a large number of compartments to represent different organs or tissues in the human body [235, 252]. PBPK models may contain enzyme information from tissues, such as CYPs, involved in the metabolism of drugs [253–255].
Both pharmacometrics and systems pharmacology have been successfully applied in adults with impaired kidney function to (1) evaluate and simplify sampling designs of studies; (2) characterize and quantify the relationships between kidney function and drug exposure or effect; (3) fine-tune dosing strategies; and (4) enhance drug labels leveraging model-based simulations [148, 256–259]. Such quantitative approaches have the potential to facilitate development and optimize utilization of drugs in neonates, infants, and children, especially those with impaired kidney function.
5.2 How to Simplify Sampling Designs of Studies in Pediatric Patients?
Design and conduct of clinical studies in pediatric patients are difficult due to enrollment constraints (especially children <6 years of age), blood volume constraints (especially in neonates and young infants), and other practical challenges of collecting pharmacokinetic or other samples [243, 260]. Pharmacometric and systems pharmacology approaches, including PBPK-based simulations, can be leveraged to (1) identify starting dose in first-in-children pharmacokinetic studies and provide rationale for dose regimen optimization; (2) simplify pharmacokinetic–pharmacodynamic (PK–PD) sampling scheme (number and timing of blood collections for pharmacokinetic and/or PK–PD analyses); and (3) optimize sample size of studies in neonates, infants, and children [243, 261–267].
Furthermore, PBPK models can also be applied in neonates, infants, and children to (1) characterize pharmacokinetic behavior of drugs; (2) identify genetic factors that influence exposure/response of drugs; (3) assess risk of drug–drug interactions; and (4) quantify the impact of impaired kidney function on drugs [268–270]. PBPK models validated in adults may be expanded to pediatric patients by incorporating identified differences in drug pharmacokinetics and response between children and adults [267, 271].
5.3 How to Characterize Drug Exposure–Response and Enhance Drug Labels?
Pharmacometric and systems pharmacology approaches, utilizing pharmacokinetic, biomarker and/or disease progression data, can be useful to (1) identify factors that influence disease progression and responses to interventions; (2) facilitate comparison of concentration–response relationships across age groups; (3) link PD/biomarker endpoints to (longer term) clinical outcome measures, which may then be used as surrogate markers for assessing efficacy in various age groups; (4) simulate treatment-related responses in pediatric patients with and without impaired kidney function [259, 264, 272–274]; and (4) enhance drug labels for pediatric patients, such as atazanavir [275], busulfan [276, 277], levofloxacin [278], argatroban [279], piperacillin-tazobactam [280], etanercept [281], and subcutaneous immunoglobulin [282–284].
5.4 How to Fine-Tune Dosing Strategies for Drugs in Pediatric Patients with Impaired Kidney Function?
In the absence of specific dosing recommendations for children with changing kidney function, pediatric doses are extrapolated from adult data [1]. A priori dosing (prior to any measurement) in neonates, infants, and children is viewed as a scaling exercise, assuming a simple linear relationship between body weight and drug pharmacokinetics. Since developmental and maturational processes in pediatric subjects are mostly nonlinear, empirical dosing recommendations may result in over- or underdosing, resulting in toxicity or therapeutic failure. After initiating therapy, an adjusted a posteriori dose, based on therapeutic drug monitoring (TDM), can be identified [285–289]. A limitation of TDM in pediatric patients is that, for a majority of drugs, target concentrations are derived from adult patients rather than defined based on pediatric data, assuming similar exposure/response and TW across age groups [290–294]. Pharmacometric approaches can be leveraged to identify predictive covariates, characterize exposure/response and TW of drugs, and provide a scientific basis for individualized dosing strategies, including Bayesian-based TDM, in neonates, infants, and children [242–245, 289, 295, 296]. Model-based approaches can also be applied to fine-tune RRT strategies in pediatric patients [296–298].
5.5 What are the Opportunities to Facilitate Development and Optimize Utilization of Drugs in Pediatric Patients?
Strategic applications of pharmacometrics and systems pharmacology have the potential to streamline development and optimize utilization of drugs in pediatric patients with and without impaired kidney function. An overview of opportunities is provided in Table 4.
6 Case Study: How to Fine-Tune Amikacin Dosing in Neonates with Impaired Kidney Function?
Neonates are known to be at high risk of infection and are exposed to antibiotic-resistant bacterial pathogens; thus, antibiotics are the class of medicines most frequently prescribed in neonates. Early appropriate antimicrobial treatment is imperative to optimize response and limit the spread of resistance [300–302]. In addition, there is an important lack of uniformity in dosing information to ensure consistent drug exposure in neonates, leading to inappropriate prescription of most antibiotics [303]. Pharmacometric approaches can be used to (1) identify and quantify covariate effects on pharmacokinetic parameters; and (2) fine-tune dosing of antibiotics in neonates with and without impaired kidney function.
In this section, we focus on the application of pharmacometrics to evaluate and fine-tune dose strategies of amikacin in neonates. After penicillins, aminoglycosides are the most commonly used drugs in the neonatal intensive care unit [304]. Amikacin is used as a short-term treatment of serious infections caused by strains of Pseudomonas sp., Escherichia coli, Klebsiella pneumoniae, Serratia sp., and Staphylococcus species [305–307]. This drug is almost exclusively eliminated by kidneys (90 %) and its clearance reflects GFR [308]. There is huge variability in the choice of neonatal dosing regimen used across the world [309]. Leroux et al. found 19 different neonatal dosing regimens proposed in the literature [44, 303, 310–323]. Amikacin use is difficult because of its toxicity and pharmacokinetic variability, and no study or specific recommendations in cases of changing kidney function were described. Potential kidney effects on pharmacokinetic parameters were tested by incorporating age and weight as indirect measures for maturation and growth, and serum creatinine as a measure of kidney function in a population pharmacokinetic model [313, 324].
Sherwin et al. recently developed a population-based pharmacokinetic model based on 70 pediatric burn patients (6 months to 17 years) receiving amikacin, and found that weight had a significant influence on amikacin clearance [325].
De Cock et al. conducted a large population pharmacokinetic modeling study using data from 874 preterm and term neonates treated with amikacin (range: GA 24–43 weeks; PNA 1–30 days) [311]. Amikacin clearance was found to be related to PNA and birth weight, with children with higher age and weight having a faster maturation of clearance. Furthermore, coadministration of ibuprofen appeared to reduce amikacin clearance, likely (at least in part) due to negative effect on kidney function [311, 326, 327]. The individual amikacin clearance can be predicted using Eq. 1, with CL i being amikacin clearance in the ith individual, CL p being the population value of amikacin clearance, and bBW being birth body weight and PNA corresponding to PNA:
Figure 7 illustrates how the predicted clearance of amikacin increases with birth body weight (representing antenatal maturation) and PNA (representing postnatal maturation), taking into account coadministration of ibuprofen.

Model-based predicted amikacin clearance values versus bBW for PNA of 0, 14, or 28 days with and without coadministration of ibuprofen, according to De Cock et al. [311]. bBW Birth body weight, PNA postnatal age, CL clearance
Predictive performance of this model was externally validated in 239 neonates. An evidence-based dosing regimen, summarized in Table 5, was proposed by performing simulations with the developed model. They demonstrated that the currently used dosing regimens for amikacin, based on reference handbooks, may increase the risk of toxicities, and should be revised [311].
The authors also performed a study to extrapolate the amikacin model to other drug compounds almost entirely eliminated through glomerular filtration and with similar physicochemical properties (netilmicin, tobramycin, vancomycin, and gentamicin). They showed that pediatric covariate models may represent physiological information on developmental changes in glomerular filtration that may be leveraged to describe kinetics of other antibiotics that are primarily eliminated by kidneys [328].
7 Discussion and Outlook
Growth, maturation, and environmental factors affect drug kinetics, response, and dosing in pediatric patients. Changes in kidney function, as a result of normal growth and development as well as underlying kidney diseases, comorbidities, medications, and environmental and genetic factors, will not only have an impact on renal clearance but also on the absorption, distribution, metabolism, and nonrenal clearance of drugs [141, 142]. Both drug exposure and response may change during childhood and impact the TW and efficacy/safety balance of drugs in neonates, infants, and children. Current markers of kidney function provide limited value in assessing and monitoring kidney function in children, especially during the first days of life and in cases of AKI [172, 173]. Therefore, new renal biomarkers are needed. ‘Omics’-based technologies, such as proteomics and metabolomics, can be leveraged to uncover novel markers in plasma and urine for kidney function during normal development, AKI, and CKD.
Motivated by challenges in conducting clinical studies in pediatric subjects, supported by regulatory agencies such as the European Medicines Agency (EMA) and the US FDA, pharmacometric and systems pharmacology have been suggested to facilitate the design and conduct of studies in pediatric subjects [329]. Strategic use of model-based quantitative approaches and biomarkers [240, 330] has the potential to streamline development and optimize utilization of drugs in pediatric patients with and without impaired kidney function by (1) informing the design of pediatric clinical trials, including sample size and first dose selection, providing rationale for the dose range to be studied, and simplifying PK–PD sampling; (2) characterizing disease progression to project long-term clinical outcomes; (3) quantifying the effects of impaired kidney function (and RRT) on drug pharmacokinetics and/or response; (4) facilitating key development decisions; and (5) providing a scientific rationale for pediatric drug labels.
The recently formed Drug Disease Model Resources (DDMoRe) consortium facilitates collaborations between pharmaceutical industries and academic partners [331]. They aim to address the lack of common tools, languages, and standards for modeling and simulation to improve model-based knowledge integration. A public drug and disease model library, supported by an open source and universally applicable framework, provides access to disease modeling tools [331, 332]. It should also be noted that online tools have been developed to facilitate evaluation and optimization of study designs in adult and pediatric subjects. For example, Mentré et al. developed a software tool known as PFIM, which is a set of R functions that evaluates and/or optimizes study designs based on the expression of the Fisher information matrix (FIM) in nonlinear mixed effects models (http://www.pfim.biostat.fr/) [333, 334].
In clinical practice, pharmacometric approaches can be applied to identify predictive covariates, such as the impact of kidney function changes on drugs, and provide a scientific basis for optimizing dosing in pediatric patients [242, 244, 245]. Bayesian-based TDM methods can leverage patient characteristics, physiological differences between adults and children, genetic and environmental factors and pharmacokinetic properties of drugs, and individualize dosing strategies in neonates, infants, and children. Decision support tools are emerging to assist clinicians at the bedside in personalized dosing of pediatric patients, including neonates, such as the EzeCHiel (http://www.ezechiel.ch/) [244, 289] and DoseMe software packages (http://www.doseme.com.au/) [335].
Table 6 outlines opportunities to overcome challenges in further streamlining development and optimizing utilization of therapeutics in neonates, infants, and children, especially those with impaired kidney function.
Collaborative efforts between clinicians and scientists in academia, industry, and regulatory agencies are required to (1) identify new renal biomarkers for early detection and enhanced monitoring of kidney injury and disease; (2) collect and share prospective pharmacokinetic, genetic, and clinical data with the goal of creating large clinical outcome databases; (3) build and evaluate integrated pharmacometric models for key diseases and therapeutics; (4) optimize and standardize dosing strategies; (5) develop user-friendly bedside decision tools for clinicians; and (6) enhance drug labels for neonates, infants, and children.
References
Cella M, Knibbe C, Danhof M, Della Pasqua O. What is the right dose for children? Br J Clin Pharmacol. 2010;70(4):597–603.
‘t Jong GW, Vulto AG, de Hoog M, Schimmel KJ, Tibboel D, van den Anker JN. A survey of the use of off-label and unlicensed drugs in a Dutch children’s hospital. Pediatrics. 2001;108(5):1089–93.
Conroy S, McIntyre J. The use of unlicensed and off-label medicines in the neonate. Semin Fetal Neonatal Med. 2005;10(2):115–22.
Conroy S, McIntyre J, Choonara I, Stephenson T. Drug trials in children: problems and the way forward. Br J Clin Pharmacol. 2000;49(2):93–7.
Baer GR, Nelson RM. Ethical challenges in neonatal research: summary report of the ethics group of the newborn drug development initiative. Clin Ther. 2006;28(9):1399–407.
Schreiner MS. Paediatric clinical trials: redressing the imbalance. Nat Rev Drug Discov. 2003;2(12):949–61.
Friis-Hansen B. Water distribution in the foetus and newborn infant. Acta Paediatr Scand Suppl. 1983;305:7–11.
Fomon SJ, Nelson SE. Body composition of the male and female reference infants. Ann Rev Nutr. 2002;22:1–17.
Fomon SJ, Haschke F, Ziegler EE, Nelson SE. Body composition of reference children from birth to age 10 years. Am J Clin Nutr. 1982;35(5 Suppl):1169–75.
Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology: drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157–67.
Blake MJ, Castro L, Leeder JS, Kearns GL. Ontogeny of drug metabolizing enzymes in the neonate. Semin Fetal Neonatal Med. 2005;10(2):123–38.
Allegaert K, Fanos V, van den Anker JN, Laer S. Perinatal pharmacology. BioMed Res Int. 2014;2014:101620.
Allegaert K, van den Anker JN. Clinical pharmacology in neonates: small size, huge variability. Neonatology. 2014;105(4):344–9.
Allegaert K, van de Velde M, van den Anker J. Neonatal clinical pharmacology. Paediatr Anaesth. 2014;24(1):30–8.
Bhimma R, Purswani MU, Kala U. Kidney disease in children and adolescents with perinatal HIV-1 infection. J Int AIDS Soc. 2013;16:18596.
Knijnenburg SL, Mulder RL, Schouten-Van Meeteren AY, Bokenkamp A, Blufpand H, van Dulmen-den Broeder E, et al. Early and late renal adverse effects after potentially nephrotoxic treatment for childhood cancer. Cochrane Database Syst Rev. 2013;10:CD008944.
Patzer L. Nephrotoxicity as a cause of acute kidney injury in children. Pediatr Nephrol. 2008;23(12):2159–73.
Pazhayattil GS, Shirali AC. Drug-induced impairment of renal function. Int J Nephrol Renovasc Dis. 2014;7:457–68.
Wood T, Thoresen M. Physiological responses to hypothermia. Semin Fetal Neonatal Med. 2015;20(2):87–96.
Wildschut ED, de Wildt SN, Mathot RA, Reiss IK, Tibboel D, Van den Anker J. Effect of hypothermia and extracorporeal life support on drug disposition in neonates. Sem Fetal Neonatal Med. 2013;18(1):23–7.
Wildschut ED, van Saet A, Pokorna P, Ahsman MJ, Van den Anker JN, Tibboel D. The impact of extracorporeal life support and hypothermia on drug disposition in critically ill infants and children. Pediatr Clin North Am. 2012;59(5):1183–204.
Treszl A, Toth-Heyn P, Kocsis I, Nobilis A, Schuler A, Tulassay T, et al. Interleukin genetic variants and the risk of renal failure in infants with infection. Pediatr Nephrol. 2002;17(9):713–7.
National Heart, Lung, and Blood Institute, National Institutes of Health, US Department of Health and Human Services. 2015. http://www.nhlbi.nih.gov/childrenandclinicalstudies/whyclinical.php. Accessed 17 June 2015.
Bechard LJ, Wroe E, Ellis K. Body composition and growth. In: Duggan CW, Watkins J B, Walker WA, editors. Nutrition in pediatrics: basic science, clinical applications. Hamilton: BC Decker Inc.; 2008. pp. 20–40.
de Wildt SN, Tibboel D, Leeder JS. Drug metabolism for the paediatrician. Arch Dis Child. 2014;99(12):1137–42.
Funk RS, Brown JT, Abdel-Rahman SM. Pediatric pharmacokinetics: human development and drug disposition. Pediatr Clin North Am. 2012;59(5):1001–16.
van den Anker JN, Schwab M, Kearns GL. Developmental pharmacokinetics. Handb Exp Pharmacol. 2011;205:51–75.
Rakhmanina NY, van den Anker JN. Pharmacological research in pediatrics: from neonates to adolescents. Adv Drug Deliv Rev. 2006;58(1):4–14.
Ilett KF, Tee LB, Reeves PT, Minchin RF. Metabolism of drugs and other xenobiotics in the gut lumen and wall. Pharmacol Ther. 1990;46(1):67–93.
Johnson TN, Thomson M. Intestinal metabolism and transport of drugs in children: the effects of age and disease. J Pediatr Gastroenterol Nutr. 2008;47(1):3–10.
Rowland IR. Factors affecting metabolic activity of the intestinal microflora. Drug Metab Rev. 1988;19(3–4):243–61.
Fernandez E, Perez R, Hernandez A, Tejada P, Arteta M, Ramos JT. Factors and mechanisms for pharmacokinetic differences between pediatric population and adults. Pharmaceutics. 2011;3(1):53–72.
Hines RN. Developmental expression of drug metabolizing enzymes: impact on disposition in neonates and young children. Int J Pharm. 2013;452(1–2):3–7.
Coulthard MG. Maturation of glomerular filtration in preterm and mature babies. Early Hum Dev. 1985;11(3–4):281–92.
Schwartz GJ, Brion LP, Spitzer A. The use of plasma creatinine concentration for estimating glomerular filtration rate in infants, children, and adolescents. Pediatr Clinic North Am. 1987;34(3):571–90.
Alcorn J, McNamara PJ. Ontogeny of hepatic and renal systemic clearance pathways in infants: part II. Clin Pharmacokinet. 2002;41(13):1077–94.
Chen N, Aleksa K, Woodland C, Rieder M, Koren G. Ontogeny of drug elimination by the human kidney. Pediatr Nephrol. 2006;21(2):160–8.
Alcorn J, McNamara PJ. Ontogeny of hepatic and renal systemic clearance pathways in infants: part I. Clinl Pharmacokinet. 2002;41(12):959–98.
Hayton WL. Maturation and growth of renal function: dosing renally cleared drugs in children. AAPS PharmSci. 2000;2(1):E3.
Akaoka K, White RH, Raafat F. Human glomerular growth during childhood: a morphometric study. J Pathol. 1994;173(3):261–8.
Goyal VK. Changes with age in the human kidney. Exp Gerontol. 1982;17(5):321–31.
van den Anker JN. Pharmacokinetics and renal function in preterm infants. Acta Paediatr. 1996;85(12):1393–9.
Alcorn J, McNamara PJ. Pharmacokinetics in the newborn. Adv Drug Deliv Rev. 2003;55(5):667–86.
Allegaert K, Anderson BJ, van den Anker JN, Vanhaesebrouck S, de Zegher F. Renal drug clearance in preterm neonates: relation to prenatal growth. Ther Drug Monit. 2007;29(3):284–91.
van den Anker JN, Hop WC, de Groot R, van der Heijden BJ, Broerse HM, Lindemans J, et al. Effects of prenatal exposure to betamethasone and indomethacin on the glomerular filtration rate in the preterm infant. Pediatr Res. 1994;36(5):578–81.
Weiss CF, Glazko AJ, Weston JK. Chloramphenicol in the newborn infant. A physiologic explanation of its toxicity when given in excessive doses. N Engl J Med. 1960;262:787–94.
Rho JM, Storey TW. Molecular ontogeny of major neurotransmitter receptor systems in the mammalian central nervous system: norepinephrine, dopamine, serotonin, acetylcholine, and glycine. J Child Neurol. 2001;16(4):271–80 (discussion 81).
Takahashi H, Ishikawa S, Nomoto S, Nishigaki Y, Ando F, Kashima T, et al. Developmental changes in pharmacokinetics and pharmacodynamics of warfarin enantiomers in Japanese children. Clin Pharmacol Ther. 2000;68(5):541–55.
Marshall JD, Kearns GL. Developmental pharmacodynamics of cyclosporine. Clin Pharmacol Ther. 1999;66(1):66–75.
Kauffman RE, Nelson MV. Effect of age on ibuprofen pharmacokinetics and antipyretic response. J Pediatr. 1992;121(6):969–73.
Johnson TN. The development of drug metabolising enzymes and their influence on the susceptibility to adverse drug reactions in children. Toxicology. 2003;192(1):37–48.
Laer S, Elshoff JP, Meibohm B, Weil J, Mir TS, Zhang W, et al. Development of a safe and effective pediatric dosing regimen for sotalol based on population pharmacokinetics and pharmacodynamics in children with supraventricular tachycardia. J Am Coll Cardiol. 2005;46(7):1322–30.
Fisher DM, O’Keeffe C, Stanski DR, Cronnelly R, Miller RD, Gregory GA. Pharmacokinetics and pharmacodynamics of d-tubocurarine in infants, children, and adults. Anesthesiology. 1982;57(3):203–8.
Stephenson T. How children’s responses to drugs differ from adults. Br J Clin Pharmacol. 2005;59(6):670–3.
Van Driest SL, McGregor TL. Pharmacogenetics in clinical pediatrics: challenges and strategies. Per Med. 2013;10(7):666–71.
Rogers HL, Bhattaram A, Zineh I, Gobburu J, Mathis M, Laughren TP, et al. CYP2D6 genotype information to guide pimozide treatment in adult and pediatric patients: basis for the U.S. Food and Drug Administration’s new dosing recommendations. J Clin Psychiatry. 2012;73(9):1187–90.
Biss TT, Avery PJ, Brandao LR, Chalmers EA, Williams MD, Grainger JD, et al. VKORC1 and CYP2C9 genotype and patient characteristics explain a large proportion of the variability in warfarin dose requirement among children. Blood. 2012;119(3):868–73.
de Wildt SN, van Schaik RH, Soldin OP, Soldin SJ, Brojeni PY, van der Heiden IP, et al. The interactions of age, genetics, and disease severity on tacrolimus dosing requirements after pediatric kidney and liver transplantation. Eur J Clin Pharmacol. 2011;67(12):1231–41.
Gijsen V, Mital S, van Schaik RH, Soldin OP, Soldin SJ, van der Heiden IP, et al. Age and CYP3A5 genotype affect tacrolimus dosing requirements after transplant in pediatric heart recipients. J Heart Lung Transplant. 2011;30(12):1352–9.
Stockmann C, Fassl B, Gaedigk R, Nkoy F, Uchida DA, Monson S, et al. Fluticasone propionate pharmacogenetics: CYP3A4*22 polymorphism and pediatric asthma control. J Pediatr. 2013;162(6):1222–7, 1227.e1–2.
Peters U, Preisler-Adams S, Hebeisen A, Hahn M, Seifert E, Lanvers C, et al. Glutathione S-transferase genetic polymorphisms and individual sensitivity to the ototoxic effect of cisplatin. Anticancer Drugs. 2000;11(8):639–43.
Oldenburg J, Kraggerud SM, Cvancarova M, Lothe RA, Fossa SD. Cisplatin-induced long-term hearing impairment is associated with specific glutathione s-transferase genotypes in testicular cancer survivors. J Clin Oncol. 2007;25(6):708–14.
Wang L, Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: insights, challenges and future directions. Oncogene. 2006;25(11):1629–38.
Adam de Beaumais T, Jacqz-Aigrain E. Pharmacogenetic determinants of mercaptopurine disposition in children with acute lymphoblastic leukemia. Eur J Clin Parmacol. 2012;68(9):1233–42.
Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui CH, Yee SW, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing: 2013 update. Clin Pharmacol Ther. 2013;93(4):324–5.
Chong KW, Chan DW, Cheung YB, Ching LK, Hie SL, Thomas T, et al. Association of carbamazepine-induced severe cutaneous drug reactions and HLA-B*1502 allele status, and dose and treatment duration in paediatric neurology patients in Singapore. Arch Dis Child. 2014;99(6):581–4.
Ganesan S, Hussain N. Question 2: Should phenytoin and carbamazepine be avoided in Asian populations with the HLA-B*1502 positive genetic variant? Arch Dis Child. 2011;96(1):104–6.
Chen P, Lin JJ, Lu CS, Ong CT, Hsieh PF, Yang CC, et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med. 2011;364(12):1126–33.
Mercaptopurine. US National Library of Medicine. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=c3b5b8b0-bc5c-4ce9-bbdc-febba60c265. Accessed 17 June 2015.
US National Library of Medicine. Imuran. Available at: http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=aaa6c540-4c84-48a0-939c-cd423134fa2a. Accessed 27 Feb 215.
Wei CY, Lee MT, Chen YT. Pharmacogenomics of adverse drug reactions: implementing personalized medicine. Hum Mol Genet. 2012;21(R1):R58–65.
US National Library of Medicine. Carbamazepine. Available at: http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=c13bc0b8-7900-4ef4-98ed-e1315a08d95d. Accessed 27 Feb 2015.
US National Library of Medicine. Stattera. 2015. Available at: http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=309de576-c318-404a-bc15-660c2b1876fb. Accessed 27 Feb 2015.
Shaw K, Amstutz U, Hildebrand C, Rassekh SR, Hosking M, Neville K, et al. VKORC1 and CYP2C9 genotypes are predictors of warfarin-related outcomes in children. Pediatr Blood Cancer. 2014;61(6):1055–62.
Vear SI, Ayers GD, Van Driest SL, Sidonio RF, Stein CM, Ho RH. The impact of age and CYP2C9 and VKORC1 variants on stable warfarin dose in the paediatric population. Br J Haematol. 2014;165(6):832–5.
Benders MJ, Van Bel F, Van de Bor M. Cardiac output and ductal reopening during phototherapy in preterm infants. Acta Paediatr. 1999;88(9):1014–9.
Benders MJ, van Bel F, van de Bor M. Haemodynamic consequences of phototherapy in term infants. Eur J Pediatr. 1999;158(4):323–8.
Ardissino G, Dacco V, Testa S, Bonaudo R, Claris-Appiani A, Taioli E, et al. Epidemiology of chronic renal failure in children: data from the ItalKid project. Pediatrics. 2003;111(4 Pt 1):e382–7.
Chan JC, Williams DM, Roth KS. Kidney failure in infants and children. Pediatr Rev. 2002;23(2):47–60.
Ardissino G, Dacco V, Testa S, Bonaudo R, Claris-Appiani A, Taioli E, et al. Epidemiology of chronic renal failure in children: data from the ItalKid project. Pediatrics. 2003;111(4 Pt 1):e382–7.
Wong CJ, Moxey-Mims M, Jerry-Fluker J, Warady BA, Furth SL. CKiD (CKD in children) prospective cohort study: a review of current findings. Am J Kidney Dis. 2012;60(6):1002–11.
Medina Villanueva A, Lopez-Herce Cid J, Lopez Fernandez Y, Anton Gamero M, Concha Torre A, Rey Galan C, et al. Acute renal failure in critically-ill children. A preliminary study [in Spanish]. An Pediatr (Barc). 2004;61(6):509–14.
Bailey D, Phan V, Litalien C, Ducruet T, Merouani A, Lacroix J, et al. Risk factors of acute renal failure in critically ill children: a prospective descriptive epidemiological study. Pediatr Crit Care Med. 2007;8(1):29–35.
Stapleton FB, Jones DP, Green RS. Acute renal failure in neonates: incidence, etiology and outcome. Pediatr Nephrol. 1987;1(3):314–20.
Allegaert K, Vanhole C, de Hoon J, Guignard JP, Tibboel D, Devlieger H, et al. Nonselective cyclo-oxygenase inhibitors and glomerular filtration rate in preterm neonates. Pediatr Nephrol. 2005;20(11):1557–61.
Faught LN, Greff MJ, Rieder MJ, Koren G. Drug-induced acute kidney injury in children. Br J Clin Pharmacol. doi:10.1111/bcp.12554 (Epub 13 Nov 2014).
Mendoza SA. Nephrotoxic drugs. Pediatr Nephrol. 1988;2(4):466–76.
Misurac JM, Knoderer CA, Leiser JD, Nailescu C, Wilson AC, Andreoli SP. Nonsteroidal anti-inflammatory drugs are an important cause of acute kidney injury in children. J Pediatr. 2013;162(6):1153–9, 11599.e1.
Destache CJ. Aminoglycoside-induced nephrotoxicity—a focus on monitoring: a review of literature. J Pharm Pract. 2014;27(6):562–6.
Wargo KA, Edwards JD. Aminoglycoside-induced nephrotoxicity. J Pharm Pract. 2014;27(6):573–7.
Laurent G, Kishore BK, Tulkens PM. Aminoglycoside-induced renal phospholipidosis and nephrotoxicity. Biochem Pharmacol. 1990;40(11):2383–92.
Kacew S, Bergeron MG. Pathogenic factors in aminoglycoside-induced nephrotoxicity. Toxicol Lett. 1990;51(3):241–59 (discussion 37–9).
Plaut ME, Schentag JJ, Jusko WJ. Nephrotoxicity with gentamicin or tobramycin. Lancet. 1979;2(8141):526–7.
Moore RD, Smith CR, Lipsky JJ, Mellits ED, Lietman PS. Risk factors for nephrotoxicity in patients treated with aminoglycosides. Ann Intern Med. 1984;100(3):352–7.
Dahlager JI. The effect of netilmicin and other aminoglycosides on renal function. A survey of the literature on the nephrotoxicity of netilmicin. Scand J Infect Dis Suppl. 1980;Suppl 23:96–102.
McCracken GH Jr. Aminoglycoside toxicity in infants and children. Am J Med. 1986;80(6b):172–8.
Meyer RD. Risk factors and comparisons of clinical nephrotoxicity of aminoglycosides. Am J Med. 1986;80(6b):119–25.
McKamy S, Hernandez E, Jahng M, Moriwaki T, Deveikis A, Le J. Incidence and risk factors influencing the development of vancomycin nephrotoxicity in children. J Pediatr. 2011;158(3):422–6.
Ragab AR, Al-Mazroua MK, Al-Harony MA. Incidence and predisposing factors of vancomycin-induced nephrotoxicity in children. Infect Dis Ther. 2013;2(1):37–46.
Sinclair EA, Yenokyan G, McMunn A, Fadrowski JJ, Milstone AM, Lee CK. Factors associated with acute kidney injury in children receiving vancomycin. Ann Pharmacother. 2014;48(12):1555–62.
Chow AW, Azar RM. Glycopeptides and nephrotoxicity. Intensive Care Med. 1994;20(Suppl 4):S23–9.
Goldman RD, Koren G. Amphotericin B nephrotoxicity in children. J Pediatr Hematol Oncol. 2004;26(7):421–6.
Sabra R, Branch RA. Amphotericin B nephrotoxicity. Drug Saf. 1990;5(2):94–108.
Schreiber R, Wolpin J, Koren G. Determinants of aciclovir-induced nephrotoxicity in children. Paediatr Drugs. 2008;10(2):135–9.
Ahmad T, Simmonds M, McIver AG, McGraw ME. Reversible renal failure in renal transplant patients receiving oral acyclovir prophylaxis. Pediatr Nephrol. 1994;8(4):489–91.
Olowu WA, Adenowo OA, Elusiyan JB. Reversible renal failure in hypertensive idiopathic nephrotics treated with captopril. Saudi J Kidney Dis Transpl. 2006;17(2):216–21.
Hanevold CD. Acute renal failure during lisinopril and losartan therapy for proteinuria. Pharmacotherapy. 2006;26(9):1348–51.
Ojo AO, Held PJ, Port FK, Wolfe RA, Leichtman AB, Young EW, et al. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med. 2003;349(10):931–40.
Rudnick MR, Goldfarb S, Wexler L, Ludbrook PA, Murphy MJ, Halpern EF, et al. Nephrotoxicity of ionic and nonionic contrast media in 1196 patients: a randomized trial. The Iohexol Cooperative Study. Kidney Int. 1995;47(1):254–61.
Patzer L, Kentouche K, Ringelmann F, Misselwitz J. Renal function following hematological stem cell transplantation in childhood. Pediatr Nephrol. 2003;18(7):623–35.
Rossi R, Kleta R, Ehrich JH. Renal involvement in children with malignancies. Pediatr Nephrol. 1999;13(2):153–62.
Skinner R, Cotterill SJ, Stevens MC. Risk factors for nephrotoxicity after ifosfamide treatment in children: a UKCCSG Late Effects Group study. United Kingdom Children’s Cancer Study Group. Br J Cancer. 2000;82(10):1636–45.
Skinner R, Pearson AD, English MW, Price L, Wyllie RA, Coulthard MG, et al. Cisplatin dose rate as a risk factor for nephrotoxicity in children. Br J Cancer. 1998;77(10):1677–82.
Kintzel PE. Anticancer drug-induced kidney disorders. Drug Saf. 2001;24(1):19–38.
Moffett BS, Goldstein SL. Acute kidney injury and increasing nephrotoxic-medication exposure in noncritically-ill children. Clin J Am Soc Nephrol. 2011;6(4):856–63.
Collins AJ, Foley RN, Herzog C, Chavers BM, Gilbertson D, Ishani A, et al. Excerpts from the US Renal Data System 2009 Annual Data Report. Am J Kidney Dis. 2010;55(1 Suppl 1):S1-420, A6-7.
Pea F, Viale P, Pavan F, Furlanut M. Pharmacokinetic considerations for antimicrobial therapy in patients receiving renal replacement therapy. Clin Pharmacokinet. 2007;46(12):997–1038.
Veltri MA, Neu AM, Fivush BA, Parekh RS, Furth SL. Drug dosing during intermittent hemodialysis and continuous renal replacement therapy: special considerations in pediatric patients. Paediatr Drugs. 2004;6(1):45–65.
Keller F, Wilms H, Schultze G, Offerman G, Molzahn M. Effect of plasma protein binding, volume of distribution and molecular weight on the fraction of drugs eliminated by hemodialysis. Clin Nephrol. 1983;19(4):201–5.
Bohler J, Donauer J, Keller F. Pharmacokinetic principles during continuous renal replacement therapy: drugs and dosage. Kidney Int Suppl. 1999;72:S24–8.
Golper TA, Bennett WM. Drug removal by continuous arteriovenous haemofiltration. A review of the evidence in poisoned patients. Med Toxicol Adverse Drug Exp. 1988;3(5):341-9.
Reidenberg MM, Drayer DE. Drug therapy in renal failure. Ann Rev Pharmacol Toxicol. 1980;20:45–54.
Joy MS, Matzke GR, Armstrong DK, Marx MA, Zarowitz BJ. A primer on continuous renal replacement therapy for critically ill patients. Ann Pharmacother. 1998;32(3):362–75.
Reetze-Bonorden P, Bohler J, Keller E. Drug dosage in patients during continuous renal replacement therapy. Pharmacokinetic and therapeutic considerations. Clin Pharmacokinet. 1993;24(5):362–79.
Nobilis A, Kocsis I, Toth-Heyn P, Treszl A, Schuler A, Tulassay T, et al. Variance of ACE and AT1 receptor gene does not influence the risk of neonatal acute renal failure. Pediatr Nephrol. 2001;16(12):1063–6.
Harding D, Dhamrait S, Marlow N, Whitelaw A, Gupta S, Humphries S, et al. Angiotensin-converting enzyme DD genotype is associated with worse perinatal cardiorespiratory adaptation in preterm infants. J Pediatr. 2003;143(6):746–9.
Vasarhelyi B, Toth-Heyn P, Treszl A, Tulassay T. Genetic polymorphisms and risk for acute renal failure in preterm neonates. Pediatr Nephrol. 2005;20(2):132–5.
Padulles A, Rama I, Llaudo I, Lloberas N. Developments in renal pharmacogenomics and applications in chronic kidney disease. Pharmgenomics Pers Med. 2014;7:251–66.
Yang HY, Lu KC, Lee HS, Huang SM, Lin YF, Wu CC, et al. Role of the functional Toll-Like receptor-9 promoter polymorphism (-1237T/C) in increased risk of end-stage renal disease: a case-control study. PloS One. 2013;8(3):e58444.
Hawwa AF, McKiernan PJ, Shields M, Millership JS, Collier PS, McElnay JC. Influence of ABCB1 polymorphisms and haplotypes on tacrolimus nephrotoxicity and dosage requirements in children with liver transplant. Br J Clin Pharmacol. 2009;68(3):413–21.
Hart SN, Zhong XB. P450 oxidoreductase: genetic polymorphisms and implications for drug metabolism and toxicity. Expert Opin Drug Metab Toxicol. 2008;4(4):439–52.
Elens L, Hesselink DA, van Schaik RH, van Gelder T. The CYP3A4*22 allele affects the predictive value of a pharmacogenetic algorithm predicting tacrolimus predose concentrations. Br J Clin Pharmacol. 2013;75(6):1545–7.
Elens L, Bouamar R, Hesselink DA, Haufroid V, van der Heiden IP, van Gelder T, et al. A new functional CYP3A4 intron 6 polymorphism significantly affects tacrolimus pharmacokinetics in kidney transplant recipients. Clin Chem. 2011;57(11):1574–83.
Gijsen VM, van Schaik RH, Elens L, Soldin OP, Soldin SJ, Koren G, et al. CYP3A4*22 and CYP3A combined genotypes both correlate with tacrolimus disposition in pediatric heart transplant recipients. Pharmacogenomics. 2013;14(9):1027–36.
Elens L, Hesselink DA, Bouamar R, Budde K, de Fijter JW, De Meyer M, et al. Impact of POR*28 on the pharmacokinetics of tacrolimus and cyclosporine A in renal transplant patients. Ther Drug Monit. 2014;36(1):71–9.
de Jonge H, Metalidis C, Naesens M, Lambrechts D, Kuypers DR. The P450 oxidoreductase *28 SNP is associated with low initial tacrolimus exposure and increased dose requirements in CYP3A5-expressing renal recipients. Pharmacogenomics. 2011;12(9):1281–91.
Gijsen VM, van Schaik RH, Soldin OP, Soldin SJ, Nulman I, Koren G, et al. P450 oxidoreductase *28 (POR*28) and tacrolimus disposition in pediatric kidney transplant recipients: a pilot study. Ther Drug Monit. 2014;36(2):152–8.
Hesselink DA, Bouamar R, Elens L, van Schaik RH, van Gelder T. The role of pharmacogenetics in the disposition of and response to tacrolimus in solid organ transplantation. Clin Pharmacokinet. 2014;53(2):123–39.
Filipski KK, Mathijssen RH, Mikkelsen TS, Schinkel AH, Sparreboom A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin Pharmacol Ther. 2009;86(4):396–402.
Hassan Y, Al-Ramahi R, Abd Aziz N, Ghazali R. Drug use and dosing in chronic kidney disease. Ann Acad Med Singap. 2009;38(12):1095–103.
Atkinson AJ Jr, Huang SM. Nephropharmacology: drugs and the kidney. Clin Pharmacol Ther. 2009;86(5):453–6.
Brater DC. Drug dosing in patients with impaired renal function. Clin Pharmacol Ther. 2009;86(5):483–9.
Naud J, Nolin TD, Leblond FA, Pichette V. Current understanding of drug disposition in kidney disease. J Clin Pharmacol. 2012;52(1 Suppl):10s–22s.
Joy MS. Impact of glomerular kidney diseases on the clearance of drugs. J Clin Pharmacol. 2012;52(1 Suppl):23s–34s.
Gabardi S, Abramson S. Drug dosing in chronic kidney disease. Med Clin North Am. 2005;89(3):649–87.
Kappel J, Calissi P. Nephrology: 3. Safe drug prescribing for patients with renal insufficiency. CMAJ. 2002;166(4):473–7.
Perazella MA, Parikh C. Pharmacology. Am J Kidney Dis. 2005;46(6):1129–39.
Nolin TD, Naud J, Leblond FA, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther. 2008;83(6):898–903.
Kober A, Sjöholm I, Borgå O, Odar-Cederlöf I. Protein binding of diazepam and digitoxin in uremic and normal serum. Biochem Pharmacol. 1979;28(7):1037–42.
Knights KM, Rowland A, Miners JO. Renal drug metabolism in humans: the potential for drug-endobiotic interactions involving cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT). Br J Clin Pharmacol. 2013;76(4):587–602.
Mutsaers HA, Wilmer MJ, Reijnders D, Jansen J, van den Broek PH, Forkink M, et al. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim Biophys Acta. 2013;1832(1):142–50.
Dreisbach AW, Lertora JJ. The effect of chronic renal failure on drug metabolism and transport. Expert Opin Drug Metab Toxicol. 2008;4(8):1065–74.
Naud J, Michaud J, Leblond FA, Lefrancois S, Bonnardeaux A, Pichette V. Effects of chronic renal failure on liver drug transporters. Drug Metab Dispos. 2008;36(1):124–8.
Yeung CK, Shen DD, Thummel KE, Himmelfarb J. Effects of chronic kidney disease and uremia on hepatic drug metabolism and transport. Kidney Int. 2014;85(3):522–8.
Leblond FA, Giroux L, Villeneuve JP, Pichette V. Decreased in vivo metabolism of drugs in chronic renal failure. Drug Metab Dispos. 2000;28(11):1317–20.
Tsujimoto M, Kinoshita Y, Hirata S, Otagiri M, Ohtani H, Sawada Y. Effects of uremic serum and uremic toxins on hepatic uptake of digoxin. Ther Drug Monit. 2008;30(5):576–82.
Klammt S, Wojak HJ, Mitzner A, Koball S, Rychly J, Reisinger EC, et al. Albumin-binding capacity (ABiC) is reduced in patients with chronic kidney disease along with an accumulation of protein-bound uraemic toxins. Nephrol Dial Transplant. 2012;27(6):2377–83.
Rosner MH, Bolton WK. Renal function testing. Am J Kidney Dis. 2006;47(1):174–83.
Stevens LA, Levey AS. Measurement of kidney function. Med Clin North Am. 2005;89(3):457–73.
Zhao W, Biran V, Jacqz-Aigrain E. Amikacin maturation model as a marker of renal maturation to predict glomerular filtration rate and vancomycin clearance in neonates. Clin Pharmacokinet. 2013;52(12):1127–34.
Stevens LA, Coresh J, Greene T, Levey AS. Assessing kidney function: measured and estimated glomerular filtration rate. N Engl J Med. 2006;354(23):2473–83.
Pierrat A, Gravier E, Saunders C, Caira MV, Ait-Djafer Z, Legras B, et al. Predicting GFR in children and adults: a comparison of the Cockcroft-Gault, Schwartz, and modification of diet in renal disease formulas. Kidney Int. 2003;64(4):1425–36.
Schwartz GJ, Haycock GB, Edelmann CM Jr, Spitzer A. A simple estimate of glomerular filtration rate in children derived from body length and plasma creatinine. Pediatrics. 1976;58(2):259–63.
Counahan R, Chantler C, Ghazali S, Kirkwood B, Rose F, Barratt TM. Estimation of glomerular filtration rate from plasma creatinine concentration in children. Arch Dis Child. 1976;51(11):875–8.
Leger F, Bouissou F, Coulais Y, Tafani M, Chatelut E. Estimation of glomerular filtration rate in children. Pediatr Nephrol. 2002;17(11):903–7.
Morris MC, Allanby CW, Toseland P, Haycock GB, Chantler C. Evaluation of a height/plasma creatinine formula in the measurement of glomerular filtration rate. Arch Dis Child. 1982;57(8):611–5.
Shull BC, Haughey D, Koup JR, Baliah T, Li PK. A useful method for predicting creatinine clearance in children. Clin Chem. 1978;24(7):1167–9.
Traub SL, Johnson CE. Comparison of methods of estimating creatinine clearance in children. Am J Hosp Pharm. 1980;37(2):195–201.
van den Anker JN, de Groot R, Broerse HM, Sauer PJ, van der Heijden BJ, Hop WC, et al. Assessment of glomerular filtration rate in preterm infants by serum creatinine: comparison with inulin clearance. Pediatrics. 1995;96(6):1156–8.
Dechaux M, Gonzalez G, Broyer M. Plasma creatinine, and clearance and urinary excretion of creatinine in children [in French]. Arch Fr Pediatr. 1978;35(1):53–62.
Doolan PD, Alpen EL, Theil GB. A clinical appraisal of the plasma concentration and endogenous clearance of creatinine. Am J Medi. 1962;32:65–79.
Fong J, Johnston S, Valentino T, Notterman D. Length/serum creatinine ratio does not predict measured creatinine clearance in critically ill children. Clin Pharmacol Ther. 1995;58(2):192–7.
Seikaly MG, Browne R, Bajaj G, Arant BS Jr. Limitations to body length/serum creatinine ratio as an estimate of glomerular filtration in children. Pediatr Nephrol. 1996;10(6):709–11.
Urakami Y, Kimura N, Okuda M, Masuda S, Katsura T, Inui K. Transcellular transport of creatinine in renal tubular epithelial cell line LLC-PK1. Drug Metab Pharmacokinet. 2005;20(3):200–5.
Lee CK, Swinford RD, Cerda RD, Portman RJ, Hwang W, Furth SL. Evaluation of serum creatinine concentration-based glomerular filtration rate equations in pediatric patients with chronic kidney disease. Pharmacotherapy. 2012;32(7):642–8.
Hogg RJ, Furth S, Lemley KV, Portman R, Schwartz GJ, Coresh J, et al. National Kidney Foundation’s Kidney Disease Outcomes Quality Initiative clinical practice guidelines for chronic kidney disease in children and adolescents: evaluation, classification, and stratification. Pediatrics. 2003;111(6 Pt 1):1416–21.
Bueva A, Guignard JP. Renal function in preterm neonates. Pediatr Res. 1994;36(5):572–7.
Feldman H, Guignard JP. Plasma creatinine in the first month of life. Arch Dis Child. 1982;57(2):123–6.
Miall LS, Henderson MJ, Turner AJ, Brownlee KG, Brocklebank JT, Newell SJ, et al. Plasma creatinine rises dramatically in the first 48 hours of life in preterm infants. Pediatrics. 1999;104(6):e76.
Allegaert K, Pauwels S, Smits A, Crevecoeur K, van den Anker J, Mekahli D, et al. Enzymatic isotope dilution mass spectrometry (IDMS) traceable serum creatinine is preferable over Jaffe in neonates and young infants. Clin Chem Lab Med. 2014;52(6):e107–9.
Gallini F, Maggio L, Romagnoli C, Marrocco G, Tortorolo G. Progression of renal function in preterm neonates with gestational age < or = 32 weeks. Pediatr Nephrol. 2000;15(1–2):119–24.
Guignard JP, Drukker A. Why do newborn infants have a high plasma creatinine? Pediatrics. 1999;103(4):e49.
Matos P, Duarte-Silva M, Drukker A, Guignard JP. Creatinine reabsorption by the newborn rabbit kidney. Pediatr Res. 1998;44(5):639–41.
Wuyts B, Bernard D, Van den Noortgate N, Van de Walle J, Van Vlem B, De Smet R, et al. Reevaluation of formulas for predicting creatinine clearance in adults and children, using compensated creatinine methods. Clin Chem. 2003;49(6 Pt 1):1011–4.
Chan MH, Ng KF, Szeto CC, Lit LC, Chow KM, Leung CB, et al. Effect of a compensated Jaffe creatinine method on the estimation of glomerular filtration rate. Ann Clin Biochem. 2004;41(Pt 6):482–4.
Drion I, Cobbaert C, Groenier KH, Weykamp C, Bilo HJ, Wetzels JF, et al. Clinical evaluation of analytical variations in serum creatinine measurements: why laboratories should abandon Jaffe techniques. BMC Nephrol. 2012;13:133.
van den Anker JN. Renal function in preterm infants. Eur J Pediatr. 1997;156(7):583–4.
Miller WG, Myers GL, Ashwood ER, Killeen AA, Wang E, Thienpont LM, et al. Creatinine measurement: state of the art in accuracy and interlaboratory harmonization. Arch Pathol Lab Med. 2005;129(3):297–304.
Neuman G, Nulman I, Adeli K, Koren G, Colantonio DA, Hellden A. Implications of serum creatinine measurements on GFR estimation and vancomycin dosing in children. J Clin Pharmacol. 2014;54(7):785–91.
Myers GL, Miller WG, Coresh J, Fleming J, Greenberg N, Greene T, et al. Recommendations for improving serum creatinine measurement: a report from the Laboratory Working Group of the National Kidney Disease Education Program. Clin Chem. 2006;52(1):5–18.
Finney H, Newman DJ, Thakkar H, Fell JM, Price CP. Reference ranges for plasma cystatin C and creatinine measurements in premature infants, neonates, and older children. Arch Dis Child. 2000;82(1):71–5.
Harmoinen A, Ylinen E, Ala-Houhala M, Janas M, Kaila M, Kouri T. Reference intervals for cystatin C in pre- and full-term infants and children. Pediatr Nephrol. 2000;15(1–2):105–8.
Kandasamy Y, Smith R, Wright IM. Measuring cystatin C to determine renal function in neonates. Pediatr Crit Care Med. 2013;14(3):318–22.
Randers E, Erlandsen EJ. Serum cystatin C as an endogenous marker of the renal function: a review. Clin Chem Lab Med. 1999;37(4):389–95.
Brou NA, Jacqz-Aigrain E, Zhao W. Cystatin C as a potential biomarker for dosing of renally excreted drugs. Br J Clin Pharmacol. doi:10.1111/bcp.12602 (Epub 5 Feb 2015).
Cataldi L, Mussap M, Bertelli L, Ruzzante N, Fanos V, Plebani M. Cystatin C in healthy women at term pregnancy and in their infant newborns: relationship between maternal and neonatal serum levels and reference values. Am J Perinatol. 1999;16(6):287–95.
Galteau MM, Guyon M, Gueguen R, Siest G. Determination of serum cystatin C: biological variation and reference values. Clin Chem Lab Med. 2001;39(9):850–7.
Martini S, Prevot A, Mosig D, Werner D, van Melle G, Guignard JP. Glomerular filtration rate: measure creatinine and height rather than cystatin C! Acta Paediatr. 2003;92(9):1052–7.
Marsenic O, Wierenga A, Wilson DR, Anderson M, Shrivastava T, Simon GA, et al. Cystatin C in children on chronic hemodialysis. Pediatr Nephrol. 2013;28(4):647–53.
Hoek FJ, Korevaar JC, Dekker FW, Boeschoten EW, Krediet RT. Estimation of residual glomerular filtration rate in dialysis patients from the plasma cystatin C level. Nephrol Dial Transplant. 2007;22(6):1633–8.
Kim SJ, Sohn YB, Park SW, Jin DK, Paik KH. Serum cystatin C for estimation of residual renal function in children on peritoneal dialysis. Pediatr Nephrol. 2011;26(3):433–40.
Filler G, Huang SH, Lindsay RM. Residual renal function assessment with cystatin C. Pediatr Nephrol. 2011;26(3):333–5.
Bolignano D, Donato V, Coppolino G, Campo S, Buemi A, Lacquaniti A, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a marker of kidney damage. Am J Kidney Dis. 2008;52(3):595–605.
Bolignano D, Lacquaniti A, Coppolino G, Donato V, Campo S, Fazio MR, et al. Neutrophil gelatinase-associated lipocalin (NGAL) and progression of chronic kidney disease. Clin J Am Soc Nephrol. 2009;4(2):337–44.
Clerico A, Galli C, Fortunato A, Ronco C. Neutrophil gelatinase-associated lipocalin (NGAL) as biomarker of acute kidney injury: a review of the laboratory characteristics and clinical evidences. Clin Chem Lab Med. 2012;50(9):1505–17.
de Geus HR, Betjes MG, Bakker J. Biomarkers for the prediction of acute kidney injury: a narrative review on current status and future challenges. Clin Kidney J. 2012;5(2):102–8.
Devarajan P. Biomarkers for the early detection of acute kidney injury. Curr Opin Pediatr. 2011;23(2):194–200.
Wasilewska A, Taranta-Janusz K, Debek W, Zoch-Zwierz W, Kuroczycka-Saniutycz E. KIM-1 and NGAL: new markers of obstructive nephropathy. Pediatr Nephrol. 2011;26(4):579–86.
Wei Q, Xiao X, Fogle P, Dong Z. Changes in metabolic profiles during acute kidney injury and recovery following ischemia/reperfusion. PloS One. 2014;9(9):e106647.
Iacovidou N, Syggelou A, Chalkias A, Atzori L, Xanthos T, Fanos V. Metabolomics applied in neonatology. Bioanalysis. 2014;6(3):403–10.
Syggelou A, Iacovidou N, Atzori L, Xanthos T, Fanos V. Metabolomics in the developing human being. Pediatr Clin North Am. 2012;59(5):1039–58.
Zhao YY, Lint RC. Metabolomics in nephrotoxicity. Adv Clin Chem. 2014;65:69–89.
Holzscheiter L, Beck C, Rutz S, Manuilova E, Domke I, Guder WG, et al. NGAL, L-FABP, and KIM-1 in comparison to established markers of renal dysfunction. Clin Chem Lab Med. 2014;52(4):537–46.
Fontanilla J, Han WK. Kidney injury molecule-1 as an early detection tool for acute kidney injury and other kidney diseases. Expert Opin Med Diagn. 2011;5(2):161–73.
Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int. 2002;62(1):237–44.
Ronco C. N-GAL: diagnosing AKI as soon as possible. Crit Care. 2007;11(6):173.
Zappitelli M, Washburn KK, Arikan AA, Loftis L, Ma Q, Devarajan P, et al. Urine neutrophil gelatinase-associated lipocalin is an early marker of acute kidney injury in critically ill children: a prospective cohort study. Crit Care. 2007;11(4):R84.
Parikh CR, Jani A, Melnikov VY, Faubel S, Edelstein CL. Urinary interleukin-18 is a marker of human acute tubular necrosis. Am J Kidney Dis. 2004;43(3):405–14.
Wu H, Craft ML, Wang P, Wyburn KR, Chen G, Ma J, et al. IL-18 contributes to renal damage after ischemia-reperfusion. J Am Soc Nephrol. 2008;19(12):2331–41.
Yamamoto T, Noiri E, Ono Y, Doi K, Negishi K, Kamijo A, et al. Renal L-type fatty acid: binding protein in acute ischemic injury. J Am Soc Nephrol. 2007;18(11):2894–902.
Susantitaphong P, Siribamrungwong M, Doi K, Noiri E, Jaber BL. Performance of urinary liver-type fatty acid-binding protein in acute kidney injury: a meta-analysis. Am J Kidney Dis. 2013;61(3):430–9.
Meersch M, Schmidt C, Van Aken H, Martens S, Rossaint J, Singbartl K, et al. Urinary TIMP-2 and IGFBP7 as early biomarkers of acute kidney injury and renal recovery following cardiac surgery. PloS One. 2014;9(3):e93460.
Spanaus KS, Kollerits B, Ritz E, Hersberger M, Kronenberg F, von Eckardstein A. Serum creatinine, cystatin C, and beta-trace protein in diagnostic staging and predicting progression of primary nondiabetic chronic kidney disease. Clin Chem. 2010;56(5):740–9.
Bhavsar NA, Appel LJ, Kusek JW, Contreras G, Bakris G, Coresh J, et al. Comparison of measured GFR, serum creatinine, cystatin C, and beta-trace protein to predict ESRD in African Americans with hypertensive CKD. Am J Kidney Dis. 2011;58(6):886–93.
Kamijo A, Sugaya T, Hikawa A, Yamanouchi M, Hirata Y, Ishimitsu T, et al. Clinical evaluation of urinary excretion of liver-type fatty acid-binding protein as a marker for the monitoring of chronic kidney disease: a multicenter trial. J Lab Clin Med. 2005;145(3):125–33.
Hanai K, Babazono T, Nyumura I, Toya K, Tanaka N, Tanaka M, et al. Asymmetric dimethylarginine is closely associated with the development and progression of nephropathy in patients with type 2 diabetes. Nephrol Dial Transplant. 2009;24(6):1884–8.
Ravani P, Tripepi G, Malberti F, Testa S, Mallamaci F, Zoccali C. Asymmetrical dimethylarginine predicts progression to dialysis and death in patients with chronic kidney disease: a competing risks modeling approach. J Am Soc Nephrol. 2005;16(8):2449–55.
Naghibi B, Ghafghazi T, Hajhashemi V, Talebi A. Vancomycin-induced nephrotoxicity in rats: is enzyme elevation a consistent finding in tubular injury? J Nephrol. 2007;20(4):482–8.
Svendsen KB, Ellingsen T, Bech JN, Pfeiffer-Jensen M, Stengaard-Pedersen K, Pedersen EB. Urinary excretion of alpha-GST and albumin in rheumatoid arthritis patients treated with methotrexate or other DMARDs alone or in combination with NSAIDs. Scand J Rheumatol. 2005;34(1):34–9.
Harrison DJ, Kharbanda R, Cunningham DS, McLellan LI, Hayes JD. Distribution of glutathione S-transferase isoenzymes in human kidney: basis for possible markers of renal injury. J Clin Pathol. 1989;42(6):624–8.
Hanna MH, Segar JL, Teesch LM, Kasper DC, Schaefer FS, Brophy PD. Urinary metabolomic markers of aminoglycoside nephrotoxicity in newborn rats. Pediatr Res. 2013;73(5):585–91.
Ding W, Mak RH. Early markers of obesity-related renal injury in childhood. Pediatr Nephrol. 2015;30(1):1–4.
Fanos V, Van den Anker J, Noto A, Mussap M, Atzori L. Metabolomics in neonatology: fact or fiction? Semin Fetal Neonatal Med. 2013;18(1):3–12.
Pfister M, D’Argenio DZ. The emerging scientific discipline of pharmacometrics. J Clin Pharmacol. 2010;50(9 Suppl):6S.
van der Graaf PH. CPT: Pharmacometrics and Systems Pharmacology. CPT Pharmacometrics Syst Pharmacol. 2012;1:e8.
Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development. Part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst Pharmacol. 2013;2:e38.
Sheiner LB. The population approach to pharmacokinetic data analysis: rationale and standard data analysis methods. Drug Metab Rev. 1984;15(1–2):153–71.
Wilbaux M, Henin E, Oza A, Colomban O, Pujade-Lauraine E, Freyer G, et al. Prediction of tumour response induced by chemotherapy using modelling of CA-125 kinetics in recurrent ovarian cancer patients. Br J Cancer. 2014;110(6):1517–24.
Wilbaux M, Henin E, Oza A, Colomban O, Pujade-Lauraine E, Freyer G, et al. Dynamic modeling in ovarian cancer: an original approach linking early changes in modeled longitudinal CA-125 kinetics and survival to help decisions in early drug development. Gynecol Oncol. 2014;133(3):460–6.
Zhang Y, Wei X, Bajaj G, Barrett JS, Meibohm B, Joshi A, et al. Challenges and considerations for development of therapeutic proteins in pediatric patients. J Clin Pharmacol. 2015;55(Suppl 3):S103–15.
Thomson AH, Whiting B. Bayesian parameter estimation and population pharmacokinetics. Clin Pharmacokinet. 1992;22(6):447–67.
Admiraal R, van Kesteren C, Boelens JJ, Bredius RG, Tibboel D, Knibbe CA. Towards evidence-based dosing regimens in children on the basis of population pharmacokinetic pharmacodynamic modelling. Arch Dis Child. 2014;99(3):267–72.
Barrett J. Pharmacometrics in pediatrics. In: Schmidt S, Derendorf H, editors. Applied pharmacometrics. Berlin: Springer; 2014. pp. 83–108.
Barrett JS. Paediatric models in motion: requirements for model-based decision support at the bedside. Br J Clin Pharmacol. 2015;79(1):85–96.
Zhao W, Leroux S, Jacqz-Aigrain E. Dosage individualization in children: integration of pharmacometrics in clinical practice. World J Pediatr. 2014;10(3):197–203.
Pfister M, Nolin TD, Arya V. Optimizing drug development and use in patients with kidney disease: opportunities, innovations, and challenges. J Clin Pharmacol. 2012;52(1 Suppl):4s–6s.
Nolin TD, Arya V, Sitar DS, Pfister M. Optimizing drug development and use in patients with kidney disease. J Clin Pharmacol. 2011;51(5):628–30.
Zhang L, Roy A, Pfister M. Pharmacometrics in chronic kidney disease. In: Schmidt S, Derendorf H, editors. Applied Pharmacometrics. Berlin: Springer; 2014. pp. 109–38.
Engle WA. American Academy of Pediatrics Committee on Fetus and Newborn. Age terminology during the perinatal period. Pediatrics. 2004;114(5):1362–4.
Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model-based drug development. CPT Pharmacometrics Syst Pharmacol. 2012;1:e6.
Upton RN, Mould DR. Basic concepts in population modeling, simulation, and model-based drug development. Part 3: introduction to pharmacodynamic modeling methods. CPT Pharmacometrics Syst Pharmacol. 2014;3:e88.
Jones H, Rowland-Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst Pharmacol. 2013;2:e63.
Achour B, Barber J, Rostami-Hodjegan A. Expression of hepatic drug-metabolizing cytochrome p450 enzymes and their intercorrelations: a meta-analysis. Drug Metab Dispos. 2014;42(8):1349–56.
Johnson TN, Salem F, Jamei M, Rostami-Hodjegan A. Prediction of voriconazole non-linear pharmacokinetics using a paediatric physiologically based pharmacokinetic modelling approach. Clin Pharmacokinet. 2015;54(5):567–8.
Zane NR, Thakker DR. A physiologically based pharmacokinetic model for voriconazole disposition predicts intestinal first-pass metabolism in children. Clin Pharmacokinet. 2014;53(12):1171–82.
Tortorici MA, Cutler D, Zhang L, Pfister M. Design, conduct, analysis, and interpretation of clinical studies in patients with impaired kidney function. J Clin Pharmacol. 2012;52(1 Suppl):109S–18S.
Zhang L, Pfister M, Meibohm B. Concepts and challenges in quantitative pharmacology and model-based drug development. AAPS J. 2008;10(4):552–9.
Zhang L, Xu N, Xiao S, Arya V, Zhao P, Lesko LJ, et al. Regulatory perspectives on designing pharmacokinetic studies and optimizing labeling recommendations for patients with chronic kidney disease. J Clin Pharmacol. 2012;52(1 Suppl):79S–90S.
Zhang L, Boulton DW, Pfister M. A pharmacometric approach to quantify the impact of chronic kidney disease and hemodialysis on systemic drug exposure: application to saxagliptin. J Clin Pharmacol. 2012;52(1 Suppl):126s–33s.
Meibohm B, Laer S, Panetta JC, Barrett JS. Population pharmacokinetic studies in pediatrics: issues in design and analysis. AAPS J. 2005;7(2):E475–87.
Barrett JS, Della Casa Alberighi O, Laer S, Meibohm B. Physiologically based pharmacokinetic (PBPK) modeling in children. Clin Pharmacol Ther. 2012;92(1):40–9.
Jadhav PR, Kern SE. The need for modeling and simulation to design clinical investigations in children. J Clin Pharmacol. 2010;50(9 Suppl):121S–9S.
Leong R, Vieira ML, Zhao P, Mulugeta Y, Lee CS, Huang SM, et al. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin Pharmacol Ther. 2012;91(5):926–31.
Tod M, Jullien V, Pons G. Facilitation of drug evaluation in children by population methods and modelling. Clin Pharmacokinet. 2008;47(4):231–43.
Vong C, Bergstrand M, Nyberg J, Karlsson MO. Rapid sample size calculations for a defined likelihood ratio test-based power in mixed-effects models. AAPS J. 2012;14(2):176–86.
Ogungbenro K, Aarons L, Graham G. Sample size calculations based on generalized estimating equations for population pharmacokinetic experiments. J Biopharm Stat. 2006;16(2):135–50.
Maharaj AR, Edginton AN. Physiologically based pharmacokinetic modeling and simulation in pediatric drug development. CPT Pharmacometrics Syst Pharmacol. 2014;3:e150.
Emoto C, Fukuda T, Johnson TN, Adams DM, Vinks AA. Development of a pediatric physiologically based pharmacokinetic model for sirolimus: applying principles of growth and maturation in neonates and infants. CPT Pharmacometrics Syst Pharmacol. 2015;4:e17.
Jiang XL, Zhao P, Barrett JS, Lesko LJ, Schmidt S. Application of physiologically based pharmacokinetic modeling to predict acetaminophen metabolism and pharmacokinetics in children. CPT Pharmacometrics Syst Pharmacol. 2013;2:e80.
Yang F, Tong X, McCarver DG, Hines RN, Beard DA. Population-based analysis of methadone distribution and metabolism using an age-dependent physiologically based pharmacokinetic model. J Pharmacokinet Pharmacodyn. 2006;33(4):485–518.
Maharaj AR, Barrett JS, Edginton AN. A workflow example of PBPK modeling to support pediatric research and development: case study with lorazepam. AAPS J. 2013;15(2):455–64.
Bellanti F, Della Pasqua O. Modelling and simulation as research tools in paediatric drug development. Eur J Clin Pharmacol. 2011;67(Suppl 1):75–86.
Lee JY, Garnett CE, Gobburu JV, Bhattaram VA, Brar S, Earp JC, et al. Impact of pharmacometric analyses on new drug approval and labelling decisions: a review of 198 submissions between 2000 and 2008. Clin Pharmacokinet. 2011;50(10):627–35.
Manolis E, Osman TE, Herold R, Koenig F, Tomasi P, Vamvakas S, et al. Role of modeling and simulation in pediatric investigation plans. Paediatr Anaesth. 2011;21(3):214–21.
Hong Y, Kowalski KG, Zhang J, Zhu L, Horga M, Bertz R, et al. Model-based approach for optimization of atazanavir dose recommendations for HIV-infected pediatric patients. Antimicrob Agents Chemother. 2011;55(12):5746–52.
Booth BP, Rahman A, Dagher R, Griebel D, Lennon S, Fuller D, et al. Population pharmacokinetic-based dosing of intravenous busulfan in pediatric patients. J Clin Pharmacol. 2007;47(1):101–11.
Nguyen L. Integration of modelling and simulation into the development of intravenous busulfan in paediatrics: an industrial experience. Fundam Clin Pharmacol. 2008;22(6):599–604.
Li F, Nandy P, Chien S, Noel GJ, Tornoe CW. Pharmacometrics-based dose selection of levofloxacin as a treatment for postexposure inhalational anthrax in children. Antimicrob Agents Chemother. 2010;54(1):375–9.
Madabushi R, Cox DS, Hossain M, Boyle DA, Patel BR, Young G, et al. Pharmacokinetic and pharmacodynamic basis for effective argatroban dosing in pediatrics. J Clin Pharmacol. 2011;51(1):19–28.
Tornoe CW, Tworzyanski JJ, Imoisili MA, Alexander JJ, Korth-Bradley JM, Gobburu JV. Optimising piperacillin/tazobactam dosing in paediatrics. Int J Antimicrob Agents. 2007;30(4):320–4.
Yim DS, Zhou H, Buckwalter M, Nestorov I, Peck CC, Lee H. Population pharmacokinetic analysis and simulation of the time-concentration profile of etanercept in pediatric patients with juvenile rheumatoid arthritis. J Clin Pharmacol. 2005;45(3):246–56.
Landersdorfer CB, Bexon M, Edelman J, Rojavin M, Kirkpatrick CM, Lu J, et al. Pharmacokinetic modeling and simulation of biweekly subcutaneous immunoglobulin dosing in primary immunodeficiency. Postgrad Med. 2013;125(6):53–61.
Rojavin M, Sidhu J, Pfister M, Hubsch A. Subcutaneous immunoglobulin loading regimens for previously untreated patients with primary antibody deficiency. Clin Exp Immunol. 2014;178(Suppl 1):146–8.
Sidhu J, Rojavin M, Pfister M, Edelman J. Enhancing patient flexibility of subcutaneous immunoglobulin G dosing: pharmacokinetic outcomes of various maintenance and loading regimens in the treatment of primary immunodeficiency. Biol Ther. doi:10.1007/s13554-014-0018-0 (Epub 14 Aug 2014).
Widmer N, Csajka C, Werner D, Grouzmann E, Decosterd LA, Eap CB, et al. Principles of therapeutic drug monitoring [in French]. Rev Med Suisse. 2008;4(165):1644–8.
Widmer N, Werner D, Grouzmann E, Eap CB, Marchetti O, Fayet A, et al. Therapeutic drug monitoring: clinical practice [in French]. Rev Med Suisse. 2008;4(165):1649–50, 1652–60.
Rousseau A, Marquet P. Application of pharmacokinetic modelling to the routine therapeutic drug monitoring of anticancer drugs. Fundam Clin Pharmacol. 2002;16(4):253–62.
Santini J, Milano G, Thyss A, Renee N, Viens P, Ayela P, et al. 5-FU therapeutic monitoring with dose adjustment leads to an improved therapeutic index in head and neck cancer. Br J Cancer. 1989;59(2):287–90.
Fuchs A, Csajka C, Thoma Y, Buclin T, Widmer N. Benchmarking therapeutic drug monitoring software: a review of available computer tools. Clin Pharmacokinet. 2013;52(1):9–22.
Fletcher CV, Brundage RC, Remmel RP, Page LM, Weller D, Calles NR, et al. Pharmacologic characteristics of indinavir, didanosine, and stavudine in human immunodeficiency virus-infected children receiving combination therapy. Antimicrob Agents Chemother. 2000;44(4):1029–34.
Grub S, Delora P, Ludin E, Duff F, Fletcher CV, Brundage RC, et al. Pharmacokinetics and pharmacodynamics of saquinavir in pediatric patients with human immunodeficiency virus infection. Clin Pharmacol Ther. 2002;71(3):122–30.
Soldin OP, Soldin SJ. Review: therapeutic drug monitoring in pediatrics. Ther Drug Monit. 2002;24(1):1–8.
Soldin SJ, Steele BW. Mini-review: therapeutic drug monitoring in pediatrics. Clin Biochem. 2000;33(5):333–5.
Pichini S, Papaseit E, Joya X, Vall O, Farre M, Garcia-Algar O, et al. Pharmacokinetics and therapeutic drug monitoring of psychotropic drugs in pediatrics. Ther Drug Monit. 2009;31(3):283–318.
Gotta V, Widmer N, Montemurro M, Leyvraz S, Haouala A, Decosterd LA, et al. Therapeutic drug monitoring of imatinib: Bayesian and alternative methods to predict trough levels. Clin Pharmacokinet. 2012;51(3):187–201.
Barker CI, Germovsek E, Hoare RL, Lestner JM, Lewis J, Standing JF. Pharmacokinetic/pharmacodynamic modelling approaches in paediatric infectious diseases and immunology. Adv Drug Deliv Rev. 2014;73:127–39.
Fissell R, Schulman G, Pfister M, Zhang L, Hung AM. Novel dialysis modalities: do we need new metrics to optimize treatment? J Clin Pharmacol. 2012;52(1 Suppl):72s–8s.
Marsenic O, Zhang L, Zuppa A, Barrett JS, Pfister M. Application of individualized Bayesian urea kinetic modeling to pediatric hemodialysis. ASAIO J. 2010;56(3):246–53.
Hariharan S, Madabushi R. Clinical pharmacology basis of deriving dosing recommendations for dabigatran in patients with severe renal impairment. J Clin Pharmacol. 2012;52(1 Suppl):119s–25s.
Leekha S, Terrell CL, Edson RS. General principles of antimicrobial therapy. Mayo Clin Proc. 2011;86(2):156–67.
Eagle H, Fleischman R, Musselman AD. Effect of schedule of administration on the therapeutic efficacy of penicillin; importance of the aggregate time penicillin remains at effectively bactericidal levels. Am J Med. 1950;9(3):280–99.
Edwards MS. Antibacterial therapy in pregnancy and neonates. Clin Perinatol. 1997;24(1):251–66.
Leroux S, Zhao W, Betremieux P, Pladys P, Saliba E, Jacqz-Aigrain E. Therapeutic guidelines for prescribing antibiotics in neonates should be evidence-based: a French national survey. Arch Dis Child. 2015;100(4):394–8.
van den Anker JN, Allegaert K. Pharmacokinetics of aminoglycosides in the newborn. Curr Pharm Des. 2012;18(21):3114–18.
Edson RS, Terrell CL. The aminoglycosides. Mayo Clin Proc. 1991;66(11):1158–64.
Karanwal AB, Parikh BJ, Goswami P, Panchal HP, Parekh BB, Patel KB. Review of clinical profile and bacterial spectrum and sensitivity patterns of pathogens in febrile neutropenic patients in hematological malignancies: a retrospective analysis from a single center. Indian J Med Paediatr Oncol. 2013;34(2):85–8.
Zhanel GG, Lawson CD, Zelenitsky S, Findlay B, Schweizer F, Adam H, et al. Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin. Expert Rev Anti Infect Ther. 2012;10(4):459–73.
Pacifici GM. Clinical pharmacokinetics of aminoglycosides in the neonate: a review. Eur J Clin Pharmacol. 2009;65(4):419–27.
van den Anker JN. How to optimize the evaluation and use of antibiotics in neonates. Early Hum Dev. 2014;90(Suppl 1):S10–2.
Setiabudy R, Suwento R, Rundjan L, Yasin FH, Louisa M, Dwijayanti A, et al. Lack of a relationship between the serum concentration of aminoglycosides and ototoxicity in neonates. Int J Clin Pharmacol Ther. 2013;51(5):401–6.
De Cock RF, Allegaert K, Schreuder MF, Sherwin CM, de Hoog M, van den Anker JN, et al. Maturation of the glomerular filtration rate in neonates, as reflected by amikacin clearance. Clin Pharmacokinet. 2012;51(2):105–17.
Abdel-Hady E, El Hamamsy M, Hedaya M, Awad H. The efficacy and toxicity of two dosing-regimens of amikacin in neonates with sepsis. J Clin Pharm Ther. 2011;36(1):45–52.
Schreuder MF, Wilhelm AJ, Bokenkamp A, Timmermans SM, Delemarre-van de Waal HA, van Wijk JA. Impact of gestational age and birth weight on amikacin clearance on day 1 of life. Clin J Am Soc Nephrol. 2009;4(11):1774–8.
Siddiqi A, Khan DA, Khan FA, Razzaq A. Therapeutic drug monitoring of amikacin in preterm and term infants. Singap Med J. 2009;50(5):486–9.
Sherwin CM, Svahn S, Van der Linden A, Broadbent RS, Medlicott NJ, Reith DM. Individualised dosing of amikacin in neonates: a pharmacokinetic/pharmacodynamic analysis. Eur J Clin Pharmacol. 2009;65(7):705–13.
Allegaert K, Cossey V, Debeer A, Langhendries JP, Van Overmeire B, de Hoon J, et al. The impact of ibuprofen on renal clearance in preterm infants is independent of the gestational age. Pediatr Nephrol. 2005;20(6):740–3.
Treluyer JM, Merle Y, Tonnelier S, Rey E, Pons G. Nonparametric population pharmacokinetic analysis of amikacin in neonates, infants, and children. Antimicrob Agents Chemother. 2002;46(5):1381–7.
Labaune JM, Bleyzac N, Maire P, Jelliffe RW, Boutroy MJ, Aulagner G, et al. Once-a-day individualized amikacin dosing for suspected infection at birth based on population pharmacokinetic models. Biol Neonate. 2001;80(2):142–7.
Wang J, Liang WQ, Wu JJ, Pan CM. Population pharmacokinetic analysis of amikacin and validation on neonates using Monte Carlo method. Acta Pharmacol Sin. 2000;21(10):954–60.
Langhendries JP, Battisti O, Bertrand JM, Francois A, Kalenga M, Darimont J, et al. Adaptation in neonatology of the once-daily concept of aminoglycoside administration: evaluation of a dosing chart for amikacin in an intensive care unit. Biol Neonate. 1998;74(5):351–62.
Botha JH, du Preez MJ, Miller R, Adhikari M. Determination of population pharmacokinetic parameters for amikacin in neonates using mixed-effect models. Eur J Clin Pharmacol. 1998;53(5):337–41.
Petersen PO, Wells TG, Kearns GL. Amikacin dosing in neonates: evaluation of a dosing chart based on population pharmacokinetic data. Dev Pharmacol Ther. 1991;16(4):203–11.
Kenyon CF, Knoppert DC, Lee SK, Vandenberghe HM, Chance GW. Amikacin pharmacokinetics and suggested dosage modifications for the preterm infant. Antimicrob Agents Chemother. 1990;34(2):265–8.
Allegaert K, Anderson BJ, Cossey V, Holford NH. Limited predictability of amikacin clearance in extreme premature neonates at birth. Br J Clin Pharmacol. 2006;61(1):39–48.
Sherwin CM, Wead S, Stockmann C, Healy D, Spigarelli MG, Neely A, et al. Amikacin population pharmacokinetics among paediatric burn patients. Burns. 2014;40(2):311–8.
Allegaert K. The impact of ibuprofen or indomethacin on renal drug clearance in neonates. J Matern Fetal Neonatal Med. 2009;22(Suppl 3):88–91.
Allegaert K, Cossey V, Langhendries JP, Naulaers G, Vanhole C, Devlieger H, et al. Effects of co-administration of ibuprofen-lysine on the pharmacokinetics of amikacin in preterm infants during the first days of life. Biol Neonate. 2004;86(3):207–11.
De Cock RF, Allegaert K, Sherwin CM, Nielsen EI, de Hoog M, van den Anker JN, et al. A neonatal amikacin covariate model can be used to predict ontogeny of other drugs eliminated through glomerular filtration in neonates. Pharm Res. 2014;31(3):754–67.
Jadhav PR, Zhang J, Gobburu JV. Leveraging prior quantitative knowledge in guiding pediatric drug development: a case study. Pharm Stat. 2009;8(3):216–24.
Bai JP, Barrett JS, Burckart GJ, Meibohm B, Sachs HC, Yao L. Strategic biomarkers for drug development in treating rare diseases and diseases in neonates and infants. AAPS J. 2013;15(2):447–54.
Harnisch L, Matthews I, Chard J, Karlsson MO. Drug and disease model resources: a consortium to create standards and tools to enhance model-based drug development. CPT Pharmacometrics Syst Pharmacol. 2013;2:e34.
Drug Disease Model Resources (DDMoRe). 2015. Availabel at: http://www.ddmore.eu/. Accessed 27 Feb 2015.
Bazzoli C, Retout S, Mentré F. Design evaluation and optimisation in multiple response nonlinear mixed effect models: PFIM 3.0. Comput Methods Programs Biomed. 2010;98(1):55–65.
Mentré F, Chenel M, Comets E, Grevel J, Hooker A, Karlsson MO, et al. Current use and developments needed for optimal design in pharmacometrics: a study performed among DDMoRe’s European Federation of Pharmaceutical Industries and Associations members. CPT Pharmacometrics Syst Pharmacol. 2013;2:e46.
Hennig S, Holthouse F, Staatz CE. Comparing dosage adjustment methods for once-daily tobramycin in paediatric and adolescent patients with cystic fibrosis. Clin Pharmacokinet. 2015;54(4):409–21.
Acknowledgments
The authors would like to thank Eveline Staub, Department of Neonatology, University Children’s Hospital Basel (UKBB), Basel, Switzerland, for her constructive critique and expertise in reviewing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Frederique Rodieux, Melanie Wilbaux, Johannes N. van den Anker and Marc Pfister have no conflicts of interest to declare.
Funding
No sources of funding were used in the preparation of this review.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rodieux, F., Wilbaux, M., van den Anker, J.N. et al. Effect of Kidney Function on Drug Kinetics and Dosing in Neonates, Infants, and Children. Clin Pharmacokinet 54, 1183–1204 (2015). https://doi.org/10.1007/s40262-015-0298-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0298-7