
Abstract
Brassica napus is the most common feedstock for biodiesel production, and its cultivation area has been rapidly increased. Thus, B. napus residues left in the field after harvest are valuable resources. However, there have been few studies on biologically active substances from B. napus plant. The objective of this study is to evaluate cytotoxicity/photodynamic activity and antioxidant activity of B. napus plant extracts. B. napus plants were sequentially extracted with organic solvents (hexane, chloroform, ethanol, and water) and then screened for antioxidant activity and cytotoxicity against leukemia U937 and human liver cancer SK-HEP-1 cells. Among the solvent extracts, the cytotoxicity was the highest when cells treated with chloroform extract and irradiated. Degree of apoptosis substantially increased in both cell types in concentration-dependent manner, and non-irradiated cells showed similar results as the control cells. For the highest concentrations (100 μg/ml), toxicity effect in U937 and SK-HEP-1 cells was 94.62 ± 0.15% and 74.16 ± 1.54%, respectively. We observed the number of cells significantly decreased, and vesicles were floating in B. napus chloroform extract (BNCE) and light condition. BNCE induced DNA laddering pattern (between 300 and 1000 bp) and caspase-3/7 activation in both U937 and SK-HEP-1 cells. Total apoptotic U937 and SK-HEP-1 cells following BNCE 100 μg/ml and light treatment were significantly increased (92.62 ± 2.07% and 59.71 ± 4.38%, respectively) compared with control. Our results showed that U937 cells were more sensitive than SK-HEP-1 cells. For the antioxidant activity, B. napus ethanol extract was the highest (IC50 = 0.52 mg/ml).
Similar content being viewed by others
Introduction
Nowadays, photodynamic therapy (PDT) is being spotlighted in advanced nations as new cancer therapy, as it can minimize side effects as well as improve the quality of life of patients [1]. In PDT, photosensitizer injected into the body is selectively accumulated in tumor tissue and then activated by irradiating light of a particular wavelength, resulting in selective destruction of cancer cells. Currently, PDT has been clinically applied in cancer and general skin disease therapy [2, 3]. But, it rarely causes cutaneous phototoxicity, and patients receiving photosensitizers are advised to stay away from direct sunlight and bright ambient light until the photosensitizer concentration in the skin tissues decreases to safe level to avoid cutaneous phototoxicity [4].
Reactions to PDT can be classified as type I or type II. In a type I reaction, the photosensitizer directly reacts with sites of photosensitizer localization to generate free radicals, causing cell toxicity through an oxidation reaction. In a type II reaction, photosensitizer in triplet state directly delivers energy to oxygen to generate singlet oxygen, resulting in damage to cell structures, including lipids and membranes, nucleic acids, and proteins [5]. Most of the photosensitizers either approved for commercialization or undergoing various stages of clinical trials are related to tetrapyrrolic system (porphyrins, chlorins, bacteriochlorins, corroles, and phthalocyanines) or the extended ring systems (texaphyrin). Significant effort is now being employed in the development of derivatives with improved activity and minimal side effects [6, 7].
Brassica napus is Brassicaceae yearly plant originated from China. It has been prescribed for the treatment of dystocia, contraception, and menstrual irregularity for hundreds of years in Korea. In addition, it contains rich amount of vitamins and is ingested as an herbal treatment for spring fever and fatigue [8]. Until now, research has demonstrated the eco-friendly control of soil nematodes using B. napus rapeseed [9], effective weed control using rapeseed green manure [10], and the anticancer activity of brassinolide separated from pollen [11]. However, there has been insufficient research into the biological activity of B. napus. Recently, usage of plant residue after harvesting has increased due to enlarged cultivation areas of biodiesel material and ornamental grass. In this study, we confirmed the photodynamic and antioxidant activities of B. napus extract and provide basic data for the development of an anticancer drug and antioxidant based on its functionality.
Materials and methods
Chemicals and reagents
Roswell Park Memorial Institute (RPMI) 1640 medium, fetal bovine serum (FBS), Dulbecco’s modified Eagle’s medium (DMEM), penicillin–streptomycin (P/S), and trypsin/EDTA were purchased from Gibco BRL (Grand Island, NY, USA). Actinomycin D and colcemid were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Caspase-3/7 activity assay kit and Cell viability assay kit were purchased from Promega (Madison, WI, USA). Annexin V and Dead cell kit was purchased from Merck Millipore (Darmstadt, Germany). 2, 2-Diphenyl-1-picrylhydrazyl (DPPH) was purchased from TCI Co. (Tokyo, Japan).
Preparation of extracts from B. napus
Brassica napus was purchased from an agricultural and marine products market. Dried powder of B. napus was extracted three times with hexane, chloroform, ethanol and water for 4 h. Solvent extraction was performed sequentially from hexane with low polarity to H2O with high polarity. Extracts were filtered through Advantech No. 5C filter paper (Toyo Roshi Kaisha, Ltd., Japan) and concentrated using a rotary evaporator and stored at 4 °C.
Cell line and cell culture
U937 human leukemia cells and SK-HEP-1 human hepatic cancer cells were obtained from the Korean Cell Line Bank (KCHB30052®, KOREA). U937 cells were cultured in RPMI medium, and SK-HEP-1 cells were cultured in DMEM medium. Medium consisted of 10% (v:v) FBS and 1% (v:v) penicillin–streptomycin. Cells were incubated at 37 °C with 5% CO2 in a humidified atmosphere. Cells were passaged every 36 h to maintain an appropriate number.
Determination of the photodynamic activity
The final concentration of DMSO did not exceed 1% (v/v) in all experiments. Cultured cells were treated with various concentrations of chloroform extract of B. napus (BNCE). After 4 h of incubation, cells were irradiated at 3250 Lux for 10 min with daylight fluorescent lamps having wavelengths of 440, 550, and 620 nm or kept in the dark. After 6 h of incubation, all assays were performed. ADCL (actinomycin D and colcemid) common apoptosis agent was treated as positive control and not light-dependent apoptosis induction [12]. The combination of ADCL concentration was 0.1 mM actinomycin D and 0.1 mM colcemid. It mixed equal volume and treated 2 μl in a well.
Monitoring of morphologic changes
To confirm photodynamic activity in cancer cells, we observed morphological changes in the light and dark groups. Cells were seeded at a density of 2 × 104 cells/well in 96-well plate and incubated for 6 h. BNCE was added at concentrations of 50 and 100 μg/ml to each well. After inducing photodynamic activity, various morphologic changes during apoptosis were observed by an IX51 inverted microscope (Olympus America Inc, Center Valley, PA, USA) (200× magnification).
Cell viability assay
To determine whether or not BNCE inhibits growth of cancer cells, cell viability was determined by Cell viability Glo 2.0 assay (Promega). Briefly, cells were seeded at a density of 1 × 104 cells/well in 96-well white plate. After 6 h of incubation, cells were treated with different concentrations of BNCE (10, 25, 50, and 100 μg/ml) in each well. After inducing photodynamic activity, cells were mixed with an equal amount of CellTiter-Glo® 2.0 reagent for 10 min. Luminescence was measured with a GloMax™ Multi+ microplate multimode reader (Promega).
Caspase activity assay
To evaluate changes in caspase activity by BNCE treatment, caspase activity was measured using Caspase-Glo 3/7 assay (Promega). Cells were seeded at a density of 1 × 104 cells/well in 96-well white plate and incubated for 6 h. BNCE was added at different doses of 10, 25, 50, and 100 μg/ml to each well. After inducing photodynamic activity, an equal volume of CellTiter-Glo® reagent was added, plates were shaken for 1 h, and luminescence measured with a GloMax™ Multi+ microplate multimode reader (Promega).
DNA fragmentation
To observe DNA fragmentation, which is biochemical hallmark of apoptosis, cells were seeded at a density of 1 × 106 cells/ml in 6-well plate and incubated for 6 h. BNCE was then added at concentrations of 50, 100, and 200 μg/ml. After inducing photodynamic activity, cells were harvested and washed with ice-cold PBS. Cell pellets were suspended in Lysis buffer (wizard SV Genomic DNA Purification system, Promega) at 4 °C for 10 min and centrifuged at 4 °C 14,000 rpm for 20 min. After collection, the supernatant was incubated with 2 μl of RNase A (10 mg/ml, Ambion) and 10 μl of proteinase K (20 mg/ml, Roche) at 37 °C for 1 h, separately. DNA was precipitated with nine volumes of isopropyl alcohol at 4 °C overnight and centrifuged at 14,000 rpm for 20 min at 4 °C. Pellets were dissolved with 0.5% TE buffer. Electrophoresis was performed on 1.6% agarose containing ethidium bromide. DNA bands were examined using a UV Transilluminator Imaging System.
Flow cytometer analysis
We used a Muse™ Annexin V and Dead cell kit (Millipore, Billerica, MA, USA) for quantitative analysis of apoptosis in cancer cells treated with BNCE. Cells were seeded at a density of 1 × 106 cells/ml in 6-well plate. After 6 h of incubation, BNCE was added at concentrations from 50, 100 μg/ml. After inducing photodynamic activity, cells were harvested, washed with ice-cold PBS, and stained according to the manufacturer’s protocol. Briefly, cell suspensions were adjusted to 2 × 104 cells/100 μl and mixed an equal volume of solution for 20 min at room temperature in the dark. The reactions are measured using a Muse™ Cell Analyzer (Millipore).
Antioxidant activity
Hydrogen-donating ability may serve as significant indicator of antioxidant activity. We assessed the antioxidant activity of each extract using 2, 2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging assay and compared results with that of vitamin C as a positive control. Each extract was applied at concentrations of 250, 500 μg, 1 mg, and 2 mg/ml in 96-well plate, followed by the addition of an equal volume of 0.2 mM DPPH solution. Plate was kept in the dark for 30 min for reaction. The absorbance was measured by an Epoch microplate reader (BioTek Instruments, Inc., USA) at 517 nm. The percentage inhibition of DPPH free radical was calculated as:
where S is the absorbance of the sample containing DPPH, S 0 is the absorbance of the sample containing ethanol, C is the absorbance of the sample solvent containing DPPH, and C 0 is the absorbance of the sample solvent containing ethanol. The IC50 value was defined as the concentration of extracts required to inhibit 50% of radical scavenger activity.
Statistical analysis
All statistical analyses were performed with SPSS (SPSS Inc, Chicago, IL, USA). Significant differences between groups were assessed by one-way analysis of variance (ANOVA). A p < 0.001 was considered to be significant. Results are expressed as the mean ± SD (standard deviation) of three independent experiments.
Results and discussion
BNCE induces morphological change of cancer cells
Cell death is mainly classified into apoptosis and necrosis, which are distinguished according to cell shape and biochemical characteristics. Apoptosis is an active process controlled by genes. On the contrary, necrosis causes inflammation by destroying the cell membrane, resulting in cell swelling and spillage of intracellular materials into the surrounding environment [13]. Apoptotic cell death is morphologically characterized by cell shrinkage, membrane blebbing, cell budding, and formation of apoptotic bodies without plasma membrane breakdown [14]. In this study, we observed photodynamic-mediated cell death by introverted microscopy. As shown in Fig. 1, U937 showed a monocyte shape during normal growth, whereas SK-HEP-1 cells implanted in the well plate were epithelial form [15]. BNCE-treated cells irradiated with light showed apoptotic bodies, and the number of cells was significantly reduced. Floating small vesicles were observed in U937 cells (Fig. 1A). SK-HEP-1 cells showed loss of adherence and cell floating after apoptosis (Fig. 1B). Further, there was no difference in cell morphology between non-irradiated BNCE-treated cells and control cells. The cytotoxicity appeared only in cells irradiated with light after BNCE treatment. Other sequentially solvent extracts did not affect morphology (data not shown). The results of the examination of the UV–Vis absorbance of BNCE showed maximum absorption band at 420, 460, and 660 nm like chlorophyll a. The BNCE may possibly contain considerable amount of porphyrin derivatives.

Morphological changes in U937 and SK-HEP-1 cells treated with BNCE and light. Cells were treated with BNCE 50 and 100 μg/ml concentrations and then irradiated at 3250 Lux for 10 min with daylight fluorescent lamps (wavelengths: 440, 550, and 620 nm) or kept in the dark. After 6 h, cells were visualized under an inverted microscope. Cell death occurred only in the well treated with BNCE and light, the number of cells significantly decreased, and vesicles were floating. Magnification, ×200. (A) U937 cells, (B) SK-HEP-1 cells
BNCE has toxicity effects in dose-dependent manner
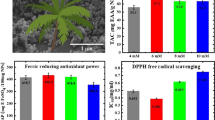
To evaluate the effects of BNCE treatment on growth inhibition in U937 and SK-HEP-1 cells, toxicity effect assay was performed. As shown in Fig. 2A, B, toxicity effect was increased in U937 and SK-HEP-1 cells, depending on light and dose. Compared to the control group, the toxicity effect increased from 16.37 ± 2.60% at a concentration of 10 μg/ml BNCE to 94.62 ± 0.15% at 100 μg/ml BNCE in U937 cells, whereas the effect increased from 14.41 ± 8.21% at 10 μg/ml BNCE to 74.16 ± 1.54% at 100 μg/ml BNCE in SK-HEP-1 cells. A significant toxicity effect was seen above 50 μg/ml BNCE in U937 and SK-HEP-1 cells (59.55 ± 5.75% and 56.07 ± 1.39%, respectively). For the low concentrations (10 and 25 μg/ml), toxicity effect is similar to DMSO treatment in SK-HEP-1 (14.41 ± 8.21% and 16.25 ± 7.36%, respectively). Results of our experiment revealed that the BNCE exhibits relatively high toxicity effect in U937 cells against SK-HEP-1 cells. This corresponds to a previous study reporting that the effects of photosensitizers depend on tissue distribution, affinity for certain cell types, penetration into various subcellular compartments, and accumulation [16]. Further, there was the difference between cells due to the various mechanisms involved in cell death [3, 17, 18]. The phototoxicity at 100 μg/ml of BNCE was higher than ADCL. U937 and SK-HEP-1 cells were not cytotoxic in the absence of light irradiation (Fig. 2).

Growth inhibitory effects of U937 and SK-HEP-1 cells by light treatment. Cells were treated with BNCE (10, 25, 50, and 100 μg/ml) for 4 h. Cells were irradiated with light for 10 min or kept in the dark, followed by incubation for 6 h. Toxicity effect was determined by CellTiter-Glo 2.0 assay. Results are expressed as the mean ± SD of three independent experiments. Statistical analysis was analyzed using one-way ANOVA p < 0.001, dark conditions not significant. Different letters denote significant differences between groups (p < 0.001). (A) U937 cells, (B) SK-HEP-1 cells
BNCE induces apoptosis via caspase-3/7 activation in U937 and SK-HEP-1 cells
Caspase is an important apoptosis inducer that exists as an inactive pro-form on the outer membranes of nuclei and mitochondria, regulated by the balance between anti-apoptotic (Bcl2, Bcl-xL) and pro-apoptotic members (Bax, Bcl-xs, Bid, Bad). Upon activation, caspase cleaves various cellular substrates such as actin, poly (ADP-ribose) polymerase, fodrin, and lamin [19]. Approximately 14 major caspases have been reported until now and are broadly categorized into initiators, effectors or executioners, and inflammatory caspases [20]. In this study, we investigated caspase-3/7 activity, which is an executioner type, according to BNCE and light treatment in U937 and SK-HEP-1 cells. In U937 cells, caspase activity increased until a concentration of 50 μg/ml in a light and BNCE dose-dependent manner. Especially, 50 μg/ml of BNCE resulted in 10.52 ± 0.67 caspase-3/7 activity higher than those of ADCL (9.30 ± 1.06). However, caspase activity was reduced at a BNCE concentration of 100 μg/ml (8.42 ± 0.32) (Fig. 3A). Reduced caspase activity at 100 μg/ml BNCE concentration was maybe due to cellular damage or caspase inactivation because of high BNCE concentration. These results are consistent with several reports that caspase-3/7 activation occurs later with lower doses of cisplatin, whereas caspase-3/7 activation occurs earlier and decreases as time passes with higher doses of cisplatin [21]. Caspase activity in SK-HEP-1 cells increased in a dose-dependent manner and reached a maximum at 100 μg/ml of BNCE. On the other hand, caspase activity of non-irradiated BNCE-treated cells was similar to that of control cells (Fig. 3B). Results indicate that the growth inhibition activity of BNCE is associated with apoptosis via a caspase-dependent mechanism. When U937 and SK-HEP-1 cells were treated with the same concentration of BNCE, caspase activity was higher in U937 cells. These results are due to differences in cell sensitivity.

Measurement of caspase-3/7 activity in U937 and SK-HEP-1 cells by BNCE and light treatment. Cells were treated with BNCE (10, 25, 50, and 100 μg/ml) for 4 h. Cells were irradiated with light for 10 min or kept in the dark, followed by incubation for 6 h. Caspase activity was determined by Caspase-Glo 3/7 assay. Results are expressed as the mean ± SD of three independent experiments. Statistical analysis was analyzed using one-way ANOVA p < 0.001, dark conditions not significant. Different letters denote significant differences between groups (p < 0.001). (A) U937 cells, (B) SK-HEP-1 cells
BNCE induces DNA fragmentation
Fragmentation of nuclear DNA into base pairs through activation of endogenous nucleases is a hallmark of apoptosis [22, 23]. Caspase activation is known to induce apoptosis, activate DNase, and cause DNA fragmentation [15]. To check whether or not inhibition of cancer cell proliferation by BNCE is related to induction of apoptosis, DNA fragmentation was observed by agarose gel electrophoresis using ethidium bromide staining and ultraviolet illumination. As shown in Fig. 4A, B, no DNA ladder pattern was observed in the control and dark. Only BNCE-treated and light-illuminated cells were markedly induced DNA laddering pattern (between 300 bp and 1000 bp) in both U937 and SK-HEP-1 cells, similar to ADCL treatment, indicating apoptotic cell death. However, U937 and SK-HEP-1 cells showed no internucleosomal (200 bp) DNA. Several research groups reported large DNA fragments during apoptosis and proposed that large DNA fragmentation occurs before internucleosomal DNA cleavage, and these large fragments serve as precursors for smaller DNA fragments [14].

Detection of DNA fragmentation in U937 and SK-HEP-1 cells by BNCE and light treatment. Cells were treated with BNCE (50, 100, and 200 μg/ml) for 4 h. Cells were irradiated with light for 10 min or kept in the dark, followed by incubation for 6 h. Cells were harvested, after which DNA was extracted and determined using electrophoresis on 1.6% agarose gel. (A) U937 cells, (B) SK-HEP-1 cells (L light, D dark)
BNCE increases apoptotic cell death
Phosphatidylserine (PS) is located in the inner leaflet of the plasma membrane. However during apoptosis, PS is translocated to the outer leaflet. Exposed PS is recognized and absorbed by phagocytes [24, 25]. External PS was stained by Annexin V and Dead reagent to quantitatively assess the degree of apoptosis using a cell analyzer. Simultaneous staining could distinguish between intact cells, early apoptosis, late apoptosis, or cell death. As a result, degree of apoptosis substantially increased in both cell types in a BNCE concentration-dependent manner, whereas non-irradiated treatment showed results similar to the control (Figs. 5A, 6A). We observed that BNCE induced cell death through apoptosis in both cell types. Total apoptotic U937 cells following BNCE (50 and 100 μg/ml) and light treatment were significantly increased (75.43 ± 4.21% and 92.62 ± 1.77%, respectively) compared with control (6.87 ± 1.36%) (Fig. 5D). For the highest BNCE concentration 100 μg/ml, total apoptosis of SK-HEP-1 cells increased 59.71 ± 4.38% compared to 6.66 ± 1.62% in control. The weakest effect (40.76 ± 3.05%) was observed in SK-HEP-1 cells treated with 50 μg/ml of BNCE for 6 h (Fig. 6D). The numbers of cells in late apoptosis and early apoptosis were similar (Fig. 6B). The total apoptosis of each cells following ADCL treatment was slightly lower than treatment 100 μg/ml BNCE and light (Figs. 5, 6). The results suggest that the relative apoptosis efficacy of 100 μg/ml BNCE in U937 cells within 6 h is substantially higher compared to 100 μg/ml BNCE in SK-HEP-1 cells. BNCE induced more late apoptosis in U937 cells than did SK-HEP-1 within 6 h. Therefore, U937 cells were more sensitive than SK-HEP-1 cells.

U937 cells viability by BNCE treatment and light irradiation. Cells were treated with BNCE (50 and 100 μg/ml) for 4 h. Irradiated with light for 10 min or kept in the dark, followed by incubation for 6 h. Cells were stained using a Muse™ Annexin V and Dead cell kit for 20 min and analyzed using a Muse™ Cell Analyzer. Results show a dose-dependent increase in the number of apoptotic U937 cells compared to dark conditions. (A), (B) Scatter plots showing distribution of Annexin V and 7-AAD staining (UL: dead cells, UR: late apoptotic/dead cells, LL: live cells, and LR: early apoptotic cells). The (C), (D) represent % of cells and the summary mean percentages ± SD of apoptosis (early and late apoptosis) of three independent experiments in dark and light. Statistical analysis was analyzed using one-way ANOVA

SK-HEP-1 cells viability by BNCE treatment and light irradiation. Cells were treated with BNCE (50 and 100 μg/ml) for 4 h. Irradiated with light for 10 min or kept in the dark, followed by incubation for 6 h. Cells were stained with Muse™ Annexin V and Dead cell kit for 20 min and analyzed using a Muse™ Cell Analyzer. Results show a dose-dependent increase in the number of apoptotic U937 cells compared to dark conditions. (A), (B) Scatter plots showing distribution of Annexin V and 7-AAD staining (UL: dead cells, UR: late apoptotic/dead cells, LL: live cells, and LR: early apoptotic cells). The (C), (D) represent % of cells and the summary mean percentages ± SD of apoptosis (early and late apoptosis) of three independent experiments in dark and light. Statistical analysis was analyzed using one-way ANOVA
Brassica napus extracts show antioxidant activity
Although excessive ROS production destroys normal cells, it plays a key role in apoptosis in cancer cells [26]. The radical scavenging activity of vitamin C was 100% at a concentration of 20 μg/ml (data not shown). In measuring the antioxidant activity of each extract based on control, IC50 values in hexane and H2O extracts were 0.63 ± 0.01 mg/ml and 0.64 ± 0.01 mg/ml, respectively. The IC50 value of EtOH extract was 0.52 ± 0.01 mg/ml and resulted in the highest activity. The IC50 value of BNCE was lower than those of other extracts at 1.47 ± 0.01 mg/ml. Antioxidant activity of H2O, EtOH, hexane, and CHCl3 extracts increased sequentially in dose-dependent manner (Fig. 7). BNCE, which most effectively suppressed cell proliferation, had the weakest antioxidant effect. Our results correspond to several reports that antioxidants increased cancer cell proliferation due to eliminating radical and ROS induced by apoptosis [27, 28]. In addition, these results were due to complex interactions among the various compounds contained in B. napus.

Antioxidant activity of sequential solvent extracts from B. napus. Antioxidant activity was evaluated by measuring reduction in DPPH radical detected at 517 nm. Vitamin C was used as a positive control. Results are expressed as the mean ± SD of three independent experiments
Apoptosis induction is the most effective means to control growth of cancer. In terms of cancer therapy, studies on the safety and efficacy of active materials derived from natural products can contribute to the development of new drugs. In this paper, we confirmed that chloroform extract of B. napus induced light-dependent apoptosis in U937 and SK-HEP-1 cells. Although BNCE is a crude extract, it has strong effect cell death at low concentrations. Further studies are needed to identify active compounds and evaluate its in vivo activity. Therefore, B. napus has sufficient potential as a photosensitizer and antioxidant source.
References
Juarranz A, Jaen P, Sanz-Rodriguez F, Cuevas J, Gonzalez S (2008) Photodynamic therapy of cancer. Basic principles and applications. Clin Transl Oncol 10:148–154
Dolmans DEJGJ, Fukumura D, Jain RK (2003) Photodynamic therapy for cancer. Nat Rev Cancer 3:380–387
Gomer CJ, Rucker N, Ferrario A, Wong S (1989) Properties and applications of photodynamic therapy. Radiat Res 120:1–18
Bromley E, Briggs B, Keltner L, Wang SS (2011) Characterization of cutaneous photosensitivity in healthy volunteers receiving talaporfin sodium. Photodermatol Photoimmunol Photomed 27:85–89
Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J (2011) Photodynamic therapy of cancer: an update. CA Cancer J Clin 61:250–281
O’Connor AE, Gallagher WM, Byrne AT (2009) Porphyrin and nonporphyrin photosensitizers in oncology: preclinical and clinical advances in photodynamic therapy. Photochem Photobiol 85:1053–1074
Pandey RK, Kessel D, Dougherty TJ (2016) Handbook of photodynamic therapy: updates on recent applications of porphyrin-based compounds. World Scientific Inc., Singapore, pp 5–24
Saeidnia S, Gohari AR (2012) Importance of Brassica napus as a medicinal food plant. J Med Plants Res 6:2700–2703
Kim HK, Ma KC, Kim MS, Bang GP, Kim JK, Park MS (2010) Environmentally-friendly control of Soil nematode by crashed-rape (Brassica napus) seed. Korean J Environ Agric 29:282–286
Boydston RA, Hang A (1995) Rapeseed (Brassica napus) green manure crop suppresses weeds in potato (Solanum tuberosum). Weed Technol 9:669–675
Wu YD, Lou YJ (2007) Brassinolide, a plant sterol from pollen of Brassica napus L., induces apoptosis in human prostate cancer PC-3 cells. Pharmazie 62:392–395
Lu DF, Wang YS, Li C, Wei GJ, Chen R, Dong DM, Yao M (2015) Actinomycin D inhibits cell proliferations and promotes apoptosis in osteosarcoma cells. Int J Clin Exp Med 8:1904–1911
Lieberthal W, Koh JS, Levine JS (1998) Necrosis and apoptosis in acute renal failure. Semin Nephrol 18:505–518
Comini LR, Fernandez IM, Rumie Vittar NB, Núñez Montoya SC, Cabrera JL, Rivarola VA (2011) Photodynamic activity of anthraquinones isolated from Heterophyllaea pustulata Hook f. (Rubiaceae) on MCF-7c3 breast cancer cells. Phytomedicine 18:1093–1095
Ha JY, Kim MK, Lee JY, Choi EB, Hong CO, Lee BW, Bae CH, Kim KK (2015) Isolation and structure identification of photosensitizer from Perilla frutescens leaves which induces apoptosis in U937. J Life Sci 25:53–61
Rosenkranz AA, Jans DA, Sobolev AS (2000) Targeted intracellular delivery of photosensitizers to enhance photodynamic efficiency. Immunol Cell Biol 78:452–464
Lin CW, Shulok JR, Kirley SD, Cincotta L, Foley JW (1991) Lysosomal localization and mechanism of uptake of Nile blue photosensitizers in tumor cells. Cancer Res 51:2710–2719
Wood SR, Holroyd JA, Brown SB (1997) The subcellular localization of Zn (II) phthalocyanines and their redistribution on exposure to light. Photochem Photobiol 65:397–402
Saraste A, Pulkki K (2000) Morphologic and biochemical hallmarks of apoptosis. Cardiovasc Res 45:528–537
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
Cregan IL, Dharmarajan AM, Fox SA (2013) Mechanisms of cisplatin-induced cell death in malignant mesothelioma cells: role of inhibitor of apoptosis proteins (IAPs) and caspases. Int J Oncol 42:444–452
Lee SG, Yu MH, Lee SP, Lee IS (2008) Antioxidant activities and induction of apoptosis by methanol extracts from avocado. J Korean Soc Food Sci Nutr 37:269–275
White E (1993) Death-defying acts: a meeting review on apoptosis. Genes Dev 7:2277–2284
Cheng YL, Chang WL, Lee SC, Liu YG, Chen CJ, Lin SZ, Tsai NM, Yu DS, Yen CY, Harn HJ (2004) Acetone extract of Angelica sinensis inhibits proliferation of human cancer cells via inducing cell cycle arrest and apoptosis. Life Sci 75:1579–1594
Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR (1995) Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med 182:1545–1556
Alvarez MG, Príncipe F, Milanesio ME, Durantini EN, Rivarola V (2005) Photodynamic damages induced by a monocationic porphyrin derivative in a human carcinoma cell line. Int J Biochem Cell Biol 37:2504–2512
Akan Z, Garip AI (2013) Antioxidants may p13rotect cancer cells from apoptosis signals and enhance cell viability. Asian Pac J Cancer Prev 14:4611–4614
Fu Y, Yang G, Zhu F, Peng C, Li W, Li H, Kim HG, Bode AM, Dong Z, Dong Z (2014) Antioxidants decrease the apoptotic effect of 5-Fu in colon cancer by regulating Src-dependent caspase-7 phosphorylation. Cell Death Dis 5:e983
Acknowledgments
This work was supported by a 2-year Research Grant of Pusan National University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Choi, E.B., Lee, M.W., Park, J.E. et al. Photodynamic apoptosis and antioxidant activities of Brassica napus extracts in U937 and SK-HEP-1 cells. Appl Biol Chem 60, 427–435 (2017). https://doi.org/10.1007/s13765-017-0295-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13765-017-0295-7