
Abstract
Chronic pain is an important health and social problem. Misuse and abuse of opioids in chronic non-cancer pain management seem to be a huge problem, in some countries. This could probably affect the normal use of such analgesics in patients in need of them. Basic and clinical researches should find the solution to mitigate the potential damage. Dysregulation of mast cell and microglia activation plays an important role in the pathogenesis and management of chronic pain. Persistent mast cell activation sensitizes nociceptors and initiates central nervous system inflammatory processes, involving microglial cell activation and sensitization of spinal somatosensory neurons. Exposure of mast cells and microglia to opioids is well known to provoke activation of these non-neuronal immune cell populations, thereby contributing to an exacerbation of pro-inflammatory and pro-nociceptive processes and promoting, over the long-term, opioid-induced hyperalgesia and tolerance. This review is intended to provide the reader with an overview of the role for these non-neuronal cells in opioid-induced chronic pain and tolerance as a consequence of prolonged exposure to these drugs. In addition, we will examine a potential strategy with the aim to modulate opioid-induced over-activation of glia and mast cells, based on endogenous defense mechanisms and fatty acid amide signaling molecules.
Similar content being viewed by others


Introduction
Chronic pain affects an estimated 20% of the world’s population and accounts for nearly one-fifth of physician visits. Pain is classified as “chronic” when it lasts beyond the normal healing time and persists or recurs for longer than 3 months. It is well distinguished from the acute warning function of the physiological nociception. Such pain often becomes the sole or predominant clinical problem in some patients [1]. For many years, it has been accepted as “a disease in its own right” [2]. Its burden on patients, on health care systems and on society has been clearly demonstrated [3]. The efficacious management of such a disease is still the topic of many discussions, especially due to its multifactorial origins.
Over the past 3 decades, following the recommendations of the WHO guideline [4], the use of opioids has been widely accepted for the treatment of cancer pain. In the last 2 decades, the simple and appealing “analgesic ladder” suggested in the WHO document has been too simplistically interpreted and has been increasingly applied also to patients with chronic non-cancer pain, as well as those with cancer [5]. This was apparently useful for the patients, because it was supposed to be of benefit in most of the chronic pain patients. Although the long-term benefit of opioid analgesia was anecdotally accepted, formal empirical evidence of such was never generated. Unfortunately, as in many other aspects of the chronic pain therapy, there are side effects and complications. The subsequent large increase in prescribing opioids, particularly in the United States, resulted in what has been identified as an “opioid epidemic”, with the consequent harm of misuse and abuse of this class of drugs [6, 7].
Regardless, chronic pain remains a major source of suffering and represents a global health priority calling for special attention and care. Even though considerable progress has been made in the classification and identification of pathophysiologic mechanisms of chronic pain, translating this knowledge into the development of an effective pain management or of improved analgesic medications specifically for chronic pain remains a challenge. At this point, a brief digression on the role of inflammation and non-neuronal cells in chronic pain states is in order. Moreover, the American claim for potential danger of the chronic opioid use has stimulated the authors to review the recent literature, in the hope to make clear the pathophysiology and the potentialities to prevent some of the phenomena consequent to such use. This article is based on previously published data and does not contain studies with human participants or animals performed by any of the authors.
Chronic Pain: The Role of Inflammation and Non-neuronal Cells
Inflammation is a natural reaction of self-protection to remove harmful stimuli [8] and is an integral component of the immune response. At the same time, there are occasions when the inflammatory response itself damages host tissue and causes organ dysfunction. For example, this occurs when there is an overly robust acute or subacute inflammatory response to pathogens or debris from damaged host cells [9]. In their review, Nathan and Ding [8] clearly note that the fundamental problem regarding inflammation is not how often it starts, but how often it fails to subside. Undoubtedly, that non-resolving inflammation is one of the principal contributors to the medical burden in industrialized societies. Inflammation is particularly insidious in cases in which the peripheral and central nervous systems are involved (‘neuroinflammation’), playing an important role in the pathogenesis of chronic pain [10], as well as chronic neurodegenerative diseases [11,12,13] and neuropsychiatric illness [14,15,16].
One of the cardinal advances in the neurosciences over the past few decades has been the discovery of an extensive bi-directional communication between the immune system and the central nervous system (CNS). Pro-inflammatory cytokines occupy a key position by regulating host responses to infection, inflammation, stress and trauma. Principal players in these actions are glia (astrocytes and microglia), and mast cells. These cell populations constitute important sources of inflammatory mediators and may have significant roles in conditions ranging from chronic pain to neurodegenerative diseases and neuropsychiatric disorders [14, 17,18,19,20,21]. This analysis will discuss the role of non-neuronal cells in chronic pain, their dysregulation in response to long-term exposure to opioids, followed by the consideration of a strategy to modulate opioid-induced over-activation of glia and mast cells, based on endogenous defense mechanisms and fatty acid amide signaling molecules.
Neuropathic pain results from damage and/or degeneration of the sensory nervous system. Central neuropathic pain accompanies spinal cord injury, multiple sclerosis, and in certain cases strokes; while painful peripheral neuropathies frequently occur with diabetes and other metabolic or infectious conditions. Neuropathic pain can also be a direct result of cancer involving peripheral nerves or as a side effect of chemotherapy. The triggering and maintenance of neuropathic pain states depends on Schwann cells, spinal microglia and astrocytes, together with elements of the peripheral immune system [22]. Under pathological conditions, dorsal horn microglia become activated with up-regulation of purinergic receptors[23, 24] that participate in neuropathic pain [23].
Upon degranulation, mast cells release molecules which activate or sensitize nociceptors, thereby contributing directly to neuropathic pain [25]. Peripheral nerve mast cells constitute the first line of activation at the site of damage, and promote the recruitment of neutrophils and macrophages [26]. Degranulation of mast cells also activates trigemino-cervical and lumbosacral pain pathways to elicit widespread tactile pain hypersensitivity [27], possibly mediated by a sensitizing effect of histamines on nociceptors. Further, the rapid release of nerve growth factor (NGF) by mast cells can sensitize nociceptors by engaging the latter’s high-affinity TrkA receptors (and indirectly via other peripheral cell types) [28]. Mast cells are important mediators of chronic visceral pain, as well [29].
Interaction Between Non-neuronal Cells and Opioids: A Painful Encounter
For many chronic pain conditions, the medical community has responded with long-term use of analgesics that were originally developed for acute pain. Among these are the opioids, which comprise natural, semi-synthetic or synthetic analgesic molecules with a pharmacological action similar to opium and endogenous endorphins. In particular, the suggestion of the WHO analgesic ladder, which legitimized the use of opioids to treat severe pain in terminally ill cancer patients, also encouraged application of the same therapeutic approach for chronic non-cancer pain, without fostering the search for solutions to limit opioids’ adverse consequences [5]. Long-term use of opioids is accompanied by decreasing levels of analgesic response not readily attributable to advancing underlying disease, necessitating dose escalation to manage pain (‘tolerance’). The latter manifestation, along with other adverse events caused by escalating doses (e.g., over-sedation, respiratory depression, abuse potential) often leaves the patient unable to continue analgesic therapy—significantly diminishing quality of life in those with chronic pain [30]. Despite recent criticisms regarding the growing climate of opiophobia and oligoanalgesia [31], such as in the United States, the use of opioids had devolved to the point of becoming an epidemic of abuse [32].
To bring attention to this situation, the American Academy of Neurology recently published a position paper in the journal Neurology with its recommendations on the prescription of these drugs, defining the emergency as “an epidemic of public health” [6]. This represented a response to publication of the results reported by a commission of experts, appointed by the US National Institutes of Health, tasked with assessing scientific evidence for the efficacy and risks of opioid analgesics. Their findings highlighted the inadequacy of most studies used to justify the prescription of opioids—which have been judged to be definitive neither in terms of chronic treatment efficacy nor safety [33, 34]. Not surprisingly, the Academy warned that risks outweigh benefits when opioids are prescribed inappropriately [6].
The most frequently reported adverse events of opioids use include constipation, nausea, vomiting, dizziness, and drowsiness. Over the longer term, and of a more serious nature, are inhibition of sex hormone production, hypogonadism, infertility, falls and fractures in older adults, cardiac problems such as QT syndrome (elongation interval between the Q and T waves on the electrocardiogram), and sleep-related breathing problems [6].
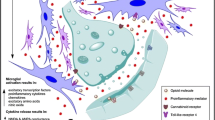
In addition to the development of dependence and tolerance, opioid use can paradoxically lead to hyperalgesia [35]. The mechanism underlying this last effect causes us to consider once again the involvement of non-neuronal cells. Opioids act primarily on neuronal circuits of the brain and spinal cord. Reciprocal signaling between immunocompetent cells in the periphery and the CNS (mast cells, microglia and astrocytes cells) is a key phenomenon underpinning chronic pain mechanisms [36]. By releasing potent pro-inflammatory agents, these non-neuronal cells participate in sensitization of neurons both in the periphery and the CNS and the resultant chronicity (non-resolving nature) of pain. This concept has important implications when discussing strategies for pharmacologically targeting chronic pain and the use of opioids. A key point in this context is that opioids such as morphine have direct effects on non-neuronal cells, in particular, mast cells and microglia. Morphine, codeine and other opioids dose-dependently activate and induce degranulation of mast cells [37,38,39,40,41]. Mast cells release numerous mediators, including pro-inflammatory cytokines (e.g., interleukin-1 and tumor necrosis factor), neuropeptides such as substance P (SP) and proteolytic enzymes such as tryptase, which can have multiple consequences on tissues affected by pain. For example, tryptase and SP act directly on nerve endings by activating and inducing further release of SP (Fig. 1) [39, 42, 43]. By acting in an autocrine/paracrine manner, SP further activates mast cells to amplify mediator release and the perception and processing of pain signals. Mast cell-derived NGF acts both directly by promoting nociceptive peptide release, and indirectly on nociceptive nerve endings by altering the mechanisms of pain, and promoting a phenotypic change in the same endings [42, 44, 45]. SP and other nociceptive neuropeptides (e.g., calcitonin gene-related peptide), as well as tryptase and NGF act on vascular structures, modifying their permeability and favoring the genesis of new blood vessels [38, 46, 47]. These results demonstrate that mast cell activation induced by morphine and other opioids contributes to the exacerbation of pro-inflammatory and pro-nociceptive processes and, in the case of cancer, activation of angiogenesis and promotion of tumor growth (Fig. 1).

Beside neurons, opioid receptors (OR) are also expressed on non-neuron cells, and their distribution is altered by chronic opioid treatment. The activation of non-neuronal cells in peripheral mast cells and spinal cord microglia and astroglia induced by chronic opioid treatment causes an up-regulation of membrane receptors and abnormal production of pro-inflammatory cytokines and chemokines. These events induce sensitization of peripheral and spinal neurons, morphine tolerance and hyperalgesia, OR opioid receptor, NK1 neurokinin 1, CGRP calcitonin gene-related peptide, TRPV1 transient receptor potential vanilloid receptor 1, and PAR2 protease-activated receptor 2
Mast cells, while coordinators of peripheral neuroinflammatory processes and a leading actor in the development of the cerebral neuroinflammation, are not the only immune cells upon which opioids act. Microglia, the primary resident innate immune system cell of the CNS is also sensitive to opioids [48,49,50,51,52,53]. Microglia subjected to chronic opioid treatment assumes an activated phenotype similar to that in chronic and neuropathic pain [54, 55]. In response to chronic morphine exposure, microglia switch from a ramified shape characteristic of a resting state to an amoeboid form, accompanied by up-regulation of cell surface markers CD11b and ionized calcium-binding adapter-1 [35, 56, 57]. These changes represent the cellular and molecular signs of microglial cell activation. Microglia express μ-and k-opioid receptors, further supporting their role as targets for opioids (Fig. 1) [58, 59]. Opioids may also indirectly activate glia via a neuron-glia signal mediated by the chemokine fractalkine [60].
Ligand engagement of opioid receptors on microglia leads to the release of chemokines (e.g., chemokine ligand 2 and CXC-chemokine ligand 1), cytokines (e.g., tumor necrosis factor-α, interleukins-1β, -6 and -10) and neurotrophic factors such as NGF and brain-derived neurotrophic factor (BDNF), and up-regulation of the purinergic receptors P2X4R and P2X7R and the pattern recognition receptor Toll-like receptor 4, all of which participate in neuroinflammation [61,62,63]. In particular, BDNF released following P2X4R activation interacts with its cognate receptor tropomyosin receptor kinase B (TrkB) on spinal somatosensory pain neurons which leads to down-regulation of co-transporter 2 of potassium chloride (KCC2) and subsequent dysregulation of Cl− homeostasis where primary afferent neurons interact with second order neurons. A critical role of the P2X4R-BDNF-KCC2 system is evident both in opioid-induced paroxysmal hyperalgesia and in post-neuronal injury hypersensitivity subsequent to neuronal cell damage [49]. Collectively, these events contribute both to the development of sensitization of spinal neurons and hyperalgesia, as well as morphine tolerance [61].
In summary, opioid analgesic action is determined by interaction with the cognate neuronal cell receptor, and predominates for short-term treatment. Many of the undesirable effects of opioids, especially hyperalgesia and tolerance, are a consequence of opioid interaction with receptors on non-neuronal cells, principally mast cells and microglia. The latter manifests initially as tolerance, with dose escalation needed to maintain efficiency, followed by hyperalgesia. As treatment time lengthens, these effects ultimately prevail over the drug’s analgesic action. An intriguing aspect of these observations is the strict parallelism between the behavior of non-neuronal cells in chronic pain and that observed after repeated administration of opioids.
The Challenge of Treating the Elderly with Opioids
Chronic pain is highly prevalent in the elderly [55, 64] and calls for special attention [65]. In the period 1995–2010 the prescription of opioids for chronic non-cancer pain in this population increased some ninefold (compared to historic levels), despite the limited number of studies conducted on the elderly [66]. Unfortunately, studies evaluating the effectiveness of analgesics often exclude elderly patients, especially those most vulnerable to pain and its impact on their lives. As such, the actual efficacy and safety profile of analgesics, especially opioids, in this population remains largely unknown—especially for long-term treatment [67]. Moreover, pharmacokinetic and metabolic changes associated with aging render the elderly vulnerable to the deleterious effects including overdosing, associated with analgesic use. Clinicians thus need to take into account changes, even physiological, that develop with age and the potential impact that they can have on therapeutic actions of analgesics.
Age-dependent changes involving non-neuronal cells negatively influence the development of more serious side effects caused by chronic opioid treatment [65]. As we age, mast cells and microglia become more sensitive to stimuli that affect their activation: both have a lower activation threshold and respond with more intense and lasting action over time [68]. These changes in reactivity are referred to as immunosenescence [69]. Immunosenescence is accompanied by an increased sensitivity to pro-inflammatory mediators with a consequent rise in mast cell degranulation. As with microglia, morphine also activates mast cells, resulting in the release of inflammatory cytokines, neuropeptides such as SP and tryptase, leading to activation of peripheral nerve terminals and increased pain [55, 70, 71]. Microglia in the elderly display what may best be described as a primed phenotype (Fig. 2), which is present also in traumatic brain injury and neurodegenerative diseases [70]. The principal difference between the two phenotypes lies in the resolution time of activation. Resting microglia undergoes brief activation intended to be beneficial and aimed at recovering tissue homeostasis. Primed microglia upon stimulation remains activated for a prolonged period with an abundant production/release of pro-inflammatory cytokines that impede the resolution and tissue homeostatic recovery. Experimental models of neurodegenerative diseases show these events to be promoters of non-resolving neuroinflammation. In these same models of neurodegenerative diseases another glial cell population, namely astrocytes—that produce cytokines, chemokines and trophic factors analogous to microglia—take on a primed behavior and respond in an exaggerated manner to noxious stimuli [70, 72]. Conceivably, these non-neuronal cells in the elderly might respond to long-term morphine treatment by developing a phenotype typical of primed cells: exaggerated, prolonged over time, and associated with non-resolving neuroinflammation. The end result may be a more rapid development of tolerance, addiction and hyperalgesia.

Reproduced with permission from Fusco et al. [125]
Resting and primed microglia: the main differences. With aging and long term of opioid therapy, the phenotype of microglia is predominantly primed. It responds in a more intense manner, producing a greater amount of pro-inflammatory mediators and for a longer time. Primed microglia induce persistent neuroinflammatory response, capable of damaging tissue integrity and neuron function.
Combination Therapy to Modulate Opioid Actions: A Pharmacological Rationale
One approach to overcome the problem of prolonged opioid use may be the addition of another agent that effectively either reduces the development of tolerance or potentiates the drug efficacy, thus allowing the use of lower doses of opioids. Classically, and even recently, multimodal analgesia has been suggested for acute pain [73, 74]. Opioids can be administered with non-steroidal anti-inflammatory drugs (NSAIDs), but also with other molecules (Table 1). There is some precedent for this type of approach. For example, agents able to modulate non-neuronal cell activity limit the development of certain side effects attributed to opium derivatives. Cromolyn, an inhibitor of mast cell degranulation, but not the opioid antagonist naltrexone, prevents granuloma formation induced by intrathecal morphine infusion [75], and also reduces toxicity caused by intravenous administration of the potent synthetic opioid fentanyl [76]. Minocycline, propentofylline and pentoxifylline reduce glial cell activation and block the development of morphine tolerance in naïve mice, as well as in a model of neuropathic pain [51, 77,78,79,80,81]. Pentoxifylline also has been found to reduce morphine consumption in patients with post-operative pain [82]. The above is highly questionable, however, given that its side effects limit their prolonged use in persistent pain conditions.
Chronic inflammatory processes trigger a program of resolution that includes the production of lipid mediators with the capacity to switch off inflammation [83]. Among these are the N-acylethanolamines, which include palmitoylethanolamide (PEA) [84]. These fatty acid amides are formed from N-acylated phosphatidylethanolamine by several pathways, but mainly involving a membrane-associated N-acylated phosphatidylethanolamine-phospholipase D that generates the respective N-acylethanolamine and phosphatidic acid [85]. This enzyme then converts N-palmitoyl-phosphatidyl-ethanolamine into PEA.
A number of observations support a PEA role in maintaining cellular homeostasis by acting as a mediator of resolution of inflammatory processes (through an Autacoid Local Injury Antagonism mechanism1[ALIA]); it is synthesized/metabolized by microglia and mast cells; it down-modulates mast cell [86,87,88] and microglia activation; [89,90,91] tissue levels of PEA are increased on demand in brain areas involved in nociception and in the spinal cord following neuropathic pain induction, in human conditions associated with pain [92, 93], as well as in settings associated with injury to nervous tissue [18, 94].
A growing body of preclinical studies demonstrates the ability of PEA to reduce inflammation and pain induced by various acute stimuli. The effect of PEA administration by different routes is dose-dependent [95,96,97]. The anti-inflammatory and analgesic effects of PEA have been confirmed in models of chronic inflammation [98] and chronic or neuropathic pain [86, 89, 99, 100]. In these models, prolonged treatment with PEA not only reduced pain but also preserved peripheral nerve morphology, reduced endoneural edema, the recruitment and activation of mast cells, and the production of pro-inflammatory mediators at the injury site [86, 89, 100]. Taken together, these data indicate that PEA, via regulation of persistent inflammatory processes and the ALIA mechanism, can directly intervene in nervous tissue alterations responsible for pain, i.e., to act as a disease-modifying agent [99]. At the clinical level PEA, in micronized and ultramicronized forms (m-PEA; um-PEA), reduces pain and disability associated with several chronic diseases [101].
These results suggest that the PEA could be a promising candidate for minimizing the risks of chronic treatment with opioids mediated by activation of non-neuronal cells. The most compelling experimental evidence to date supporting the premise that concomitant use of PEA and opioids could be beneficial is the demonstration of delayed development of tolerance to the anti-nociceptive effects of morphine in the setting of repeated administration of both compounds [102]. Concomitant administration of m-PEA doubled the number of days (from 5 to 10) of morphine treatment efficacy in two rat pain models (mechanical and thermal pain threshold) [102]. As the authors point out: “In spite of the potency and efficacy of morphine, its clinical application for chronic persistent pain is limited by the development of tolerance to the anti-nociceptive effect.” Further, these authors used immunohistochemical techniques to show that development of tolerance to morphine was accompanied by an increase in glial cell number in the spinal cord. Glial cells release large amounts of pro-inflammatory cytokines that sensitize pain transmission neurons, and thus gradually counteract morphine’s anti-nociceptive effects. PEA markedly prevented the increase in glial cells and thus prolonged the efficacy of morphine. The authors concluded their study, stating that “Multiple properties of PEA converge to an interaction with signals evoked by morphine. The evidence of a delayed development of tolerance to the anti-nociceptive effects of morphine in the presence of PEA suggests a possible application of this endogenous compound in opioid-based therapies” [102]. Clinically, m-PEA administered with generally ineffective doses of oxycodone produced good pain control with excellent tolerability, suggesting the possibility of combining the two molecules in order to reduce opioid dose and consequent undesirable effects [103].
What is the molecular basis underlying the effects of PEA? PEA acts on many cell types, including immune-derived non-neuronal cells—in particular, microglia and mast cells—which are involved in pain signaling but not directly in the transmission of pain perception [18]. A number of studies point to PEA being a ligand for peroxisome proliferator activated receptor (PPAR)α, one of a group of nuclear receptor proteins that function as transcription factors regulating the expression of genes. The primary function of PPAR-α is as a fatty acid sensor that regulates lipid and lipoprotein metabolism and energy homeostasis through the activation of several target genes. PPAR-α- and γ-isoforms in particular are associated with pro-inflammatory events. PEA anti-inflammation [104], anti-nociception/anti-neuropathology [99, 105, 106] and neuroprotection [107,108,109,110] were either absent in PPAR-α null mice or blocked by PPAR-α antagonists. An ‘entourage effect’ has also been hypothesized to explain the pharmacological actions of PEA, whereby PEA enhances the anti-inflammatory and anti-nociceptive activity of other endogenous compounds by potentiating their affinity for a receptor or by inhibiting their metabolic degradation [111, 112]. Anandamide (AEA) is a candidate molecule, as it possesses anti-inflammatory and anti-nociceptive effects. AEA and its congeners like PEA have in common the transient potential vanilloid type 1 (TRPV1) receptor action. The TRPV1 receptor, a non-selective cation channel expressed in small diameter sensory neurons, is activated by noxious heat, low pH and capsaicin. AEA itself is a TRPV1 receptor agonist, and PEA enhances AEA stimulation of the human TRPV1 receptor [113] in a cannabinoid CB2 receptor antagonist-sensitive fashion (although PEA shows no appreciable affinity for either CB1 or CB2 receptors)—which could be interpreted as PEA acting indirectly by potentiating AEA actions [96]. Interestingly, mast cells [114], and cortical[115] and spinal cord [116] microglia all reportedly express TRPV1 receptors. As the above receptors/pathways may also be influenced by opioids’ actions, it is not unreasonable to propose that PEA, acting through the same receptors (CB1, PPAR, TRPV1) has the potential to be a relevant modulator of opioid activity. PEA may also enhance the levels of the endocannabinoid 2-arachidonoylglycerol and potentiate its actions at TRPV1 channels, observations which effectively widens the ‘entourage’ effect of PEA [117].
The potential for PEA to interact with the opioid system may lie in its ability to modulate the endocannabinoid system (ECS)—which in turn influences the response to opioids through the opioid system. Elements of the ECS comprise the cannabinoid receptors CB1 and CB2, a family of nascent lipid ligands, the ‘endocannabinoids’ (exemplified by AEA and 2-arachidonoylglycerol) and the machinery for their biosynthesis and metabolism. PEA appears to be a ligand for peroxisome proliferator activated receptor (PPAR)α [104], one of a group of nuclear receptor proteins that function as transcription factors regulating the expression of genes. PPAR-α- and γ-isoforms in particular are associated with pro-inflammatory events. Interest in PPARs as potential targets for treating drug addiction arose from study of the ECS, which is thought to be involved in the addictive properties of drugs [118]. Recently, Paterniti et al. [119] demonstrated that PPAR-γ and PPAR-δ are also targets for PEA in protecting the spinal cord against pro-inflammatory insults, not by a direct action but probably mediated by cannabinoid CB1 receptor-dependent changes in PPAR expression. As the above receptors/pathways may also be influenced by opioid actions, it is not unreasonable to propose that PEA, acting through the same receptors, has the potential to be a relevant modulator of opioid activity. Concerning PPAR-γ modulation of opioids effects, recent studies [120] showed that oral administration of the PPAR-γ agonist pioglitazone attenuated morphine-induced tolerance, an effect attributed, at least in part, to its anti-inflammatory properties against morphine. Pioglitazone action was reversed by a selective PPAR-γ antagonist. These findings support the notion that PPAR-γ interaction may be relevant with regard to both tolerance and dependence. A role for PPAR-γ cannot be discounted in view of a report describing that PEA may act also as an agonist of PPAR-γ in protecting the spinal cord against pro-inflammatory insults [119].
Both the phytocannabinoid Δ9-tetrahydrocannabinol (Δ9-THC) [121] and the synthetic CB1 receptor agonist CB-55940 [122] were determined to attenuate the development of oral morphine tolerance. Further, blocking N-methyl-d-aspartate (NMDA) glutamate receptors in the presence of Δ9-THC was more effective than Δ9-THC alone in reducing the development of tolerance, which may have been due to pre-synaptic CB1 receptor agonists acting to reduce glutamate transmission, a reduction amplified when NMDA receptors are blocked [122]. The CB1 and NMDA receptor systems may thus play a key role in the behavioral plasticity associated with chronic morphine administration.
The notion that the endocannabinoid and opioid systems are closely linked is supported also by data demonstrating that the l-arginine/nitric oxide (NO)/cyclic guanosine monophosphate (cGMP) pathway participates in peripheral anti-nociception produced by μ-, κ- or δ-opioid receptor agonists, non-steroidal analgesics, and α-(2C)-adrenoceptor agonists [95]. In a rat paw model of hyperalgesia induced by intra-plantar injection of prostaglandin E2, the local peripheral anti-nociceptive effect PEA was antagonized by a selective neuronal NO synthase inhibitor and by a non-selective NOS inhibitor. Exogenously administered PEA was able to induce NO release, while the cGMP-phosphodiesterase inhibitor zaprinast potentiated the anti-nociceptive effect of low-dose PEA. Thus, PEA appears able to activate neuronal NO synthase, thus initiating the NO/cGMP pathway and inducing peripheral anti-nociceptive effects—a pathway common to that elicited by μ-, κ- or δ-opioid receptor agonists.
Concluding Remarks
The three major opioid receptor families, μ, δ and κ are activated by endogenous opioid peptides (enkephalins, endorphins, and dynorphins), and the opioid drugs. In addition to pain modulation and addiction, opioid receptors are widely involved in various physiological and pathophysiological processes, including the regulation of membrane ion homeostasis, cell proliferation, emotional response, epileptic seizures, immune function, feeding, obesity, respiratory and cardiovascular control as well as some neurodegenerative disorders [123].
The ECS plays a major role in the control of pain as well as in mood regulation, reward processing and the development of addiction. Both opioid and cannabinoid receptors are coupled to G proteins expressed throughout the brain’s reinforcement circuitry. Given the central role that PEA may play in the ECS, it is not surprising that it can influence the effects of opioids. The existing literature clearly demonstrates that PEA either directly or indirectly affects a variety of receptors and pathways known to modify the responses to opioids such as morphine.
Because opioid-induced neuroinflammation is implicated in the development of tolerance to these analgesic substances, intervention in pathways that lead to neuroinflammation and its resolution is an attractive strategy for pain therapeutics. PEA has been extensively studied in this setting of neuroinflammation [124]. Current evidence supports the concept that by acting on mast cells and glia, PEA promotes the body’s capacity for self-defense against (neuro)inflammation (ALIA mechanism). Indeed, initial investigations provide evidence favoring the use of PEA in conjunction with opioids [102]. Pharmacological studies demonstrating the ability of PEA to affect opioid and cannabinoid receptors/pathways, coupled with opioid effects being modulated by the ECS, allow us to infer that opioid analgesic effects may be potentiated or last longer with reduced tolerance development. Building on two basic observations, namely, that opioids reduce chronic/neuropathic pain perception and PEA modifies the underlying cause(s) of this pain, we are drawn to the conclusion that usage of PEA and opioids, either in fixed combinations or by concomitant administration, merit larger clinical trials to confirm these observations in humans. Considering the benefit/risk of the PEA, these results suggest that PEA, possibly associated with antioxidant molecules, can be an innovative therapeutic tool to enhance the effects of opioid analgesics and impede the development of opioid tolerance and hyperalgesia.
References
Treede RD, Rief W, Barke A, Aziz Q, Bennett MI, Benoliel R, Cohen M, Evers S, Finnerup NB, First MB, Giamberardino MA, Kaasa S, Kosek E, Lavand’homme P, Nicholas M, Perrot S, Scholz J, Schug S, Smith BH, Svensson P, Vlaeyen JW, Wang SJ. A classification of chronic pain for ICD-11. Pain. 2015;156(6):1003–7. https://doi.org/10.1097/j.pain.0000000000000160.
Niv D, Devor M, Erdine S, Breivik H, and Varrassi G. Europe Against Pain. https://www.researchgate.net/publication/313472969_Europe_Against_Pain. 2001; https://doi.org/10.13140/rg.2.2.34590.95047
Langley P, Muller-Schwefe G, Nicolaou A, Liedgens H, Pergolizzi J, Varrassi G. The impact of pain on labor force participation, absenteeism and presenteeism in the European Union. J Med Econom. 2010;13(4):662–72.
World Health Organization. Cancer pain relief. Geneva: World Health Organization; 1986.
Ballantyne JC, Kalso E, Stannard C. WHO analgesic ladder: a good concept gone astray. BMJ. 2016;352:i20. https://doi.org/10.1136/bmj.i20.
Franklin GM, American Academy of Neurology. Opioids for chronic noncancer pain: a position paper of the American Academy of Neurology. Neurology. 2014;83(14):1277–84.
Wilkerson RG, Kim HK, Windsor TA, Mareiniss DP. The Opioid Epidemic in the United States. Emerg Med Clin North Am. 2016;34(2):e1–23. https://doi.org/10.1016/j.emc.2015.11.002 (Epub 2016 Feb 17).
Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–82.
Castellheim A, Brekke OL, Espevik T, Harboe M, Mollnes TE. Innate immune responses to danger signals in systemic inflammatory response syndrome and sepsis. Scand J Immunol. 2009;69:479–91.
Myers RR, Campana WM, Shubayev VI. The role of neuroinflammation in neuropathic pain: mechanisms and therapeutic targets. Drug Discov Today. 2006;11:8–20.
Freeman LC, Ting JP. The pathogenic role of the inflammasome in neurodegenerative diseases. J Neurochem. 2016;136(Suppl 1):29–38. https://doi.org/10.1111/jnc.13217.
McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126:479–97.
Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808.
Zis P, Daskalaki A, Bountouni I, Sykioti P, Varrassi G, Paladini A. Depression and chronic pain in the elderly: Links and management challenges. Clin Interv Aging. 2017;12:709–20.
Castanon N, Luheshi G, Layé S. Role of neuroinflammation in the emotional and cognitive alterations displayed by animal models of obesity. Front Neurosci. 2015;9:229. https://doi.org/10.3389/fnins.2015.00229.
Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O. Neuroinflammation and psychiatric illness. J Neuroinflamm. 2013;10:43. https://doi.org/10.1186/1742-2094-10-43.
Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D, van Noort JM. Innate and adaptive immune responses in neurodegeneration and repair. Immunology. 2014;142(2):151–66.
Skaper SD, Facci L, Giusti P. Mast cells, glia and neuroinflammation: partners in crime? Immunology. 2014;141(3):314–27. https://doi.org/10.1111/imm.12170.
Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90.
Silver R, Curley JP. Mast cells on the mind: new insights and opportunities. Trends Neurosci. 2013;36:513–21.
Thacker MA, Clark AK, Marchand F, McMahon SB. Pathophysiology of peripheral neuropathic pain: immune cells and molecules. Anesth Analg. 2007;105:838–47.
DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6.
Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, Grahames CB, Casula MA, Yiangou Y, Birch R, Anand P, Buell GN. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–96.
Tsuda M, Kuboyama K, Inoue T, Nagata K, Tozaki-Saitoh H, Inoue K. Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Mol Pain. 2009;5:28. https://doi.org/10.1186/1744-8069-5-28.
Xanthos DN, Gaderer S, Drdla R, Nuro E, Abramova A, Ellmeier W, Sandkühler J. Central nervous system mast cells in peripheral inflammatory nociception. Mol Pain. 2011;7:42. https://doi.org/10.1186/1744-8069-7-42.
Zuo Y, Perkins NM, Tracey DJ, Geczy CL. Inflammation and hyperalgesia induced by nerve injury in the rat: a key role of mast cells. Pain. 2003;105:467–79.
Levy D, Kainz V, Burstein R, Strassman AM. Mast cell degranulation distinctly activates trigemino-cervical and lumbosacral pain pathways and elicits widespread tactile pain hypersensitivity. Brain Behav Immun. 2012;26:311–7.
Koda H, Mizumura K. Sensitization to mechanical stimulation by inflammatory mediators and by mild burn in canine visceral nociceptors in vitro. J Neurophysiol. 2002;87:2043–51.
Wood D. Visceral pain: spinal afferents, enteric mast cells, enteric nervous system and stress. Curr Pharm Des. 2011;17:1573–5.
Kane-Gill SL, Rubin EC, Smithburger PL, Buckley MS, Dasta JF. The cost of opioid-related adverse drug events. J Pain Palliat Care Pharmacother. 2014;28:282–93.
Schatman ME, Ziegler SJ. Pain Management, prescription opioid mortality, and the CDC: is the devil in the data? J Pain Res. 2017;10:2489–95.
O’Donnell JK, Halpin J, Mattson CL, Goldberger BA, Gladden RM. Deaths involving fentanyl, fentanyl analogs, and U-47700—10 States, July-December 2016. MMWR Morb Mortal Wkly Rep. 2017;66:1197–202.
Reuben DB, Alvanzo AA, Ashikaga T, Bogat GA, Callahan CM, Ruffing V, Steffens DC. National Institutes of Health Pathways to Prevention Workshop: the role of opioids in the treatment of chronic pain. Ann Intern Med. 2015;162(4):295–300.
Chou R, Turner JA, Devine EB, Hansen RN, Sullivan SD, Blazina I, Dana T, Bougatsos C, Deyo RA. The effectiveness and risks of long-term opioid therapy for chronic pain: a systematic review for a National Institutes of Health Pathways to Prevention Workshop. Ann Intern Med. 2015;162(4):276–86.
Ferrini F, Trang T, Mattioli TA, Laffray S, Del’Guidice T, Lorenzo LE, Castonguay A, Doyon N, Zhang W, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu JM, Cahill CM, Salter MW, De Koninck Y. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl- homeostasis. Nat Neurosci. 2013;16(2):183–92.
Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14:217–31.
Kan L, Mutso AA, McGuire TL, Apkarian AV, Kessler JA. Opioid signaling in mast cells regulates injury responses associated with heterotopic ossification. Inflamm Res. 2014;63(3):207–15.
Nguyen J, Luk K, Vang D, Soto W, Vincent L, Robiner S, Saavedra R, Li Y, Gupta P, Gupta K. Morphine stimulates cancer progression and mast cell activation and impairs survival in transgenic mice with breast cancer. Br J Anaesth. 2014;113(Suppl 1):i4–13.
Vincent L, Vang D, Nguyen J, Gupta M, Luk K, Ericson ME, Simone DA, Gupta K. Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood. 2013;122(11):1853–62.
Blunk JA, Schmelz M, Zeck S, Skov P, Likar R, Koppert W. Opioid-induced mast cell activation and vascular responses is not mediated by mu-opioid receptors: an in vivo microdialysis study in human skin. Anesth Analg. 2004;98(2):364–70.
Levy JH, Brister NW, Shearin A, Ziegler J, Hug CC Jr, Adelson DM, Walker BF. Wheal and flare responses to opioids in humans. Anesthesiology. 1989;70(5):756–60.
Skaper SD. Nerve growth factor: a neuroimmune crosstalk mediator for all seasons. Immunology. 2017. https://doi.org/10.1111/imm.12717.
Fitzgerald JJ, Ustinova E, Koronowski KB, de Groat WC, Pezzone MA. Evidence for the role of mast cells in colon-bladder cross organ sensitization. Auton Neurosci. 2013;173(1–2):6–13.
Hirose M, Kuroda Y, Murata E. NGF/TrkA signaling as a therapeutic target for pain. Pain Pract. 2015. https://doi.org/10.1111/papr.12342.
Bergman E, Ulfhake B, Fundin BT. Regulation of NGF-family ligands and receptors in adulthood and senescence: correlation to degenerative and regenerative changes in cutaneous innervation. Eur J Neurosci. 2000;12(8):2694–706.
Chen WH, Mao CQ, Zhuo LL, Ong JL. Beta-nerve growth factor promotes neurogenesis and angiogenesis during the repair of bone defects. Neural Regen Res. 2015;10(7):1159–65.
Ribatti D, Ranieri G. Tryptase, a novel angiogenic factor stored in mast cell granules. Exp Cell Res. 2015;332(2):157–62.
Giron SE, Griffis CA, Burkard JF. Chronic pain and decreased opioid efficacy: an inflammatory link. Pain Manag Nurs. 2015;16(5):819–31.
Trang T, Al-Hasani R, Salvemini D, Salter MW, Gutstein H, Cahill CM. Pain and poppies: the good, the bad, and the ugly of opioid analgesics. J Neurosci. 2015;35(41):13879–88.
Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Dis. 2014;13:533–48.
Horvath RJ, Romero-Sandoval EA, De Leo JA. Inhibition of microglial P2X4 receptors attenuates morphine tolerance, Iba1, GFAP and mu opioid receptor protein expression while enhancing perivascular microglial ED2. Pain. 2010;150(3):401–13.
Ji RR. Targeting microglial purinergic signaling to improve morphine analgesia. Pain. 2010;150(3):377–8.
Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends Neurosci. 2005;28(12):661–9.
Loggia ML, Chonde DB, Akeju O, Arabasz G, Catana C, Edwards RR, Hill E, Hsu S, Izquierdo-Garcia D, Ji RR, Riley M, Wasan AD, Zürcher NR, Albrecht DS, Vangel MG, Rosen BR, Napadow V, Hooker JM. Evidence for brain glial activation in chronic pain patients. Brain. 2015;138(Pt 3):604–15.
Paladini A, Fusco M, Coaccioli S, Skaper SD, Varrassi G. Chronic pain in the elderly: the case for new therapeutic strategies. Pain Phys. 2015;18(5):E863–76.
Mattioli TA, Leduc-Pessah H, Skelhorne-Gross G, Nicol CJ, Milne B, Trang T, Cahill CM. Toll-like receptor 4 mutant and null mice retain morphine-induced tolerance, hyperalgesia, and physical dependence. PLoS One. 2014;9(5):e97361.
Schwarz JM, Smith SH, Bilbo SD. FACS analysis of neuronal-glial interactions in the nucleus accumbens following morphine administration. Psychopharmacology. 2013;230(4):525–35.
Mika J, Popiolek-Barczyk K, Rojewska E, Makuch W, Starowicz K, Przewlocka B. Delta-opioid receptor analgesia is independent of microglial activation in a rat model of neuropathic pain. PLoS One. 2014;9(8):e104420. https://doi.org/10.1371/journal.pone.0104420.
Chao CC, Gekker G, Sheng WS, Hu S, Tsang M, Peterson PK. Priming effect of morphine on the production of tumor necrosis factor-alpha by microglia: implications in respiratory burst activity and human immunodeficiency virus-1 expression. J Pharmacol Exp Ther. 1994;269(1):198–203.
Cao H, Zhang YQ. Spinal glial activation contributes to pathological pain states. Neurosci Biobehav Rev. 2008;32(5):972–83.
Mélik Parsadaniantz S, Rivat C, Rostène W, Réaux-Le Goazigo A. Opioid and chemokine receptor crosstalk: a promising target for pain therapy? Nat Rev Neurosci. 2015;16(2):69–78.
Coller JK, Hutchinson MR. Implications of central immune signaling caused by drugs of abuse: mechanisms, mediators and new therapeutic approaches for prediction and treatment of drug dependence. Pharmacol Ther. 2012;134(2):219–45.
Zhou D, Chen ML, Zhang YQ, Zhao ZQ. Involvement of spinal microglial P2X7 receptor in generation of tolerance to morphine analgesia in rats. J Neurosci. 2010;30(23):8042–7.
Weiner DK. Office management of chronic pain in the elderly. Am J Med. 2007;120(4):306–15.
Varrassi G, Fusco M, Coaccioli S, Paladini A. Chronic pain and neurodegenerative processes in elderly people. Pain Practice. 2015;15:1–3.
Olfson M, Wang S, Iza M, Crystal S, Blanco C. National trends in the office-based prescription of schedule II opioids. J Clin Psychiatry. 2013;74(9):932–9.
Veal FC, Peterson GM. Pain in the frail or elderly patient: does tapentadol have a role? Drugs Aging. 2015;32(6):419–26.
Mate I, Madrid JA, De la Fuente M. Chronobiology of the neuroimmunoendocrine system and aging. Curr Pharm Des. 2014;20:4642–55.
Tsuboi I, Harada T, Hirabayashi Y, et al. Age-related decline of mast cell regeneration in senescence-accelerated mice (SAMP1) after chemical myeloablation due to senescent stromal cell impairment. Exp Biol Med. 2012;237:1289–97.
Norden DM, Muccigrosso MM, Godbout JP. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology. 2015;96(Pt A):29–41.
von Bernhardi R, Eugenín-von Bernhardi L, Eugenín J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci. 2015;7:124.
Hennessy E, Griffin ÉW, Cunningham C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1β and TNF-α. J Neurosci. 2015;35(22):8411–22.
Kehlet H, Dahl JB. Anaesthesia, surgery, and challenges in postoperative recovery. Lancet. 2003;362(9399):1921–28.
Varrassi G, Hanna M, Macheras G, Montero A, Montes Perez A, Meissner W, Perrot S, Scarpignato C. Multimodal analgesia in moderate-to-severe pain: a role for a new fixed combination of dexketoprofen and tramadol. Curr Med Res Opin. 2017;33(6):1165–73. https://doi.org/10.1080/03007995.2017.1310092 (Epub 2017 Apr 7).
Yaksh TL, Allen JW, Veesart SL, Horais KA, Malkmus SA, Scadeng M, Steinauer JJ, Rossi SS. Role of meningeal mast cells in intrathecal morphine-evoked granuloma formation. Anesthesiology. 2013;118(3):664–78.
Agarwal A, Azim A, Ambesh S, Bose N, Dhiraj S, Sahu D, Singh U. Salbutamol, beclomethasone or sodium chromoglycate suppress coughing induced by iv fentanyl. Can J Anaesth. 2003;50(3):297–300.
Fukagawa H, Koyama T, Kakuyama M, Fukuda K. Microglial activation involved in morphine tolerance is not mediated by toll-like receptor 4. J Anesth. 2013;27(1):93–7.
Horvath RJ, DeLeo JA. Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J Neurosci. 2009;29(4):998–1005.
Cui Y, Liao XX, Liu W, Guo RX, Wu ZZ, Zhao CM, Chen PX, Feng JQ. novel role of minocycline: attenuating morphine antinociceptive tolerance by inhibition of p38 MAPK in the activated spinal microglia. Brain Behav Immun. 2008;22(1):114–23.
Mika J, Osikowicz M, Makuch W, Przewlocka B. Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur J Pharmacol. 2007;560:142–9.
Raghavendra V, Tanga FY, DeLeo JA. Attenuation of morphine tolerance, withdrawal-induced hyperalgesia, and associated spinal inflammatory immune responses by propentofylline in rats. Neuropsychopharmacology. 2004;29:327–34.
Lu CH, Chao PC, Borel CO, Yang CP, Yeh CC, Wong CS, Wu CT. Preincisional intravenous pentoxifylline attenuating perioperative cytokine response, reducing morphine consumption, and improving recovery of bowel function in patients undergoing colorectal cancer surgery. Anesth Analg. 2004;99:1465–71.
Buckley CD, Gilroy DW, Serhan CN. The resolution of inflammation. Nat Rev Immunol. 2013;58:389–462.
Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462.
Leung D, Saghatelian A, Simon GM, Cravatt BF. Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry. 2006;45:4720–6.
Bettoni I, Comelli F, Colombo A, Bonfanti P, Costa B. Non-neuronal cell modulation relieves neuropathic pain: efficacy of the endogenous lipid palmitoylethanolamide. CNS Neurol Disord Drug Targets. 2013;12(1):34–44.
Cerrato S, Brazis P, della Valle MF, Miolo A, Puigdemont A. Effects of palmitoylethanolamide on immunologically induced histamine, PGD2 and TNFalpha release from canine skin mast cells. Vet Immunol Immunopathol. 2010;133(1):9–15.
Facci L, Dal Toso R, Romanello S, Buriani A, Skaper SD, Leon A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc Natl Acad Sci USA. 1995;92:3376–80.
Luongo L, Guida F, Boccella S, Bellini G, Gatta L, Rossi F, de Novellis V, Maione S. Palmitoylethanolamide reduces formalin-induced neuropathic-like behaviour through spinal glial/microglial phenotypical changes in mice. CNS Neurol Disord Drug Targets. 2013;12(1):45–54.
Loría F, Petrosino S, Mestre L, Spagnolo A, Correa F, Hernangómez M, Guaza C, Di Marzo V, Docagne F. Study of the regulation of the endocannabinoid system in a virus model of multiple sclerosis reveals a therapeutic effect of palmitoylethanolamide. Eur J Neurosci. 2008;28(4):633–41.
Franklin A, Parmentier-Batteur S, Walter L, Greenberg DA, Stella N. Palmitoylethanolamide increases after focal cerebral ischemia and potentiates microglial cell motility. J Neurosci. 2003;23:7767–75.
Ghafouri N, Ghafouri B, Larsson B, Stensson N, Fowler CJ, Gerdle B. Palmitoylethanolamide and stearoylethanolamide levels in the interstitium of the trapezius muscle of women with chronic widespread pain and chronic neck-shoulder pain correlate with pain intensity and sensitivity. Pain. 2013;154:1649–58.
Sarchielli P, Pini LA, Coppola F, Rossi C, Baldi A, Mancini ML, Calabresi P. Endocannabinoids in chronic migraine: CSF findings suggest a system failure. Neuropsychopharmacology. 2007;32:1384–90.
Esposito E, Cuzzocrea S. Palmitoylethanolamide is a new possible pharmacological treatment for inflammation associated with trauma. Mini Rev Med Chem. 2013;13:237–55.
Romero TR, Duarte ID. N-palmitoyl-ethanolamine (PEA) induces peripheral antinociceptive effect by ATP-sensitive K + -channel activation. J Pharmacol Sci. 2012;118:156–60.
Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394(6690):277–81.
Jaggar SI, Hasnie FS, Sellaturay S, Rice AS. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain. 1998;76:189–99.
De Filippis D, Luongo L, Cipriano M, Palazzo E, Cinelli MP, de Novellis V, Maione S, Iuvone T. Palmitoylethanolamide reduces granuloma-induced hyperalgesia by modulation of mast cell activation in rats. Mol. Pain. 2011;7:3. https://doi.org/10.1186/1744-8069-7-3.
Di Cesare Mannelli L, D’Agostino G, Pacini A, Russo R, Zanardelli M, Ghelardini C, Calignano A. Palmitoylethanolamide is a disease-modifying agent in peripheral neuropathy: pain relief and neuroprotection share a PPAR-alpha-mediated mechanism. Mediators Inflamm. 2013; p. 328797. https://doi.org/10.1155/2013/328797.
Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: involvement of CB(1), TRPV1 and PPARgamma receptors and neurotrophic factors. Pain. 2008;139:541–50.
Paladini A, Fusco M, Cenacchi T, Schievano C, Piroli A, Varrassi G. Micronized and ultra-micronized palmitoylethanolamide in the treatment of chronic pain: a pooled data meta-analysis. Pain Phys. 2016;19(2):11–24.
Di Cesare Mannelli L, Corti F, Micheli L, Zanardelli M, Ghelardini C. Delay of morphine tolerance by palmitoylethanolamide. Biomed Res Int. 2015;2015:894732.
Desio P. Associazione dell’ossicodone a lenta titolazione con Palmitoiletanolamide per il trattamento del low back pain. Anestesia e Medicina critica (AMC). 2011;1(2):63–71.
Lo Verme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, Piomelli D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67(1):15–9.
de Novellis V, Luongo L, Guida F, Cristino L, Palazzo E, Russo R, Marabese I, D’Agostino G, Calignano A, Rossi F, Di Marzo V, Maione S. Effects of intra-ventrolateral periaqueductal grey palmitoylethanolamide on thermoceptive threshold and rostral ventromedial medulla cell activity. Eur J Pharmacol. 2012;676:41–50.
D’Agostino G, La Rana G, Russo R, Sasso O, Iacono A, Esposito E, Raso GM, Cuzzocrea S, Lo Verme J, Piomelli D, Meli R, Calignano A. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-alpha agonist, modulates carrageenan-induced paw edema in mice. J Pharmacol Exp Ther. 2007;322:1137–43.
D’Agostino G, Russo R, Avagliano C, Cristiano C, Meli R, Calignano A. Palmitoylethanolamide protects against the amyloid-β25-35-induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology. 2012;37:1784–92.
Esposito E, Impellizzeri D, Mazzon E, Paterniti I, Cuzzocrea S. Neuroprotective activities of palmitoylethanolamide in an animal model of Parkinson’s disease. PLoS One. 2012;7(8):e41880. https://doi.org/10.1371/journal.pone.0041880.
Scuderi C, Valenza M, Stecca C, Esposito G, Carratù MR, Steardo L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-α. J. Neuroinflamm. 2012;9:49. https://doi.org/10.1186/1742-2094-9-21.
Raso GM, Esposito E, Vitiello S, Iacono A, Santoro A, D’Agostino G, Sasso O, Russo R, Piazza PV, Calignano A, Meli R. Palmitoylethanolamide stimulation induces allopregnanolone synthesis in C6 Cells and primary astrocytes: involvement of peroxisome-proliferator activated receptor-α. J Neuroendocrinol. 2011;23:591–600.
Smart D, Jonsson KO, Vandevoorde S, Lambert DM, Fowler CJ. ‘Entourage’ effects of N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and upon anandamide metabolism. Brit. J. Pharmacol. 2002;136:452–8.
Okine BN, Madasu MK, McGowan F, Prendergast C, Gaspar JC, Harhen B, Roche M, Finn DP. N-palmitoylethanolamide in the anterior cingulate cortex attenuates inflammatory pain behaviour indirectly via a CB1 receptor-mediated mechanism. Pain. 2016;157(12):2687–96.
Petrocellis De. Davis, JB, Di Marzo V. Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett. 2001;506:253–6.
Bíró T, Maurer M, Modarres S, Lewin E, Brodie C, Acs G, Acs P, Paus R, Blumberg PM. Characterization of functional vanilloid receptors expressed by mast cells. Blood. 1998;91(4):1332–40.
Kim SR, Kim SU, Oh U, Jin BK. Transient receptor potential vanilloid subtype 1 mediates microglial cell death in vivo and in vitro via Ca2 + -mediated mitochondrial damage and cytochrome c release. J. Immunol. 2006;177:4322–9.
Katsura H, Obata K, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J Neurosci. 2006;26:8680–90.
Petrosino S, Schiano Moriello A, Cerrato S, Fusco M, Puigdemont A, De Petrocellis L, Di Marzo V. The anti-inflammatory mediator palmitoylethanolamide enhances the levels of 2-arachidonoyl-glycerol and potentiates its actions at TRPV1 cation channels. Br J Pharmacol. 2016;173(7):1154–62. https://doi.org/10.1111/bph.13084.
Merritt LL, Martin BR, Walters C, Lichtman AH, Damaj MI. The endogenous cannabinoid system modulates nicotine reward and dependence. J Pharmacol Exp Ther. 2008;326:483–92.
Paterniti I, Impellizzeri D, Crupi R, Morabito R, Campolo M, Esposito E, Cuzzocrea S. Molecular evidence for the involvement of PPAR-δ and PPAR-γ in anti-inflammatory and neuroprotective activities of palmitoylethanolamide after spinal cord trauma. J. Neuroinflamm. 2013;10:20. https://doi.org/10.1186/1742-2094-10-20.
Ghavimi H, Charkhpour M, Ghasemi S, Mesgari M, Hamishehkar H, Hassanzadeh K, Arami S, Hassanzadeh K. Pioglitazone prevents morphine antinociceptive tolerance via ameliorating neuroinflammation in rat cerebral cortex. Pharmacol. Rep. 2015;67:78–84.
Cichewicz DL, Welch SP. Modulation of oral morphine antinociceptive tolerance and naloxone-precipitated withdrawal signs by oral Delta-tetrahydrocannabinol. J Pharmacol Exp Ther. 2003;305:812–7.
Fischer BD, Ward SJ, Henry FE, Dykstra LA. Attenuation of morphine anti-nociceptive tolerance by a CB(1) receptor agonist and an NMDA receptor antagonist: Interactive effects. Neuropharmacology. 2010;58:544–50.
Feng Y, He X, Yang Y, Chao D, Lazarus LH, Xia Y. Current research on opioid receptor function. Curr Drug Targets. 2012;13:230–46.
Skaper SD, Facci L, Barbierato M, Zusso M, Bruschetta G, Impellizzeri D, Cuzzocrea S, Giusti P. N-Palmitoylethanolamine and Neuroinflammation: a novel therapeutic strategy of resolution. Mol Neurobiol. 2015;52(2):1034–42.
Fusco M, Paladini A, Skaper SD, Varrassi G. Sindrome del dolore cronico e neuropatico nell’anziano: basi fisiopatologiche e prospettive di terapia razionale. Pain Nursing Magazine. 2014; 3:94–104. http://www.painnursing.it/rassegna/sindrome-del-dolore-cronico-e-neuropatico-nellanziano-basi-fisiopatologiche-e-prospettive-di-terapia-razionale
Acknowledgements
Funding
The paper has been supported in part by MIUR, “PON Ricerca e Competitività 2007–2013, Project PON 1_02512”. No funding was received for the article processing charges.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Mariella Fusco is an employee of the Epitech Group srl. Giustino Varrassi, Stephen D. Skaper, Daniele Battelli, Panagiotis Zis, Stefano Coaccioli, Maria Caterina Pace and Antonella Paladini have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously published data and does not contain studies with human participants or animals performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to https://doi.org/10.6084/m9.figshare.5972698.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Varrassi, G., Fusco, M., Skaper, S.D. et al. A Pharmacological Rationale to Reduce the Incidence of Opioid Induced Tolerance and Hyperalgesia: A Review. Pain Ther 7, 59–75 (2018). https://doi.org/10.1007/s40122-018-0094-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40122-018-0094-9