
Abstract
The field of tumor immunology has faced many complex challenges over the last century, but the approval of immune checkpoint inhibitors (anti-cytotoxic T-lymphocyte-associated protein 4 [CTLA4] and anti-programmed cell death-1 [PD-1]/PD-ligand 1 [PD-L1]) and talimogene laherparepvec (T-VEC) for the treatment of metastatic melanoma have awakened a new wave of interest in cancer immunotherapy. Additionally, combinations of vaccines and oncolytic viral therapies with immune checkpoint inhibitors and other systemic agents seem to be promising synergistic strategies to further boost the immune response against cancer. These combinations are undergoing clinical investigation, and if successful, will hopefully soon become available to patients. Here, we review key basic concepts of tumor-induced immune suppression in malignant melanoma, the historical perspective around vaccine development in melanoma, and advances in oncolytic viral therapies. We also discuss the emerging role for combination approaches with different immunomodulatory agents as well as new developments in personalized immunization approaches.
Similar content being viewed by others
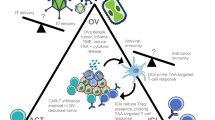


Many different types of vaccines have been tested in the treatment of malignant melanoma, but no single agent has yet shown a significant survival benefit in clinical trials. |
Oncolytic viruses, with their ability to selectively enter and replicate in cancer cells, are being used as a type of vaccine for the treatment of malignant melanoma, but only one such oncolytic virus, talimogene laherparepvec (T-VEC) has been approved by the US FDA for the treatment of advanced melanoma as a single agent. |
It is becoming evident that combinations of vaccines/oncolytic viral treatments with approaches that harness complementary aspects of the cancer immune response will likely be needed to improve the effectiveness of these therapies in malignant melanoma. |
1 Introduction
Cancer vaccination is not a new concept, and its application to malignancies antedates even our understanding of the immune system. Perhaps the first known instance dates as far back as 1891, with the development of the Coley toxin by Dr. William B. Coley, who inoculated patients who had solid tumors with toxins from streptococcal organisms and in some cases observed tumor regression [1, 2]. Furthermore, in the 1960s, Morton et al. [3] observed impressive regression of melanoma lesions after injection of Bacillus Calmette-Guérin (BCG) intratumorally, with responses noted at both injected and non-injected sites. Since these earlier attempts to treat cancer by enhancing anti-tumor immunity, our increasing understanding of both the innate and the adaptive immune system and their interaction with the tumor microenvironment has allowed us to make significant advances in developing cancer immunotherapies. However, it is becoming evident that rational combinations of immune agents targeting complementary or synergistic aspects in the cancer immunity cycle will likely be needed to maximize the efficacy of these therapies [4].
We review key basic concepts of tumor-induced immune suppression in malignant melanoma, the historical perspective of vaccine development in melanoma and advances in oncolytic viral therapies. We also discuss the rationale and emerging role for combination approaches with different immune checkpoint agents and the recent development of novel and personalized neoantigen vaccines.
2 Anti-Tumor Immune Responses and Tumor-Induced Immune Suppression
Extensive preclinical studies in numerous animal models have demonstrated that clinically effective anti-tumor immunity is largely due to a type I (Th1)-driven immune response [5]. This consists of a characteristic immune infiltrate with a high density of infiltrating T cells, including cluster of differentiation (CD)-8+ and memory CD4+ cells, and a low density of immune-suppressive cells such as T regulatory cells (Tregs), Th2 cells, tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs). This immunologically active infiltrate was demonstrated to be associated with improved survival and a decreased risk of relapse in a number of tumors, independent of stage, disease burden, and other risk factors [5].
An efficient immune response against a tumor involves complex interactions between the innate and adaptive immune systems that make up the cells comprising the “immune synapse” [6]. In this junction between T cells and antigen-presenting cells (APCs) and their targets, CD4+ T cells play a crucial role in cell-mediated immunity and inflammation by secretion of type 1 cytokines, such as interferon (IFN)-γ, which in turn activate CD8+ cytotoxic T cells through upregulation of granzyme B in cytolytic granules. These cytotoxic granules are then released, leading to effective killing of tumor cells [7]. In addition, natural killer (NK) cells target cells with low major histocompatibility complex (MHC) class 1 expression for elimination [8].
Other cells are involved in the suppressive immune response, including CD4+FoxP3+CD25+ regulatory T cells, and MDSCs [9]. Moreover, macrophages can mature into different phenotypes, with M1 macrophages leading to phagocytosis and release of IFN-γ, and M2 macrophages releasing inhibitory cytokines such as interleukin (IL)-4, IL-10, and transforming growth factor (TGF)-ß [10].
Furthermore, the immunological synapse is formed by a T-cell receptor (TCR) complex, which is controlled by both stimulatory and inhibitory receptors, and the first step in this interaction involves the presentation of self and non-self antigens to T cells by APCs, consisting primarily of dendritic cells (DCs). For effective CD8+ T-cell activation, the TCR binds a peptide tumor antigen/MHC complex, and the concurrent activation of other costimulatory signals leads to an effective T-cell response [11]. Known co-stimulatory signals such as CD28/B7-1 on the T cell and APC, respectively, and other agonist receptors such as GITR, OX40, and ICOS help enhance the stimulatory response; whereas inhibitory signals, including the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death-1 (PD-1), TIM3, and LAG3, are responsible for inhibition of T-cell function [12].
Tumors can evade immune recognition through different mechanisms: chronic tolerance, leading to anergy; by promoting an immune-tolerant environment through increased concentration of Tregs, MDSCs, and inhibitory cytokines; and by upregulation of immune checkpoint inhibitory molecules such as CTLA-4, PD-1 and PD-ligand 1 (PD-L1) on the APC [13]. These latter receptors promote T-cell exhaustion and inhibition of effector T-cell function. To bypass these resistance mechanisms, different approaches have been developed in attempts to enhance the immune response against melanoma, including vaccines, oncolytic viruses, and their combinations with checkpoint inhibitors.
3 Vaccine Strategies in Melanoma
The understanding of the function of antibodies in combating infection led to the idea that antitumor antibodies could be harnessed to treat cancer [14]. This new approach to cancer therapy led to the approval of the first therapeutic vaccine, Sipuleucel-T, against prostate cancer in 2010 [15]. Since then, vaccine therapies have been extensively studied in other cancer subtypes, with melanoma at the forefront of many of these efforts.
Vaccines can be characterized by the source of antigen used and the adjuvants or immune modulators administered with the antigen [16]. The ideal antigen source has been the subject of much debate, and antigens can vary from simple peptides or proteins, DNA/RNA, gangliosides, lysates, or even whole tumor cells. Each antigen type has its advantages and limitations, with a general rule being that smaller and simpler antigens such as peptides will be easier to prepare, store, administer, and monitor but tend to have a limited range of target population cells that are activated and thus a less diverse immune response [16]. More complex vaccines, on the other hand, have a broader and more varied antigen spectrum and are more generally relevant to a diverse patient population but are associated with challenges related to production, storage, administration, and monitoring of the immune response in patients [16]. Furthermore, the antigen source can be either autologous-derived or allogeneic (i.e., from a reserve of melanoma cell lines). The former may be advantageous in that the approach is more individualized with adequate antigen presentation of proteins unique to a particular patient. Unfortunately, autologous vaccines can frequently be complex and time consuming to derive and require tumor tissue samples for processing and production. In contrast, allogeneic vaccines can be prepared ahead of time and be readily available to patients, without a delay in treatment. Allogeneic vaccines also obviate the need for invasive procedures to obtain tumor tissue and can be preselected for antigens with high expression [16]. We now review the more relevant phase II/III trials of melanoma vaccines and discuss their results and limitations (Table 1).
3.1 Autologous Vaccines
3.1.1 HSPPC-96 (Vitespen)
Several autologous vaccine approaches have been tested in phase III clinical trials, including the vitespen vaccine, a heat shock protein gp96 peptide complex (HSPPC-96) vaccine derived from autologous tumor cells [17]. This protein is believed to play a role as a chaperone in antigen presentation through its interaction with CD91, a heat shock protein receptor on APCs, helping to facilitate MHC class I presentation and eventual effector T-cell responses [18]. The phase III trial enrolled 322 patients with stage IV melanoma across the globe and randomized patients in a 2:1 fashion to receive vitespen or physician’s choice (dacarbazine, temozolomide, IL-2, or complete resection). This trial failed to show any significant benefit in overall survival (OS) for the vaccine, although post hoc analyses showed a survival advantage in M1a and M1b patients receiving more than ten doses of the vaccine compared with those receiving fewer treatments. However, the efficacy is likely not clinically significant [17].
3.1.2 M-Vax
M-vax, a whole-cell autologous vaccine composed of the patient’s irradiated melanoma cells modified with the hapten dinitrophenyl (DNP) and mixed with BCG as an immune adjuvant was evaluated in the adjuvant setting in an earlier phase trial of 214 patients with stage III melanoma [19]. This phase II study reported an OS rate of 44% at 5 years, but the phase III trial was suspended because of difficulties with vaccine preparation, and results have never been published.
3.1.3 Dendritic Cell Vaccines
DCs are a powerful adjuvant in vaccine therapy through their role as APCs, and tumor lysates can be pulsed onto DCs to produce autologous vaccines [20,21,22]. Several trials are currently evaluating the safety and efficacy of these types of autologous vaccines.
3.1.3.1 DC-TC or CLBS20 or (NBS20/Eltrapuldencel-T)
The use of immune adjuvants in boosting the immune response has been evaluated in vaccine development, and novel DC-based products have shown promising results in early phase trials in advanced melanoma [23]. The vaccine CLBS20 (NBS20/eltrapuldencel-T) is a melanoma cell vaccine using the patient’s own monocyte-derived DCs loaded with antigens from irradiated tumor cells derived from autologous melanoma cell lines (DC-TC) thus making the vaccine patient specific. The vaccine is injected subcutaneously in a suspension with 500 μg of granulocyte-macrophage colony stimulating factor (GM-CSF) [24]. The vaccine showed very minimal toxicity and encouraging responses in two phase II trials of advanced melanoma [25]. The first trial was a single-arm study of 54 patients that revealed a 5-year survival of 50%. The second study was a randomized phase II trial of 42 patients receiving the vaccine versus injections of autologous irradiated tumor cells. Results of the trial reported a 2-year survival rate of 72% for the vaccine versus 31% for control patients (hazard ratio [HR] 0.27; p = 0.007) [25]. The phase III study, INTUS (NCT01875653), with projected accrual of 250 patients with metastatic melanoma, randomized patients 2:1 to receive either DC-TC or autologous mononuclear cells [25]. The study was terminated, but final results were not published as of late 2017.
3.2 Allogeneic Vaccines
3.2.1 Melacine
There are currently more phase III data available on allogeneic vaccine approaches in melanoma, and two major examples include Melacine and Canvaxin. Melacine is a lysate of two melanoma cell lines combined with mycobacterial components used as immunologic adjuvants [26]. A large phase III trial of 689 patients conducted by the Southwest Oncology Group (SWOG) randomized patients with intermediate-thickness node-negative disease to Melacine for 2 years versus observation in the adjuvant setting. Results from this trial (SWOG 9035) showed no statistically significant difference in disease-free survival (DFS) [26]. In an initially unplanned subgroup analysis of the same study, however, evaluation of human leukocyte antigen (HLA)-A2-positive and/or HLA-C3-positive patients revealed the 5-year DFS for vaccinated patients was 77 versus 64% (p = 0.004) in the observation group [27]. The trial was never confirmed because of a lack of interest by industry, but Melacine has been approved in Canada for stage IV melanoma based on quality-of-life measures.
3.2.2 Canvaxin
Canvaxin is composed of three irradiated melanoma cell lines (Canvaxin; CancerVax; Carlsbad, CA, USA) that were selected for antigen expression and had shown encouraging results in earlier phase trials with patients with melanoma [28]. Two phase III trials randomized patients to Canvaxin versus placebo, with both arms receiving BCG as an immune booster for the adjuvant treatment of melanoma after resection [29, 30]. About 1160 patients with stage III melanoma and 496 patients with stage IV melanoma rendered disease free with surgery were enrolled, but the trials were stopped early after interim analysis demonstrated no survival benefit. Of note, 5-year survival times for both control and vaccine arms were markedly longer than historical controls: 42.3% for stage IV and 63.4% for stage III patients. However, the control arms for both stages had minimally longer, although non-significant, survival than the vaccine-treated arms. The potential deleterious effect of multiple vaccinations leading to tolerance to tumor antigens was discussed as playing a possible role in these results. This landmark study represented an enormous setback for cancer vaccine development, and enthusiasm for the field may have waned after these results.
3.2.3 Ganglioside Vaccines
Gangliosides are glycosphingolipids found in cell membranes and are overexpressed in tissues arising from the neuroectodermal layer, including melanomas, sarcomas, neuroblastomas, and astrocytomas [31]. The GM2 ganglioside is a tumor-associated antigen (TAA) found in most melanomas but rarely in normal melanocytes and normal tissues [32]. Moreover, it can induce a humoral response, with immunoglobulin (Ig)-G and IgM production in patients with melanoma, making it a potential target for immunotherapy treatment in this disease [33]. Two large phase III trials have evaluated the efficacy of the GM2-KLH/QS-21 vaccine, composed of the ganglioside antigen GM2 combined with keyhole limpet hemocyanin (KLH) and the immune adjuvant QS-21, in the adjuvant setting. The intergroup trial ECOG 1694 enrolled 880 patients with stage IIB–III melanoma and randomized them to receive either the GM2 vaccine for 3 years or high-dose IFNα-2b (HDI) for 1 year with relapse-free survival (RFS) as a primary endpoint [34]. Unfortunately, the trial closed after interim analysis because of inferiority in the vaccine arm, with HDI demonstrating a statistically significant improvement in both RFS and OS.
The second trial, EORTC 18961, randomized only stage II patients (T3-4N0M0) after primary resection to receive 3 years of treatment with the GM2 vaccine versus observation [35]. This trial enrolled 1314 patients and closed at the second interim analysis, after 1.8 years of follow-up, after failure to meet its endpoint of improved RFS and even showing a detrimental effect on OS in the vaccine versus observation arms. Thus, a harmful effect from the vaccine was not ruled out, and speculation over whether repeated vaccinations could have played a part in this result was mentioned. Of note, at a longer 4-year follow-up analysis of the study, both RFS and OS were similar but clearly not better than placebo. In light of current aggregate results from large trials of adjuvant IFN in melanoma, the benefit shown by interferon versus GM2 vaccine in ECOG 1694 should not be extrapolated to infer an overall positive impact of interferon but may actually suggest a deleterious effect of the vaccine instead.
3.3 Peptide Vaccines
Protein or peptide fragments have become a common source of antigens for melanoma vaccines, with several ongoing and completed phase III melanoma studies [36,37,38]. These compounds are easy to produce, store, and administer, and—because of their narrow spectrum of possible immune targets—the immune response they elicit can also be monitored.
3.3.1 Gp 100
The gp100 vaccine, composed of HLA*A0201-restricted peptides from the melanoma antigen glycoprotein 100 (gp100), was the first peptide vaccine to show a clinical benefit. In a randomized phase III study, 185 patients with advanced or unresectable HLA*A0201-restricted melanoma were randomized to receive either gp100 vaccine plus incomplete Freund’s adjuvant followed by high-dose IL-2 or IL-2 alone [38]. The observed overall response rate (ORR) was 16 versus 6% (p = 0.03) and progression-free survival (PFS) was 2.2 versus 1.6 months (p = 0.008) in favor of the vaccine plus IL-2. The median OS was superior in the combination arm but did not reach statistical significance (17.8 vs. 11.1 months; p = 0.06).
Unfortunately, better results were not observed in a phase III trial of the gp100 vaccine when tested in combination with the anti-CTLA-4 drug ipilimumab compared with ipilimumab monotherapy [36]. In this trial, which led to the US FDA approval of ipilimumab, patients were treated with either the gp100 vaccine alone, the combination of ipilimumab plus gp100, or with ipilimumab alone. Contrary to what was expected, the gp100 vaccine did not impact survival, with median OS of about 10.0 months for the ipilimumab-based arms regardless of gp100 vaccination and 6.4 months for the gp100 alone arm. The lack of benefit in combination with ipilimumab compared with IL-2 was neither obvious nor explained. However, the trial was a landmark in immunotherapy and led to the approval of ipilimumab. This opened the door for immune checkpoint-based therapy in melanoma.
3.3.2 Other Peptide Vaccines
A multiepitope peptide vaccine (PV), comprised of tyrosinase, gp100, and MART-1 peptides recognized only by HLA-A2-positive recipients, was studied in a phase III trial in the adjuvant setting for stage III and IV melanoma. Patients were grouped based on their HLA-A2 status [37]. HLA-A2-positive patients were treated with either PV alone, GM-CSF alone, or a combination of the PV plus GM-CSF or placebo, and HLA-A2-negative patients were treated with either GM-CSF or placebo. This trial enrolled 815 patients, and results were negative for all arms, including no significant difference in OS or RFS for either PV or GM-CSF, alone or in combination. Although GM-CSF has shown an ability to increase the numbers of monocyte/macrophages and their antitumor activity in retrospective and in vitro studies, this large prospective study further demonstrated the lack of benefit of GM-CSF as an adjuvant treatment for melanoma.
3.4 Limitations of Current Vaccine Therapy Strategies in Melanoma
As noted, single-agent vaccine-based immunotherapies have yielded a great number of disappointing results in the past, with many factors playing a role in their limited clinical success. Some reasons include clinical trial design issues and lack of demonstrable efficacy in the chosen primary endpoints for these trials. In addition, issues with development of immunological tolerance and evasion by the tumor, exhaustion of T-cell populations and the choice of patient populations studied in these trials (patients with advanced disease and a more compromised immunologic system) all likely played a role in the overwhelming failure of many of these studies. Also of note, while immune monitoring in the peripheral blood may be technically feasible with these vaccines, the chosen immunologic markers in past trials have not necessarily correlated with clinical responses, suggesting that other biomarkers are needed to measure both clinical and immune responses to these therapies [39].
Some of the limitations of PVs, for instance, are that they normally contain a single epitope, offering a limited spectrum of targets for the immune system, and must also match their patients in HLA compatibility. In addition, many melanoma PVs use HLA-A2 restriction, which is the most common allele in patients from North America but still may limit patient eligibility. Therefore, there is reason to suggest that greater optimization of peptide/protein/cellular vaccines, along with other strategies to render the tumor microenvironment more favorable to T-cell activation, are needed before further development of peptide and other cancer vaccine therapies.
Another important issue has been the availability of TAAs. Since these antigens can frequently be expressed in both cancer and normal cells, there is a potential for autoimmunity and, hence, collateral damage to normal tissues through increased levels of circulating T cells specific to that antigen [40]. This highlights the need to identify target antigens that are uniquely expressed in tumor and not normal cells. Fortunately, it is likely that these older approaches to selecting cancer antigens will change with the growing interest in the study of “neoantigens” [41]. These are peptides produced by somatic non-synonymous mutations in cancer cells, which are expressed only by tumor and not normal cells and can now be identified easily through genetic sequencing of a patient’s tumor and may be eventually targeted immunologically [41]. This is discussed in further detail in Sect. 6.1.
4 Oncoviral Therapy in Melanoma
Oncolytic viruses can replicate inside tumor cells and activate the immune system to fight cancer, and oncoviral therapy can employ either native or attenuated live viruses [42]. Their mode of action and antitumor activity is believed to stem from two distinct mechanisms. The first is the selective entry of the virus and replication inside the tumor cell, which in turn leads to direct lysis of tumor and release of viral particles into the milieu, resulting in expansion of the lytic effect and further tumor destruction. The second mechanism involves the induction of a host immune response triggered by the release of tumor antigens and other cytokines into the microenvironment [42]. In addition, emerging data show evidence of tumor-specific immunity, which may amplify tumor destruction by recruitment of other cells not infected by the virus [42].
Viruses that can selectively enter and replicate in cancer cells have been evaluated as candidates for oncolytic viral therapies, including herpesvirus, poxvirus, picornavirus, adenovirus, paramyxovirus, parvovirus, reovirus, Newcastle disease virus (NDV), and rhabdovirus [42, 43]. Although these viruses can enter both healthy and cancer cells, there are critical factors in normal cell responses to stress and homeostasis that are abnormal in cancer cells, making it difficult for tumor cells to identify and clear these viruses. For instance, protein kinase R (PKR), a critical molecule in clearing intracellular infections, may be absent or attenuated in cancer cells, thereby permitting viral replication [42, 43]. We review the main oncolytic viruses that have been evaluated in clinical trials in melanoma thus far (Table 2).
4.1 Herpes Simplex Viruses
The herpes simplex virus, type 1 (HSV-1) is an alpha-herpes virus that has been well studied in human pathogenesis and has been evaluated in oncolytic viral therapy as it can enter a wide variety of host cells [44, 45]. Replication-competent HSV-1 vectors have been mutated in genes that affect viral replication, neuropathogenicity, and immune evasiveness. They have been developed and tested for their safety and efficacy in preclinical models, and examples of two of these agents that are currently FDA approved or undergoing clinical development in melanoma are talimogene laherparepvec (T-VEC) and HF10, respectively [45].
4.1.1 T-VEC
Over six decades after the discovery of the oncolytic potential of viruses, a randomized clinical trial using an oncolytic virus was able to demonstrate clinical benefit in patients with cancer. This came with the development of an attenuated HSV-1-engineered oncolytic virus named T-VEC. This compound selectively replicates in tumors, secretes human GM-CSF, and is the first oncolytic immunotherapy agent approved by the FDA for cancer treatment, specifically in melanoma [46, 47]. The virus is modified by the deletion of two genes (ICP34.5 and ICP 47), which play a part in the neurovirulence of the pathogen and the suppression of antigen presentation, respectively. The addition of the GM-CSF gene leads to local production of GM-CSF, which acts in recruitment of APCs into the tumor microenvironment [47]. The phase III OPTiM trial randomized 436 patients with injectable but unresectable melanoma to either intratumoral T-VEC or subcutaneous GM-CSF in a 2:1 ratio, with a primary endpoint of durable response rate (DRR) of at least 6 months [48]. The trial met its primary endpoint, with a significantly better DRR reported in the T-VEC arm of 16.3 versus 2.1% in the GM-CSF arm. The ORR was also higher for T-VEC (26.4 vs. 5.7% for GM-CSF). In addition, a survival advantage was also observed in the T-VEC arm (23.3 vs. 18.9 months; HR 0.79; 95% confidence interval [CI] 0.62–1.00; p = 0.051). The drug was very well tolerated, with the most frequent adverse events (AEs) being fatigue, chills, and pyrexia. Grade 3 and 4 cellulitis was seen in <2% of patients. The efficacy and duration of response were both greater in patients without visceral metastases and in those receiving the agent as first-line therapy. The former may be explained by the lack of a robust systemic immune response from an otherwise local treatment, although an impressive reduction of at least 50% in size was observed in up to 15% of non-injected, visceral lesions in the T-VEC-injected cohort. These data indicate that T-VEC is beneficial in select melanoma patients and lends support to systemic immune checkpoint combinations with T-VEC, which are currently underway and are discussed in Sect. 5.1.
4.1.2 HF10
TBI-1401 (HF10) is a spontaneously mutated HSV-1 and a potent oncolytic agent with its genome modified at two loci, as well as loss of UL-56, which attenuates viral neuroinvasiveness [49]. A phase I study of HF10 in patients with recurrent breast cancer reported tumor shrinkage [49]. Phase I and II trials (NCT02428036 and NCT02272855) examining HF10 (both as a single agent and in combination with ipilimumab) in patients with melanoma are ongoing.
4.2 Poxviruses
Poxviruses are large double-stranded DNA viruses, with vaccinia virus serving as the archetypal poxvirus used in the smallpox vaccine administered to millions of people worldwide in the past. Pexa-vac (JX-594) is a targeted recombinant oncolytic poxvirus encoding GM-CSF and has been tested in two phase I clinical trials in stage IV melanoma [50, 51]. Both were extremely small trials, including seven and ten patients, respectively. They observed two complete responses (CRs), two partial responses (PRs), and several mixed responses as well as progression of disease (PD). In addition, JX-594 was found to replicate successfully and achieve lysis of tumor cells when blood samples and biopsies were analyzed for activity (GM-CSF and β-galactosidase expression), viral replication, and induction of systemic immunity [51].
Another recombinant vaccinia virus that was tested in a phase I trial in patients with metastatic melanoma uses a virus expressing the costimulatory molecule B7.1 (CD80) [52]. Of 12 patients evaluated in the study, one had a PR and two experienced stable disease (SD). Both systemic and local immunity were observed with treatment, measured by T-cell responses. In another phase I study by Kaufman et al. [53], a different vaccinia virus expressing three costimulatory molecules, B7.1, ICAM-1, and LFA-3 (rV-TRICOM), was investigated in 13 patients with metastatic melanoma [53]. The trial observed a 31% ORR, including one CR. An inverse association was observed in anti-vaccinia antibody and anti-vaccinia T-cell responses. These studies confirm the safety and tolerability of intratumoral injection of vaccinia viruses with encouraging results, but the small size of the trials means confirmation in much larger cohorts of patients is required. A phase I study is accruing patients for treatment with GL-ONC1, a vaccinia virus in solid tumor patients before surgery (NCT02714374).
4.3 Coxsackievirus
Coxsackievirus A21 is a naturally occurring “common cold” enterovirus observed to express the viral capsid receptors for intercellular adhesion molecule (ICAM-1) and decay-accelerating factor (DAF). Both of these molecules are also endogenously overexpressed on the surface of melanoma cells, which facilitates viral entry into the melanoma cell, leading to tumor lysis [54]. CAVATAK (CVA21), a genetically unmodified wild-type human enteroviral therapy, was studied in a phase I trial and demonstrated acceptable safety [55]. This was followed by the phase II CALM study in 57 patients with advanced melanoma who were evaluable for response, with the primary endpoint of immune-related PFS (irPFS) at 6 months of treatment [56]. An interim efficacy analysis of the first 21 patients enrolled showed that treatment was well tolerated and more than three objective responses had been achieved. The study was completed in June 2015, and an extension of the trial presented at the Society for Immunotherapy of Cancer (SITC) meeting reported that the original study achieved its primary endpoint, with 38.6% durable responses noted [57]. A 13-patient extension of the trial showed an increase in effector CD8+ T cells and PD-L1+ expression levels in the tissue microenvironment, and RNA NanoString confirmed a Th1-gene shift in the immune cell infiltrate. In addition, the extension study showed an ability of the virus to resensitize previously resistant lesions to immune checkpoint blockade, supporting the development of combinations with these therapies. A clinical trial combining CVA21 with anti-CTLA4 is underway and discussed in Sect. 5.
4.4 Reovirus
Reovirus is a nonenveloped double-stranded RNA virus that causes mild infections in humans, which are normally limited to the gastrointestinal and upper respiratory tracts [58]. The virus has shown a specificity for replication in cells with an activated Ras signaling pathway, making it an appealing candidate for treatment in melanoma where this pathway is almost uniformly activated [59]. In a phase II trial of Reovirus Serotype 3-Dearing Strain (Reolysin), the therapy was administered intravenously in 21 patients with metastatic melanoma [58]. The trial did not observe any objective responses, although treatment was very well tolerated without dose reductions, and viral replication was noted in 2 of 13 patients analyzed with tumor biopsies. This study provided the basis for a phase II combination with paclitaxel and carboplatin in patients with melanoma (NCT00984464). The trial has been completed, but results are still pending.
4.5 Limitations of Vaccine/Oncolytic Viral Approaches as Monotherapy
As evidenced by a plethora of negative trials, it seems that most vaccines and oncoviral approaches have somewhat limited activity when used as single agents in melanoma and other cancers. A major issue that has been identified is the type of vaccine antigen selected for therapy. For instance, instead of selecting from a pool of existing candidate protein antigens, one recent approach to choosing antigens could be the evaluation of somatic mutations found throughout the genome of cancer cells, which would allow the identification of mutation-derived neoantigens capable of being processed and presented for vaccine development [60]. Neoantigen-based vaccines are currently undergoing clinical development and are discussed in Sect. 6.1 [41]. Another challenge in vaccine development is the consideration of the exact nature of the immune adjuvants to boost immunologic responses. For instance, DC vaccines, despite showing variable results in clinical trials, remain an important part of the immunologic response that is only now being better understood [23].
One of the crucial challenges for further development of immunotherapy agents is identifying markers predictive of clinical benefit. Immune markers used in trials thus far have not been reliable surrogates of clinical response when expanded to larger studies, specifically in melanoma. For instance, in the case of the PV gp100, preclinical studies showed a potentiation of effect in combination with anti-CTLA-4, but this did not translate to efficacy in the phase III trial [36]. Another instance is the case of GM-CSF, which is known to mount immunologic responses, but several trials have failed to show improvements in clinical outcomes [61].
Therefore, development of more reliable markers is needed, and it may be of more value to assess the tumor infiltrate itself for its immunologic phenotype or look at the peripheral/systemic immune infiltrate that may be associated with circulating tumor cells, for instance. Regardless of the choice of antigen, oncoviral agent, or immune adjuvant, it is becoming clear that strategies in combination with other treatments such as immune checkpoint inhibitors, cytotoxic chemotherapy, radiation, and targeted therapies are more promising therapeutic strategies, and these are being actively studied.
5 Combination Approaches with Immune Checkpoints and Other Agents
Given the relative safety and tolerability of these immune agents, as well as the ease of administration and lower likelihood of competing toxicities, approaches involving combinations of vaccines and oncolytic viruses with other treatments are a potential strategy to improve response to melanoma therapies. The rationale for these approaches would be to eliminate residual tumor by potentiating an anti-tumor immune response, e.g., in the adjuvant setting, or perhaps prior to standard metastatic therapies by releasing neoantigens into the milieu and potentially rendering other immunologic agents more effective. In the case of immune checkpoints for instance, IFNγ is known to upregulate PD-L1 expression on the surface of tumor cells, and preclinical data show oncolytic viruses can often increase levels of IFNγ in the local stroma, making this combination particularly attractive [62]. We now review some of the current available trial data with combinations of vaccines/oncoviral therapies and other immunologic agents in the treatment of melanoma (Tables 3 and 4).
5.1 T-VEC Combinations
In a phase Ib/II study of the anti-CTLA-4 drug ipilimumab in combination with T-VEC for patients with previously untreated stage IIIB–IV melanoma, 19 patients were included in the safety analysis and no dose-limiting toxicities (DLTs) were observed [63]. The reported ORR was 50%, with 44% durable responses lasting at least 6 months and 18-month OS of 67%. Results showed 26.3% of patients experienced grade 3 or 4 AEs, similar to single-agent ipilimumab, but only 15.8% were attributed to T-VEC, and no new emerging toxicities were observed. Although patient numbers are still very small, these are very encouraging results given that the single-agent response rate with ipilimumab is around 20% and that of T-VEC about 26%, and no competing or added toxicities were observed with the combination [36, 48]. Moreover, increased levels of activated CD8+ T cells were found in patients with confirmed responses after T-VEC alone, suggesting a role for T-VEC in inducing a systemic immune response. The large randomized phase II portion of the trial presented at the American Society of Clinical Oncology (ASCO) 2017 annual meeting by Chesney et al. [64] reported results for 198 patients randomized 1:1 to either T-VEC plus ipilimumab or ipilimumab monotherapy. In this part of the trial, the ORR was 38.8% for the combination and 18% for ipilimumab alone (p = 0.002; odds ratio [OR] 2.9), with a median follow-up time of 68 weeks for the combination and 58 weeks for the single-agent arm. About 89 versus 83% of patients in the combination versus single-agent arms, respectively, showed continued response. The most common reported AEs were fatigue in 59% receiving combination versus 42% of patients receiving the single agent, chills (53 vs. 3%), and diarrhea (42 vs. 35%). Grade 3 or higher treatment-related AEs occurred in 28% of those receiving the combination and 18% receiving the single agent. Three deaths were reported in the combination arm, all unrelated to treatment: one from myocardial infarction and two from disease progression. The randomized phase II portion of this study met its primary endpoint, confirming the improvement in response with the addition of T-VEC to anti-CTLA-4 therapy in melanoma, and lending further support to the synergistic combination of an oncolytic virus and immune checkpoint inhibitor.
MASTERKEY-265 (NCT02263508) is a phase Ib/III study evaluating T-VEC in combination with the anti-PD1 agent pembrolizumab in unresected and advanced melanoma and is currently accruing patients [65]. An early efficacy analysis of 16 evaluable patients in another study reported that the combination seemed tolerable, with an unconfirmed ORR of 56% [65]. The large phase III trial is still accruing but should provide a definitive answer to whether T-VEC can enhance the efficacy of a PD-1 checkpoint inhibitor. Therefore, the combination of T-VEC and immune checkpoint inhibitors in patients with advanced melanoma seems promising, and larger trials will hopefully confirm their efficacy and lead to further development.
5.2 Coxsackievirus Combinations
CAVATAK is being studied in the phase Ib MITCI trial in combination with ipilimumab in 26 patients with advanced melanoma [66]. An interim report of 16 patients showed no reported DLTs and only one grade 3 AE of fatigue, attributed to ipilimumab. There were four confirmed objective responses out of seven evaluable patients, with activity noted in both injected and non-injected lesions [66].
5.3 Newcastle Disease Virus Combinations
NDV is a paramyxoma avian virus causing deadly infections in poultry and other bird species [67]. Its selectivity for replication in human malignant cells was confirmed and believed to be secondary to V protein restriction by the host cell as well as production of inflammatory cytokines by the virus [68]. The virus can also induce both innate and adaptive immune responses, and recombinant strains are being developed for the treatment of human malignancies [67]. One clinical trial showed the vaccine to be active after administration to 83 patients with stage II melanoma after complete resection, with a reported survival of 60% after 10 years of follow-up [68]. Interestingly, preclinical data demonstrated NDV administration was able to overcome resistance to CTLA-4 checkpoint blockade therapy in B16 melanoma cell lines, supporting the development of this agent in the setting of resistance to checkpoint blockade or in combination with these agents [69].
5.4 Vaccine Combinations
5.4.1 Dorgenmeltucel-L (HyperAcute Melanoma)
Other combinations of vaccines that are also further along in clinical trial development include dorgenmeltucel-L, which consists of genetically modified allogeneic melanoma cells engineered to express the murine carbohydrate α(1,3)Gal, primarily responsible for the hyperacute rejection of foreign tissue and for which humans have an inherent pre-existing immunity [70]. Phase I and II studies of the vaccine have shown it to be safe and well tolerated as a single agent and in combination with pegylated IFN. Activation of the host immune system was reported, measured through development of autoimmune antibodies in all evaluable patients; four patients developed vitiligo correlating with either CR or durable response after complete resection. In an ongoing randomized phase IIb study, the vaccine is being tested in combination with either ipilimumab, nivolumab, or pembrolizumab in patients with metastatic melanoma (NCT02054520).
5.4.2 Vigil or (EATC-Mel)
Another vaccine in early clinical development is the engineered autologous tumor cell (EATC-Mel) or Vigil vaccine, previously called FANG. This vaccine consists of irradiated autologous tumor transfected with a dual DNA plasmid modified to express GM-CSF and bi-shRNA furin components. It was observed that knockdown of furin downregulates both downstream TGFβ1 and TGFβ2 expression, and the vaccine showed tumor-specific systemic immune response and was well tolerated in a pilot trial of 12 patients with Ewing’s sarcoma [71]. It is now being studied in combination with pembrolizumab in patients with advanced melanoma (NCT02574533).
5.4.3 Astuprotimut-R or (recMAGE-A3 + AS15 ASCI)
Other vaccines, such as astuprotimut-R have now commenced phase III development. This is a recombinant form of human melanoma antigen A3 (MAGE-A3), a protein originally discovered in melanoma and commonly found in metastatic cancer, combined with a proprietary adjuvant composed of the immunostimulant AS15 [72]. In contrast to other peptide vaccines, recMAGE-A3 plus AS15 ASCI (Antigen-Specific Cancer Immunotherapeutic) can induce both MHC I and II responses in immune cells and may produce a more potent immune response [72]. In a phase I study of the vaccine testing two immune adjuvants (AS15 and AS02B) in metastatic melanoma in the first-line setting, 75 MAGE-A3-positive patients were treated. Tolerance to therapy was acceptable, and four patients experienced objective responses in the AS15 arm, including three CRs [72]. Moreover, anti-MAGE-A3 antibodies were three times higher in the AS15 group, with a 6-month PFS of 25 versus 14% and median OS of 33 versus 19.9 months. The vaccine has undergone further testing in melanoma, although the phase III trials in melanoma and lung cancer noted a lack of efficacy (NCT00796445), and other studies are ongoing, including a phase II trial in combination with high-dose IL-2 (NCT01266603).
6 Future Directions and Closing Remarks
6.1 Neoantigen or Neo-epitope Vaccines
To obviate the known issues of autoimmunity with TAAs present in both tumor and healthy tissue, antigens arising solely through tumor-specific mutations needed to be isolated for ideal vaccine development. These neoantigens or neo-epitopes are peptides specific to the tumor in question and lead to greater immune responses and antitumor activity [73]. Fortunately, the greater availability of massive parallel-sequencing techniques has now rendered the identification of these tumor-specific peptides more feasible. In addition, the fine tuning of algorithms predicting which mutated antigens will bind to the patient’s HLA proteins with the highest affinity has made it easier to select these antigens or epitopes for vaccine development. These rapid advances in technology have made it possible to design a vaccine that is unique to each patient and one that will potentially elicit the greatest anti-tumor immune response.
Over the last 2 years, a robust amount of data now shows that neoantigens derived from the mutanome of the cancer genome are strong candidates for vaccine development, driving the field toward the development of fully individualized vaccines. Multiple studies are underway using single-agent neoantigen-based vaccines as well as combinations with immune checkpoint inhibitors, with the first published study reported in 2015 [74]. In this study of a DC vaccine with high affinity to patient-specific tumor-derived mutant peptides, three patients with advanced melanoma were treated, and all patients developed enhanced T-cell immunity specific to their neoantigen repertoire. Clinical activity could not be assessed because of a lack of measurable disease [74].
Another neoantigen-based vaccine undergoing clinical investigation is IVAC Mutanome, a synthetic RNA vaccine produced by a process integrating next-generation sequencing based cancer mutanome mapping and target antigen selection into a process referred to as MERIT (mutanome engineered RNA immunotherapy) [75, 76]. The vaccine is engineered by first identifying the non-synonymous mutations in each patient’s tumor via whole exome and RNA sequencing of tissue and blood, which is then followed by selection of ten mutations per patient based on algorithms predicting high affinity to both autologous HLA class II and class I binding. The ten selected mutations are then engineered into synthetic RNAs. This vaccine has shown confirmed activity in preclinical studies, demonstrating uptake by lymph nodes and translation by DCs of the target peptides contained in the RNA [75]. Results from the phase I first-in-human testing in melanoma (NCT02035956) have now been published [77]. In total, 13 patients with stage III and IV melanoma were treated with 20 doses of the vaccine without serious adverse reactions and with the majority mounting exclusively CD4+ T-cell responses. Eight of the 13 patients without lesions at the start of therapy remained free of disease throughout the follow-up period of 12–23 months. The other five patients had relapses soon after enrollment. Of these five patients, one had CR with only vaccine therapy, another had PR followed by progression and death, and yet another initially had SD, then quick progression, followed by a CR and durable response after treatment with PD-1 therapy.
Investigators from the Dana-Farber institute recently reported a phase I study (NCT01970358) of a neoantigen vaccine selected by algorithms predicting HLA class I binding and targeting up to 20 neoantigens per patient [78]. Six patients with previously untreated stage III and IV melanoma after surgical resection received seven vaccine doses. After 25 months of follow-up, four demonstrated no disease recurrence (all had stage III melanoma), whereas the remaining two patients (with stage IV melanoma) had recurrence of their disease. However, these two patients developed durable CRs after subsequent treatment with anti-PD-1 therapy. As seen with the prior neoantigen vaccine, an overwhelmingly greater CD4+ T-cell response was elicited, and samples collected before and after treatment with PD-1 therapy demonstrated an expansion of the repertoire of neoantigen-specific T cells. Yet another personalized vaccine is NEO-PV-01, a synthetic peptide(s) neoantigen vaccine undergoing phase I testing in combination with nivolumab and the investigational adjuvant Poly-ICLC (Hiltonol) for patients with advanced melanoma and other solid tumors (NCT02897765). These promising results support further development of this technology in larger randomized trials of single agents and other immunomodulatory combinations with minimal added toxicity.
6.2 Concluding Remarks
Although the field of tumor immunology is not new, it has faced many complex challenges over the last century. As we increase our knowledge of the complexities of the immune system and its interaction with tumor cells and their microenvironment we can gain new insights into selecting better targets for cancer vaccine development and further refining our trial design strategies to identify predictive biomarkers and optimize clinical endpoints. Furthermore, combinations of vaccine/oncoviral approaches with immune checkpoint inhibitors and other systemic therapies seem promising and are currently undergoing clinical investigation to potentially become treatment options for patient care in the near future. It is likely that only through combination therapy will vaccines find their place in the treatment of melanoma. Meanwhile, with the widespread availability of whole-exome sequencing technology, fully personalized vaccine approaches are finally becoming feasible and show encouraging early results.
References
Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. Clin Orthop Relat Res. 1893;1991(262):3–11.
McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J. 2006;26:154–8.
Morton DL, et al. BCG immunotherapy of malignant melanoma: summary of a seven-year experience. Ann Surg. 1974;180(4):635–43.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10.
Mittal D, et al. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16–25.
Dustin ML. The immunological synapse. Cancer Immunol Res. 2014;2(11):1023–33.
Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ. 2010;17(4):616–23.
Gras Navarro A, Bjorklund AT, Chekenya M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front Immunol. 2015;6:202.
Savage PA, Leventhal DS, Malchow S. Shaping the repertoire of tumor-infiltrating effector and regulatory T cells. Immunol Rev. 2014;259(1):245–58.
Laoui D, et al. Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front Immunol. 2014;5:489.
Dustin ML. Cell adhesion molecules and actin cytoskeleton at immune synapses and kinapses. Curr Opin Cell Biol. 2007;19(5):529–33.
Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–42.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–9.
Schwartz RS. Paul Ehrlich’s magic bullets. N Engl J Med. 2004;350(11):1079–80.
Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22.
Ozao-Choy J, Lee DJ, Faries MB. Melanoma vaccines: mixed past, promising future. Surg Clin North Am. 2014;94(5):1017–30 (viii).
Testori A, et al. Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician’s choice of treatment for stage IV melanoma: the C-100-21 Study Group. J Clin Oncol. 2008;26(6):955–62.
Tosti G, et al. HSPPC-96 vaccine in metastatic melanoma patients: from the state of the art to a possible future. Expert Rev Vaccines. 2009;8(11):1513–26.
Berd D, et al. Immunopharmacologic analysis of an autologous, hapten-modified human melanoma vaccine. J Clin Oncol. 2004;22(3):403–15.
Dillman R, et al. Phase I/II trial of melanoma patient-specific vaccine of proliferating autologous tumor cells, dendritic cells, and GM-CSF: planned interim analysis. Cancer Biother Radiopharm. 2004;19(5):658–65.
Dillman RO, Selvan SR, Schiltz PM. Patient-specific dendritic-cell vaccines for metastatic melanoma. N Engl J Med. 2006;355(11):1179–81.
Dillman RO, et al. Phase II trial of dendritic cells loaded with antigens from self-renewing, proliferating autologous tumor cells as patient-specific antitumor vaccines in patients with metastatic melanoma: final report. Cancer Biother Radiopharm. 2009;24(3):311–9.
Zhang S, Wang Q, Miao B. Review: dendritic cell-based vaccine in the treatment of patients with advanced melanoma. Cancer Biother Radiopharm. 2007;22(4):501–7.
Javed A, Sato S, Sato T. Autologous melanoma cell vaccine using monocyte-derived dendritic cells (NBS20/eltrapuldencel-T). Future Oncol. 2016;12(6):751–62.
Dillman RO, BM Langford JE et al. Phase III multicenter trial of eltrapuldencel-T: Autologous dendritic cells loaded with irradiated autologous tumor cells (DC-TC) in granulocyte-macrophage colony stimulating factor (GM-CSF) in patients with metastatic melanoma (INTUS trial). In: ASCO. 2015. Chicago, IL: J Clin Oncol 33, 2015 (suppl; abstr TPS9082).
Sondak VK, et al. Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: overall results of a randomized trial of the Southwest Oncology Group. J Clin Oncol. 2002;20(8):2058–66.
Sosman JA, et al. Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: impact of HLA class I antigen expression on outcome. J Clin Oncol. 2002;20(8):2067–75.
Morton DL, et al. Prolonged survival of patients receiving active immunotherapy with Canvaxin therapeutic polyvalent vaccine after complete resection of melanoma metastatic to regional lymph nodes. Ann Surg. 2002;236(4):438–48 (discussion 448-9).
Morton DMN, Thompson J, et al. An international, randomized, doubleblind, phase 3 study of the specific active immunotherapy agent, Onamelatucel-L (Canvaxin), compared to placebo as post-surgical adjuvant in AJCC stage IV melanoma. 2006. Ann Surg Oncol. 2006;13(Suppl 2):5s.
Morton DLMN, Thompson JF, et al. An international, randomized, phase III trial of bacillus Calmette-Guerin (BCG) plus allogeneic melanoma vaccine (MCV) or placebo after compete resection of melanoma metastatic to regional or distant sites. J Clin Oncol. 2007;25(18S):8508 (abstract).
Livingston P. Ganglioside vaccines with emphasis on GM2. Semin Oncol. 1998;25(6):636–45.
Tsuchida T, et al. Gangliosides of human melanoma. J Natl Cancer Inst. 1987;78(1):45–54.
Livingston PO, et al. Vaccines containing purified GM2 ganglioside elicit GM2 antibodies in melanoma patients. Proc Natl Acad Sci U S A. 1987;84(9):2911–5.
Kirkwood JM, et al. High-dose interferon alfa-2b significantly prolongs relapse-free and overall survival compared with the GM2-KLH/QS-21 vaccine in patients with resected stage IIB-III melanoma: results of intergroup trial E1694/S9512/C509801. J Clin Oncol. 2001;19(9):2370–80.
Eggermont AM, et al. Adjuvant ganglioside GM2-KLH/QS-21 vaccination versus observation after resection of primary tumor > 1.5 mm in patients with stage II melanoma: results of the EORTC 18961 randomized phase III trial. J Clin Oncol. 2013;31(30):3831–7.
Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Lawson DH, et al. Randomized, placebo-controlled, phase III trial of yeast-derived granulocyte-macrophage colony-stimulating factor (GM-CSF) versus peptide vaccination versus GM-CSF plus peptide vaccination versus placebo in patients with no evidence of disease after complete surgical resection of locally advanced and/or stage IV melanoma: a trial of the Eastern Cooperative Oncology Group-American College of Radiology Imaging Network Cancer Research Group (E4697). J Clin Oncol. 2015;33(34):4066–76.
Schwartzentruber DJ, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364(22):2119–27.
Rosenberg SA, et al. Impact of cytokine administration on the generation of antitumor reactivity in patients with metastatic melanoma receiving a peptide vaccine. J Immunol. 1999;163(3):1690–5.
Amos SM, et al. Autoimmunity associated with immunotherapy of cancer. Blood. 2011;118(3):499–509.
Katsnelson A. Mutations as munitions: Neoantigen vaccines get a closer look as cancer treatment. Nat Med. 2016;22(2):122–4.
Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–62.
Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res. 2016;22(5):1048–54.
Dharmadhikari N, Mehnert JM, Kaufman HL. Oncolytic virus immunotherapy for melanoma. Curr Treat Options Oncol. 2015;16(3):326.
Varghese S, Rabkin SD. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 2002;9(12):967–78.
Hu JC, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12(22):6737–47.
Liu BL, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303.
Andtbacka RH, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–8.
Sahin TT, et al. Impact of novel oncolytic virus HF10 on cellular components of the tumor microenviroment in patients with recurrent breast cancer. Cancer Gene Ther. 2012;19(4):229–37.
Hwang TH, et al. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol Ther. 2011;19(10):1913–22.
Mastrangelo MJ, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6(5):409–22.
Kaufman HL, et al. Targeting the local tumor microenvironment with vaccinia virus expressing B7.1 for the treatment of melanoma. J Clin Invest. 2005;115(7):1903–12.
Kaufman HL, et al. Local delivery of vaccinia virus expressing multiple costimulatory molecules for the treatment of established tumors. Hum Gene Ther. 2006;17(2):239–44.
Shafren DR, et al. Coxsackievirus A21 binds to decay-accelerating factor but requires intercellular adhesion molecule 1 for cell entry. J Virol. 1997;71(6):4736–43.
Shafren DR, et al. Systemic therapy of malignant human melanoma tumors by a common cold-producing enterovirus, coxsackievirus a21. Clin Cancer Res. 2004;10(1 Pt 1):53–60.
Andtbacka RHIH, Daniels GA, Spitler LE, Lutzky J, Hallmeyer S, et al. CALM study: a phase II study of intratumoral coxsackievirus A21 in patients with stage IIIc and stage IV malignant melanoma. In: ASCO. 2013. Chicago, IL: J Clin Oncol. 2013;31 (abstr TPS3128).
Andtbacka RHCB, Hallmeyer S, et al. Phase II calm extension study: Coxsackievirus A21 delivered intratumorally to patients with advanced melanoma induces immune-cell infiltration in the tumor microenvironment. in SITC. J Immunother Cancer. 2015;3(Suppl 2):P343.
Galanis E, et al. Phase II trial of intravenous administration of Reolysin((R)) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol Ther. 2012;20(10):1998–2003.
Strong JE, et al. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17(12):3351–62.
Snyder A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99.
Hoeller C, et al. Systematic review of the use of granulocyte-macrophage colony-stimulating factor in patients with advanced melanoma. Cancer Immunol Immunother. 2016;65(9):1015–34.
Bellucci R, et al. Interferon-gamma-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015;4(6):e1008824.
Puzanov I, et al. Talimogene Laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol. 2016;34(22):2619–26.
Chesney JAPI, Ross MI et al. Primary results from a randomized (1:1), open-label phase II study of talimogene laherparepvec (T) and ipilimumab (I) vs I alone in unresected stage IIIB- IV melanoma. In: ASCO Annual Meeting. 2017. Chicago, IL: J Clin Oncol 35, no. 15_suppl (May 2017):9509–9509.
Long GVDR, Ribas A. Primary analysis of MASTERKEY-265 phase 1b study of talimogene laherparepvec (T-VEC) and pembrolizumab (pembro) for unresectable stage IIIB-IV melanoma. in Society for Melanoma Congress. 2015. San Francisco, CA: J Immunother. Cancer. 2015;3(Suppl 2):P181.
Curti BRJ, Faries M et al. The MITCI (phase Ib) study: a novel immunotherapy combination of coxsackievirus A21 and ipilimumab in patients with advanced melanoma. In: ESMO. 2016. Copenhagen, Denmark: Annals of Oncology 2016;27 (Supplement 6); vi359–vi378.
Zhao L, Liu H. Newcastle disease virus: a promising agent for tumour immunotherapy. Clin Exp Pharmacol Physiol. 2012;39(8):725–30.
Lam HY, et al. Safety and clinical usage of newcastle disease virus in cancer therapy. J Biomed Biotechnol. 2011;2011:718710.
Zamarin D, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6(226):226ra32.
Riker AIRG, Vahanian NN et al. NLG-0304: A phase 2B study of ipilimumab with or without dorgenmeltucel-L (HyperAcute-Melanoma) immunotherapy for patients with stage IV melanoma. In: ASCO. 2014. Chicago, IL: J Clin Oncol 2014;32:5s (suppl; abstr TPS9115).
Ghisoli MBA, Schneider R et al. Pilot trial of vigil immunotherapy in Ewing’s sarcoma. in ASCO. Chicago, IL: J Clin Oncol 2015;33:10522 (suppl; abstr).
Kruit WH, et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J Clin Oncol. 2013;31(19):2413–20.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74.
Carreno BM, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–8.
Castle JC, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72(5):1081–91.
Vormehr M, et al. Mutanome engineered RNA immunotherapy: towards patient-centered tumor vaccination. J Immunol Res. 2015;2015:595363.
Sahin U, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–6.
Ott PA, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used to conduct this study or prepare this manuscript.
Conflict of interest
Dr. Jeffrey Sosman has received consulting fees from Bristol Myers Squibb, Merck, and Array Pharmaceuticals. Dr. Sunandana Chandra has received consulting fees from Bristol Myers Squibb, EMD Serono, Regeneron, and Biodesix and has been awarded research funding from Bristol Myers Squibb. Ludimila Cavalcante and Akansha Chowdhary have no conflicts of interest that are directly relevant to the content of this manuscript.
Rights and permissions
About this article

Cite this article
Cavalcante, L., Chowdhary, A., Sosman, J.A. et al. Combining Tumor Vaccination and Oncolytic Viral Approaches with Checkpoint Inhibitors: Rationale, Pre-Clinical Experience, and Current Clinical Trials in Malignant Melanoma. Am J Clin Dermatol 19, 657–670 (2018). https://doi.org/10.1007/s40257-018-0359-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-018-0359-4