
Abstract
Apolipoprotein E (apoE) is an essential protein in lipid and cholesterol metabolism. Although the three common isoforms in humans differ only at two sites, their consequences in Alzheimer’s disease (AD) are dramatically different: only the ε4 allele is a major genetic risk factor for late-onset Alzheimer’s disease. The isoforms exist as a mixture of oligomers, primarily tetramer, at low μM concentrations in a lipid-free environment. This self-association is involved in equilibrium with the lipid-free state, and the oligomerization interface overlaps with the lipid-binding region. Elucidation of apoE wild-type (WT) structures at an oligomeric state, however, has not yet been achieved. To address this need, we used native electrospray ionization and mass spectrometry (native MS) coupled with ion mobility (IM) to examine the monomer and tetramer of the three WT isoforms. Although collision-induced unfolding (CIU) cannot distinguish the WT isoforms, the monomeric mutant (MM) of apoE3 shows higher stability when submitted to CIU than the WT monomer. From ion-mobility measurements, we obtained the collision cross section and built a coarse-grained model for the tetramer. Application of electron-capture dissociation (ECD) to the tetramer causes unfolding starting from the C-terminal domain, in good agreement with solution denaturation data, and provides additional support for the C4 symmetry structure of the tetramer.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Apolipoprotein E (apoE) proteins are separately expressed in liver and the central nervous system to regulate lipid homeostasis via lipid transport and cholesterol metabolism [1,2,3]. The APOE gene is a major genetic factor for late-onset Alzheimer’s disease (LOAD); among the three allelic variants in human, ε4 is the major risk factor, ε3 appears neutral, whereas ε2 is suggested to be protective [4,5,6]. The differences between the three isoforms are single-site changes to 299 constituent amino acids. ApoE2 has cysteines at sites 112 and 158, whereas the prevalent isoform apoE3 has arginine at site 158, and apoE4 has arginines at both sites. Isoform-dependent biochemical properties and functions of apoE are presumably caused by these single amino acid polymorphisms [2, 3].
In a lipid-free environment, apoE exists with two independent-folded domains connected by a protease-sensitive hinge region [2]. The N-terminal domain, which contains a receptor-binding region, is a four-helix-bundle [7]. High-resolution structures of the lipid-binding C-terminal domain or the wild-type (WT) full-length protein are unknown owing to the disorder and propensity to self-associate of the C-terminal domain [2, 8,9,10,11,12]. Lipid-free apoE exists as a mixture, primarily tetramer at low micromolar concentrations, but will dissociate to the monomer when the concentration is lowered [8,9,10,11,12,13]. Thus far, only one NMR structure exists of an apoE3 monomeric mutant (MM), for which five residues in the C-terminal domain were substituted to prevent self-association [14].
The physiological concentrations of apoE are 40–70 μg/mL (1–2 μM) in plasma and 3–5 μg/mL (~ 0.1 μM) in cerebrospinal fluid [6]. Lipid-bound apoE, the prominent species in vivo, is likely in equilibrium with lipid-free apoE [15]. The presence of lipid-poor or lipid-free reservoirs of apoE in vivo likely makes its self-association important for function [12] especially given that the lipid-binding region overlaps with the self-association region [2, 12], whereas only the monomeric form binds lipid [13]. Sedimentation methods [8, 9, 15], fluorescence correlation spectroscopy [16] for the equilibrium constants, and mass spectrometry (MS)-based footprinting [17,18,19] can interrogate equilibrium and conformational changes among the three isoforms and between the WT and MM. Although footprinting coupled with bottom-up MS analysis can report on changes in this heterogeneous system, at the peptide and even residue level, they do not directly provide compositional information of the solution species [17,18,19,20,21,22,23].
Native electrospray ionization MS (native MS) introduces proteins and protein assemblies into the gas phase in a native or near-native state, preserving many non-covalent interactions [24,25,26]. Native MS can, thus, provide an overall view of the protein state ranging from small peptides [27] to large mega-Dalton complexes [28, 29]. Additional low-resolution information about protein shape and structure comes from native MS when it is coupled with ion mobility (IM) and coarse-grained modeling [30,31,32,33]. Submitting the protein to increasing collision energy prior to IM causes collision-induced unfolding (CIU) to report on protein stability in the gas phase and indirectly in solution [34,35,36,37,38,39]. Combining collisional activation with top-down electron-capture dissociation (ECD) provides sequence information on flexible regions [37, 40, 41] and on exposed protein surfaces [42].
In this study, we examined the oligomeric compositions of the three WT apoE isoforms and apoE3 MM [14] by native MS. We compared the conformation and stability of each isoform at monomeric and tetrameric states based on their collision cross sections (CCS) and collision-induced unfolding (CIU). These results, when combined, support a coarse-grained model of the apoE tetramer that is consistent with the top-down ECD results.
Materials and Methods
Materials
Three apoE WT isoforms and one MM of apoE3 were expressed and purified as described earlier [43]. Mutations of apoE3, introduced into the c-DNA using Agilent QuikChange site-directed mutagenesis kit (Santa Clara, CA), were accomplished as previously reported (PDB: 2L7B) [14]. Ammonium acetate (≥ 99.999% trace metals basis) and LC/MS-grade water were obtained from MilliporeSigma (St. Louis, MO).
Sample Preparation for Native MS
ApoE protein solutions were buffer exchanged with 200 mM ammonium acetate (pH = 7) with a Vivaspin 10 kDa molecular weight cut off filter (Sartorius AG, Goettingen, Germany) at 4 °C and diluted to desired concentrations as needed with the same buffer (5 or 15 μM). About 5-μL sample solution was loaded in a nanoESI emitter (Thermo Fisher, Waltham, MA) and injected into mass spectrometers.
Ion Mobility MS Analysis
Mass spectra for the native MS and ion mobility were collected on a Waters hybrid ion-mobility quadrupole time-of-flight mass spectrometer SYNAPT G2 HDMS (Milford, MA) with the following settings: capillary voltage 1.6–1.7 kV, sampling cone 20 V, extraction cone 10 V, and source temperature 30 °C. The backing pressure was 5.6–5.8 mBar for transferring intact protein ions. Nitrogen was used as the carrier gas in ion mobility experiments at a flow rate of 35 mL/min. IMS wave velocity and wave height were 500 m/s and 20 V, respectively. Collision voltages in trap region were varied between 10 and 200 V to unfold protein oligomers [44], while the transfer region was maintained at 20 V. The ion mobility measurements of CCS were calibrated as previously reported [37]. Data were exported from Masslynx (v 4.1) and Driftscope software (Water Inc., Milford, MA). Native mass spectra were processed with UniDec [45]. The collision cross-section (CCS) of apoE3MM (PDB file: 2L7B) was calculated using the IMPACT projection approximation with a scaling factor of 1.14. CIU fingerprint contour plots were constructed by plotting CCS against trap collision voltages with normalized intensities by using OriginPro 9.0 (OriginLab Corp., Northampton, MA).
ECD MS Analysis
ECD product-ion mass spectra were acquired on a Bruker Daltonics 12 T FTICR mass spectrometer (Bremen, Germany). The source settings were: capillary voltage 1.5–1.7 kV, drying gas temperature 25 °C, and drying gas flow rate 2.0 L/min. The skimmer 1 voltage was varied between 20 and 200 V to pre-activate ions. Other ion-optics parameters were adjusted to optimize signal intensities. Ion accumulation time was 2 s, and the time of flight was 2.5 ms. All charge states in a narrow distribution were subjected to ECD with a pulse length of 0.04 s, bias of 0.4 V, lens of 10 V, and hollow-cathode current of 1.6 A. Peak picking was performed with SNAP centroid peak on Bruker Data Analysis. Data were processed with Bruker Daltonics BioTools and manually verified by comparing to theoretical fragment ions predicted by Protein Prospector (University of California–San Francisco MS facility).
Coarse-Grained Modeling
Coarse-grained modeling was performed using a Monte Carlo annealing algorithm built within the Integrative Modeling Platform library in Python. Experimental data were input in the form of spherical radii corresponding to measured collision cross sections, and in some cases, distance or connectivity restraints between multiple spheres that optimized agreement with experimental cross sections. Ensembles of 10,000 models were generated according to scoring functions with and without C4 symmetry constraints, and models with tetrahedral symmetry were generated by applying the appropriate transformation to the C4 models. For each ensemble, the CCS values were calculated using the IMPACT projection approximation and considered only models within ± 3% of the experimental cross section. Models agreeing with all CCS datasets were then clustered into an average-linkage hierarchy by using a pairwise RMSD matrix, and the probability density function of each cluster was estimated using Gaussian kernels.
Results and Discussion
Native MS Reveals Heterogeneity of ApoE
Native mass spectra obtained on the IM-QToF instrument at the lowest collision energy (10 V) reveal the oligomeric heterogeneity of WT apoE solution at 5 μM for all three isoforms (Figure 1). For apoE2WT (Figure 1a), the most abundant species is a tetramer with a narrow charge-state distribution from 24+ to 19+ centered at approximately m/z 6500 (M.W. = 138.2 kDa). The second most abundant species that displays three charge states 11+ to 9+ from m/z 3000 to 4000 is a monomer (M.W. = 34.35 kDa). Small amounts of dimer spanning 16+ to 14+ (M.W. = 68.80 kDa) and trimer from 20+ to 17+ (M.W. = 103.5 kDa) are also observed. The ion at m/z = 5750 represents both trimer at 18+ and tetramer at 24+, according to the resolved arrival time distributions (ATDs) of ions with this m/z in the IM experiment, and supported by the clear presence of a trimer at 17+. Mass spectra are simplified, and the relative abundances of monomer, dimer, and trimer are reduced when protein concentrations are increased to 15 μM (Figure S1). This concentration-dependent behavior is consistent with chemical equilibrium in solution and suggests that the oligomers in the gas phase are relevant to those in solution. Furthermore, the presence of dimer and trimer may not be easily identified by other biophysical methods because their relative abundances are too low.

Native mass spectra of (a) apoE2WT, (b) apoE3WT, (c) apoE4WT, and (d) apoE3MM at 5 μM. Monomer (blue), dimer (green), trimer (yellow), and tetramer (red) are labeled, respectively
In contrast to the WT protein, we saw only peaks representing the monomer for native MS of the apoE3MM (Figure 1d). This is a good negative control establishing that the oligomers are not formed by non-specific interactions in solution or gas phase. The charge distribution of the monomer in apoE3MM is the same as that of WT, ranging from 11+ to 9+. The native MS data not only establish the different oligomeric compositions of WT and MM but also provide direct evidence of different oligomeric species that agree with the previously proposed apoE self-association process [8, 9, 16].
Ion Mobility
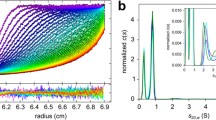
It is important to ensure minimal gas-phase unfolding of protein structure when using native MS. We first converted the ATDs of the monomeric species (6–9 ms) and the tetrameric species (11–16 ms) of the three WT isoforms at presumably their lowest energy (i.e., collisional activation voltage is at the lowest value, 10 V) into CCS distributions (Figure 2a for apoE3WT; also see Figure S2 for apoE2WT and apoE4WT; median CCS values of all recorded charge states are provided in Table S1). For the monomeric species, the three charge states show almost identical widths and median values of the CCS distributions. Median CCS values of the tetrameric species at different charge states are similar (standard deviations are less than 2%), yet with a trend that the median CCS values gradually decrease as charge increases. Overall, the CCS distributions of both monomers and tetramers are not charge-state dependent, and their peak shapes indicate the presence of one major conformation. The median value of the CCS distribution for apoE3MM at 10+ (2440 Å2) under the same experimental conditions is ~ 6.5% smaller than a theoretically calculated CCS of the solution NMR structure (2610 Å2), suggesting slight compaction of the structure owing to gas phase collapse [30] and no significant gas-phase activation at the ground state. Interestingly, the width of the CCS distribution of the tetrameric species, approximately 1000 Å2, is only about 2-fold broader than that of monomeric species, and relatively narrow compared to some similarly sized proteins [46]. This indicates that the tetrameric species also has a nearly singular and stable conformation.

(a) The CCS distributions of WT apoE3 monomeric (left) and tetrameric species (right) for different charge states. (b) The CCS distributions of the monomeric species (M) of all isoforms at charge state 10+, and the tetrameric species (T) of three WT isoforms at charge state 22 +
The CCS distributions at the most abundant charge states (i.e., 10+ for monomer (M) and 22+ for tetramer (T)) are the same for the three WT isoforms (Figure 2b), indicating their conformational similarities. Furthermore, ApoE3MM also possesses a similar conformation as the WT monomeric species because they display the same CCS distributions. This conclusion contradicts previous reports that apoE3MM and WT are significantly different, particularly in the C-terminal domain [17, 18]. This is because those studies pertain to a solution structure [17, 18] that is a weighted average of all the oligomeric species of the WT. In contrast, we extracted information from the WT monomer and directly compared it to the MM. That the three apoE WT isoforms have similar structures according to the gas-phase CCS is consistent with previous reports [17, 18].
Collision-Induced Unfolding
It is difficult to resolve closely resembled structures in IM at the ground state. To evaluate further the higher-order structure of apoE, we applied a CIU strategy [34,35,36,37,38] on the monomeric and tetrameric species, respectively. The CIU contour plots for the monomeric species of apoE3WT at 10+ (Figure 3a), apoE3MM at 10+ (Figure 3b), and the tetrameric species of apoE3WT at 22+ (Figure 3c) show that all selected protein ions adopt more extended structures with increasing trap collision voltages. We chose to characterize CIU of monomeric species at 10+ charge state because not only is this charge state the most abundant but also an optimal charge-state choice for CIU of a 35 kDa protein according to the power-law correlation [47]. By comparing the centroid CCS at the ground state (10 V) to that at the highest energy activation state in our experiment (160 V), we observed approximately a 23% structural extension. Three main features, including the initial native state, an intermediate partial unfolded state, and a stable major unfolded state, were observed for the WT monomer over the experimental activation frame. Our observation of the three features supports the two-domain structure proposal of Chen et al. [14] because there is a general correlation between protein gas-phase unfolding intermediates and domain structures in solution, as proposed by Zhong et al. [47].

CIU fingerprints of (a) apoE3WT monomer, (b) apoE3MM monomer, (c) apoE3WT tetramer. The scale of normalized ion intensities is labeled on the right
Although the monomeric species of apoE3WT and apoE3MM are iso-cross-sectional at the ground state, apoE3MM produces a different CIU fingerprint than does apoE3WT. Transition from the first feature to the second feature occurs when the collision energy is adjusted corresponding to 50 V for both isoforms. The first feature, however, is more abundant for apoE3MM compared to that of apoE3WT at 55 V, indicating that apoE3MM is more stable than apoE3WT. An additional fourth feature (CCS ~ 3100 Å2) for apoE3MM appears at 55 V, peaks at 65 V, and co-exists with the major unfolded state (CCS ~ 2900 Å2) but with gradual decreasing relative abundance at higher energies. In contrast to a proposed WT structure [2], the NMR structure of apoE3MM shows extensive salt-bridging and hydrogen bonding between the N-terminal and C-terminal domains [14]. Highly intermingled domain structures may induce additional features in the CIU fingerprint [47], possibly explaining the fourth feature and the broadened CCS distribution at higher collision voltages in the CIU of apoE3MM. To check whether CIU fingerprints only report CCS changes of ions within a specific m/z range, we examined the full m/z range of the mass spectra recorded at different CID activation energies. A fraction of higher charge state ions at approximately m/z = 1500, corresponding to the partially denatured protein ions, are observed for three WT isoforms in the range corresponding to 20 to 120 V, but this is not significant for apoE3MM (Figure S3). These observations additionally support the hypothesis that the monomeric species of apoE3MM is more stable than the WT. As a result, the fourth CIU feature corresponding to a conformation that is more extended than the major unfolding state may survive and exist for apoE3MM. In contrast, for the WT isoforms, any further energy increases may cause protein denaturation or fragmentation so that the protein ions at the original m/z range are no longer tractable.
The tetramer remains assembled while a fraction of the protein undergoes unfolding as the collision voltage increases, displaying four main features and one low-intensity fifth feature. Centroid CCS of the fourth (~ 8800 Å2) and fifth (~ 9500 Å2) features increased about 17% and 27%, respectively, compared to that of the ground-state conformation (~ 7500 Å2). The tetramer does not undergo a large extent of unfolding [36]. This is in part because the tetramers have already a large CCS or undergo dissociation before further unfolding under these experimental conditions. All three WT isoforms have essentially the same CIU fingerprints for both monomeric and tetrameric species (see Figure S4 for apoE2WT and apoE4WT). The unfolding differences for the three WT isoforms in solution are found in the N-terminal domain [48]. The structural difference in the N-terminal domain was also revealed by MS-based footprinting [19]. We hypothesize that gas-phase unfolding of the N-terminal domain does not occur in our CIU experiment owing to the range of activation energies applied in this experiment. As a result, three WT isoforms display similar CIU patterns at both monomeric and tetrameric states. An ECD experiment, in which unfolding regions can be localized, would help test our hypothesis (discussed later).
Coarse-Grained Model of ApoE Tetramer
ApoE has a high propensity to tetramerize in a lipid-free environment via the C-terminal domain [2, 8, 9, 17, 18], making it difficult to obtain structures for any WT oligomeric state by NMR or X-ray crystallography. The only available full-length structure for an ApoE monomer is of apoE3MM by solution NMR [14]. There are currently no reported structures of any apoE oligomers. Nevertheless, with CCS constraints from both monomeric and tetrameric species, we can construct a model for the tetramer by using a coarse-grained approach [32, 33].
In coarse-grained modeling, one globular protein chain is usually represented as one sphere whereas a protein complex would consist of several overlapping spheres [32, 33, 49]. Our initial attempts to model the apoE tetramer using one sphere per monomer, however, resulted in no models in agreement with both basic connectivity [50, 51] requirements and the experimentally derived CCS restraints. We hypothesized that the assumption of sphericity, which holds for many globular, single-domain proteins, is not appropriate for the dynamic two-domain structure of apoE. Supporting this hypothesis is evidence that tetrameric apoE cannot be calibrated as a globular protein on the basis of size-exclusion chromatography [15].
Therefore, we developed a two-domain model for the monomeric subunit based on the proposed structures for the WT [2, 52], which agrees with our experimental CCS restraints. To build our coarse-grained model, we started with a computational model from Hsieh [52] featuring two distinct domains, in contrast to the NMR structure of apoE3MM where the domains are intermeshed [14] (note also that the mutant, designed to be a monomer, has a considerably different structure than the WTs). The two-domain structure was split at residue 183 to make a two-body structure, and the CCS of each domain was computed by using the scaled projection approximation function in IMPACT. Next, each domain was represented as a sphere with radii corresponding to the computed CCS values. A linear search was performed to verify that the two spheres could be combined with a physically relevant overlap value to yield a CCS value in agreement with experiment; however, these values were not used to bias the tetramer modeling efforts.
Next, we assembled four copies of this two-domain monomer by applying a connectivity restraint between the C-terminal domains. Our model assumed symmetry, which is very common within homomeric protein complexes, and drastically reduces variability in the ensemble of putative structures derived from a Monte Carlo search [32]. We first assumed a C4 symmetry, which combined with the connectivity restraints between the C-terminal domains yields a planar arrangement of C-terminal domains in a square geometry. The free model parameters that are explored in the Monte Carlo search are (1) the distance between adjacent C-terminal domains, (2) the distance between C-terminal and N-terminal domains within a monomer, and (3) the angle created by an N-terminal domain from the plane of the C-terminal domains. These parameters were sampled from spherical overlap values of 15% to 45% (in 1 and 2) or from 0 to 180° (in 3). The resulting ensemble revealed two well-defined structural families that agreed with all the experimental data (Figure 4a). The most prominent family, cluster 1 (green), accounts for over 80% of the possible solutions in our simulation (mean structures of the two families sampled in our experiments are shown in Figure 4b). The favored cluster 1 is non-planar compared to the cluster 2.

Coarse-grained model of an apoE tetramer. (a) An examination of the average-linkage hierarchy of all models: the dominant cluster 1 is colored in green, and cluster 2 in red. (b) The average structures of the cluster 1, cluster 2, and the tetrahedron symmetry. The C-terminal domain is colored in gray, and the N-terminal domain in green. A visual depiction of the degrees of freedom searched to give the model is shown in SI, Figure S6. (C) The average CCSs of each cluster. Deviations compared to the averaged experimental values are in parenthesis
We also explored the possibility of tetrahedral structures but found that the CCSs of these structures are much larger than the experimental cross sections (Figure 4c) and, therefore, not likely to represent the native or gas-phase conformation of ApoE. Models generated without any symmetry requirements appear to converge around the same structural families found in the C4 ensemble; however, the clusters are more poorly defined. We note that although we have included error values comparing the monomer CCS within the tetramer with our experimental results for the monomer, we cannot rule out stabilization or structural rearrangement of the monomer within the tetramer. Therefore, the error values presented for the monomer do not necessarily inform the quality of the model.
ECD of ApoE Tetramer
Both the IM-MS results and the coarse-grained model are at low resolution. To improve the modeling, one can incorporate collision-induced dissociation (CID) in the mass spectrometer to obtain topology [32]. Using CID for the order of subunits is not very informative for a homo-oligomer. Therefore, we turned to ECD to obtain more structural information to support our model for the tetramer.
We followed the same idea as in CIU, gradually activating the tetrameric complex by increasing skimmer I voltage, and allowing ECD to locate unfolding regions that cannot be identified in CIU and IM [37, 41, 42]. We submitted the entire narrow charge-state distribution of the tetramer to ECD because the charge state envelope of the tetramer from 20+ to 25+, centered at m/z = 6000, is at the upper isolation limit of the instrument [41] (see Figure 5a for ECD spectra at varying skimmer I voltages for apoE3). Fragment ions from ECD are below m/z = 2000 whereas a series of charge-reduced precursor ions fall into the high m/z region. The density of fragments below m/z = 2000 becomes greater with increases in the skimmer I potential (from 30 to 160 V). Interestingly, another tetrameric complex with slightly lower mass appears when the skimmer I voltage becomes 160 V (Figure S5). This new tetramer arises by the loss of C-terminal tail 293–299 from the original tetramer, considering that the mass difference between the two tetramers is approximately 760 Da, and we see an abundant y7 ion at m/z 765.3473. Consequently, the second tetramer, when submitted to ECD, produces a series of z ions that are 765.35 Da less than the corresponding z ions from original tetramer. Assignment of z ions from the second tetramer is not included in the text unless otherwise mentioned.

(a) ECD mass spectra of apoE3WT tetramer at different in-source (IS) pre-activation voltages of 30, 60, 80, 120, and 160 V. (b) Assignment of ECD fragment ions in the N-terminal domain (NT; upper panel) and the C-terminal domain (CT; bottom panel)
The ECD fragment ions that we identified based on their accurate mass show that the C- and N-termini have different fragmentation patterns (Figure 5b). Fragment ions from the C-terminus are observed starting at the minimum activation energy required for detectable signal intensities of the precursor ions. At low activation voltages (i.e., 30 V and 60 V), z ions from residues 275–299 already form, strongly suggesting that this region is highly flexible [14]. With a pre-activation voltage higher than 80 V, fragmentation occurs deeper along the backbone from the C-terminus until residue 221. The new tetramer formed by a loss of the C-terminal tail 293–299 from the original tetramer, fragments to z ions even deeper into the “hinge” region to residue 192, suggesting more unfolding of this new tetramer. There are no c ions from the N-terminus until the pre-activation voltage is advanced to 120 V. There are fewer c ions, and their relative abundances are much lower compared to the z ions arising from the C-terminus.
We also found this ECD fragmentation pattern for the apoE2 and apoE4 tetramers. The overall trend is very similar among three isoforms except a few more z ions form from the C-terminus of apoE4 at low pre-activation voltages (Figure S7). Our gas-phase unfolding data showing that most ECD fragmentation ions are from the C-terminal domain agree with solution unfolding results [48]. The two domains are independently unfolded in solution; the four-helix-bundle N-terminal domain unfolds at a much higher denaturant concentration than the C-terminal domain [48].
We have proposed, based on studies of model systems, that the extent of ECD fragmentation correlates well with protein flexibility (i.e., with X-ray structural B-factors) [37, 41]. Li et al. [42] reported that ECD fragmentation originates not only from flexible regions but also from surface-exposed regions. We hypothesize that ECD can validate coarse-grained models of oligomers. Our observations that the highly flexible C-terminal tail (293–299) fragments upon applying low to medium pre-activation voltages (30–80 V) whereas no fragmentation occurs at the highly structured and stable N-terminal domain are consistent with our proposed correlation between ECD fragmentation and B-factors.
When activating a protein complex by CID, asymmetric charge partitioning occurs whereby one monomer subunit is ejected with disproportionate amount of charge relative to its mass [53]. For apoE, the pre-activation voltage at the source region does not sufficiently energize the complex to eject a monomer. We suggest that one monomer subunit starts to unfold while the tetrameric complex remains integral as the pre-activation voltage (ion energy) increases. When the C-terminal domain continues to unfold, more z ions are produced. Although the C-terminal domain is at the tetramer interface [2, 8, 9, 17], the arrangement in our model provides the opportunity for the C-terminal domain to interact readily with electrons. We found fewer c ions, suggesting that the N-terminal domain does not extensively unfold, as is also supported by the CIU/IM experiment. In a tetrahedron arrangement, on the contrary, the C-terminal domain is protected somewhat by the N-terminal domain. Thus, fewer z ions would be formed owing to the limited access to electrons, and more c ions from the N-terminal would be expected. Thus, the ECD results showing that fragmentation continues deeper along the backbone from the C-terminal than the N-terminus further confirms the proposed model of the apoE tetramer.
Of note are z ions from residues 221–299 of the original tetramer and 192–299 of the second tetramer that lose the C-terminus 293–299 at the highest pre-activation voltage. There must be regions other than 221–299 for the original tetramer and 192–299 for the new, truncated tetramer responsible for holding the tetrameric complex together. Although most studies suggest that the critical oligomerization interface is located in the C-terminus [2, 8, 9, 17], Gau et al. [18] found that 183–205 in the “hinge” region may be also involved. Our ECD study also supports Gau’s hypothesis by showing that tetramer remains intact when region 221–299 has unfolded in the gas phase.
Conclusions
Investigating ApoE structure and self-association behavior can bring insight for understanding its function in vivo. Higher order structure, however, is difficult to approach by using high-resolution methods because the protein is oligomeric and heterogeneous in solution, explaining that only one full-length structure of apoE3MM exists [14]. MS-based structural proteomics [17,18,19] and several conventional biophysical methods [2, 48] afford valuable yet averaged information in solution about the structure and biophysical properties of apoE. Native MS coupled with IM can follow the conformation and gas-phase unfolding of the monomeric and tetrameric species. The CCSs of the ground state and the CIU results show that the monomer and tetramer of the three WT isoforms are structurally similar in a lipid-free environment, as was reported in other structural proteomics studies [17, 18]. The narrow CCS distribution of the ground-state tetramer suggests that the tetramer is stable and less dynamic than the monomer. The ground-state monomer of apoE3MM has a similar CCS, but it shows increased stability upon activation compared to that of WT, which may be attributed to the extensive interactions of the C- and N-terminal domains of the apoE3MM. The CCSs of the WT monomer and tetramer, and the fact that the C-terminal domain is involved in self-association allows building a low-resolution, coarse-grained model for the tetramer with a C4 symmetry, an approach that may be effective for other oligomers. That the tetramer has a non-planar arrangement is consistent with the majority of the models that we calculated. To test the symmetry of the model (here C4), one can utilize native MS coupled with ECD; here, we found that most fragment ions arise from the C-terminal domain. After collisionally activating the ions prior to ECD, we found that the N-terminus starts to produce fragment ions with low abundancies. Both these results indicate that the tetramer starts to unfold from the C-terminal domain, making the denatured C-terminus easily accessible to the bombarding electrons used for ECD. The C4 symmetry, not the tetrahedron arrangement, is in good agreement with our ECD results.
This strategy, building low-resolution coarse-grained model from IM and seeking additional support from ECD, is reported here for the first time and is recommended for systems for which oligomerization hinders structure elucidation.
References
Mahley, R.W., Apolipoprotein, E.: Cholesterol transport protein with expanding role in cell biology. Science (New York, N.Y.). 240, 622–630 (1988)
Hatters, D.M., Peters-Libeu, C.A., Weisgraber, K.H.: Apolipoprotein E structure: insights into function. Trends Biochem. Sci. 31, 445–454 (2006)
Liu, C.C., Kanekiyo, T., Xu, H., Bu, G.: Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118 (2013)
Corder, E.H., Saunders, A.M., Strittmatter, W.J., Schmechel, D.E., Gaskell, P.C., Small, G.W., Roses, A.D., Haines, J.L., Pericak-Vance, M.A.: Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science (New York, N.Y.). 261, 921–923 (1993)
Chartier-Harlin, M.C., Parfitt, M., Legrain, S., Perez-Tur, J., Brousseau, T., Evans, A., Berr, C., Vidal, O., Roques, P., Gourlet, V., et al.: Apolipoprotein E, epsilon 4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer's disease: analysis of the 19q13.2 chromosomal region. Hum. Mol. Genet. 3, 569–574 (1994)
Yamazaki, Y., Painter, M.M., Bu, G., Kanekiyo, T.: Apolipoprotein E as a therapeutic target in Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs. 30, 773–789 (2016)
Wilson, C., Wardell, M.R., Weisgraber, K.H., Mahley, R.W., Agard, D.A.: Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science (New York, N.Y.). 252, 1817–1822 (1991)
Perugini, M.A., Schuck, P., Howlett, G.J.: Self-association of human apolipoprotein E3 and E4 in the presence and absence of phospholipid. J. Biol. Chem. 275, 36758–36765 (2000)
Barbier, A., Clement-Collin, V., Dergunov, A.D., Visvikis, A., Siest, G., Aggerbeck, L.P.: The structure of human apolipoprotein E2, E3 and E4 in solution 1. Tertiary and quaternary structure. Biophys Chem. 119, 158–169 (2006)
Choy, N., Raussens, V., Narayanaswami, V.: Inter-molecular coiled-coil formation in human apolipoprotein E C-terminal domain. J. Mol. Biol. 334, 527–539 (2003)
Westerlund, J.A., Weisgraber, K.H.: Discrete carboxyl-terminal segments of apolipoprotein E mediate lipoprotein association and protein oligomerization. J. Biol. Chem. 268, 15745–15750 (1993)
Hatters, D.M., Zhong, N., Rutenber, E., Weisgraber, K.H.: Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J. Mol. Biol. 361, 932–944 (2006)
Garai, K., Baban, B., Frieden, C.: Dissociation of apolipoprotein E oligomers to monomer is required for high-affinity binding to phospholipid vesicles. Biochemistry. 50, 2550–2558 (2011)
Chen, J., Li, Q., Wang, J.: Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc. Natl. Acad. Sci. U. S. A. 108, 14813–14818 (2011)
Yokoyama, S., Kawai, Y., Tajima, S., Yamamoto, A.: Behavior of human apolipoprotein E in aqueous solutions and at interfaces. J. Biol. Chem. 260, 16375–16382 (1985)
Garai, K., Frieden, C.: The association-dissociation behavior of the ApoE proteins: kinetic and equilibrium studies. Biochemistry. 49, 9533–9541 (2010)
Huang, R.Y., Garai, K., Frieden, C., Gross, M.L.: Hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry determine the interface and dynamics of apolipoprotein E oligomerization. Biochemistry-Us. 50, 9273–9282 (2011)
Gau, B., Garai, K., Frieden, C., Gross, M.L.: Mass spectrometry-based protein footprinting characterizes the structures of oligomeric apolipoprotein E2, E3, and E4. Biochemistry. 50, 8117–8126 (2011)
Frieden, C., Wang, H., Ho, C.M.W.: A mechanism for lipid binding to apoE and the role of intrinsically disordered regions coupled to domain-domain interactions. Proc. Natl. Acad. Sci. U. S. A. 114, 6292–6297 (2017)
Garai, K., Mustafi, S.M., Baban, B., Frieden, C.: Structural differences between apolipoprotein E3 and E4 as measured by (19)F NMR. Protein Sci. 19, 66–74 (2010)
Xu, G., Chance, M.R.: Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 107, 3514–3543 (2007)
Engen, J.R.: Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal. Chem. 81, 7870–7875 (2009)
Fitzgerald, M.C., West, G.M.: Painting proteins with covalent labels: what's in the picture? J. Am. Soc. Mass Spectrom. 20, 1193–1206 (2009)
Loo, J.A.: Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 16, 1–23 (1997)
Marcoux, J., Robinson, C.V.: Twenty years of gas phase structural biology. Structure (London, England : 1993). 21, 1541–1550 (2013)
Heck, A.J.: Native mass spectrometry: a bridge between interactomics and structural biology. Nat. Methods. 5, 927–933 (2008)
Shi, L., Holliday, A.E., Shi, H., Zhu, F., Ewing, M.A., Russell, D.H., Clemmer, D.E.: Characterizing intermediates along the transition from polyproline I to polyproline II using ion mobility spectrometry-mass spectrometry. J. Am. Chem. Soc. 136, 12702–12711 (2014)
Snijder, J., Rose, R.J., Veesler, D., Johnson, J.E., Heck, A.J.: Studying 18 MDa virus assemblies with native mass spectrometry. Angew. Chem. (Int. ed. Engl.). 52, 4020–4023 (2013)
van de Waterbeemd, M., Fort, K.L., Boll, D., Reinhardt-Szyba, M., Routh, A., Makarov, A., Heck, A.J.: High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nat. Methods. 14, 283–286 (2017)
Ruotolo, B.T., Benesch, J.L., Sandercock, A.M., Hyung, S.J., Robinson, C.V.: Ion mobility-mass spectrometry analysis of large protein complexes. Nat. Protoc. 3, 1139–1152 (2008)
Uetrecht, C., Rose, R.J., van Duijn, E., Lorenzen, K., Heck, A.J.: Ion mobility mass spectrometry of proteins and protein assemblies. Chem. Soc. Rev. 39, 1633–1655 (2010)
Hall, Z., Politis, A., Robinson, C.V.: Structural modeling of heteromeric protein complexes from disassembly pathways and ion mobility-mass spectrometry. Structure (London, England : 1993). 20, 1596–1609 (2012)
Pukala, T.L., Ruotolo, B.T., Zhou, M., Politis, A., Stefanescu, R., Leary, J.A., Robinson, C.V.: Subunit architecture of multiprotein assemblies determined using restraints from gas-phase measurements. Structure (London, England : 1993). 17, 1235–1243 (2009)
Shelimov, K.B., Jarrold, M.F.: Conformations, unfolding, and refolding of Apomyoglobin in vacuum: an activation barrier for gas-phase protein folding. J. Am. Chem. Soc. 119, 2987–2994 (1997)
Boeri Erba, E., Ruotolo, B.T., Barsky, D., Robinson, C.V.: Ion mobility-mass spectrometry reveals the influence of subunit packing and charge on the dissociation of multiprotein complexes. Anal. Chem. 82, 9702–9710 (2010)
Ruotolo, B.T., Hyung, S.J., Robinson, P.M., Giles, K., Bateman, R.H., Robinson, C.V.: Ion mobility-mass spectrometry reveals long-lived, unfolded intermediates in the dissociation of protein complexes. Angew. Chem. (Int. ed. Engl.). 46, 8001–8004 (2007)
Cui, W., Zhang, H., Blankenship, R.E., Gross, M.L.: Electron-capture dissociation and ion mobility mass spectrometry for characterization of the hemoglobin protein assembly. Protein Sci. 24, 1325–1332 (2015)
Hopper, J.T., Oldham, N.J.: Collision induced unfolding of protein ions in the gas phase studied by ion mobility-mass spectrometry: the effect of ligand binding on conformational stability. J. Am. Soc. Mass Spectrom. 20, 1851–1858 (2009)
Eschweiler, J.D., Rabuck-Gibbons, J.N., Tian, Y., Ruotolo, B.T.: CIUSuite: a quantitative analysis package for collision induced unfolding measurements of gas-phase protein ions. Anal. Chem. 87, 11516–11522 (2015)
Zhang, H., Cui, W., Wen, J., Blankenship, R.E., Gross, M.L.: Native electrospray and electron-capture dissociation in FTICR mass spectrometry provide top-down sequencing of a protein component in an intact protein assembly. J. Am. Soc. Mass Spectrom. 21, 1966–1968 (2010)
Zhang, H., Cui, W., Wen, J., Blankenship, R.E., Gross, M.L.: Native electrospray and electron-capture dissociation FTICR mass spectrometry for top-down studies of protein assemblies. Anal. Chem. 83, 5598–5606 (2011)
Li, H., Wolff, J.J., Van Orden, S.L., Loo, J.A.: Native top-down electrospray ionization-mass spectrometry of 158 kDa protein complex by high-resolution Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 86, 317–320 (2014)
Mondal, T., Wang, H., DeKoster, G.T., Baban, B., Gross, M.L., Frieden, C.: ApoE: in vitro studies of a Small molecule effector. Biochemistry. 55, 2613–2621 (2016)
Hyung, S.J., Robinson, C.V., Ruotolo, B.T.: Gas-phase unfolding and disassembly reveals stability differences in ligand-bound multiprotein complexes. Chem. Biol. 16, 382–390 (2009)
Marty, M.T., Baldwin, A.J., Marklund, E.G., Hochberg, G.K., Benesch, J.L., Robinson, C.V.: Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem. 87, 4370–4376 (2015)
Pacholarz, K.J., Porrini, M., Garlish, R.A., Burnley, R.J., Taylor, R.J., Henry, A.J., Barran, P.E.: Dynamics of intact immunoglobulin G explored by drift-tube ion-mobility mass spectrometry and molecular modeling. Angew. Chem. (Int. ed. Engl.). 53, 7765–7769 (2014)
Zhong, Y., Han, L., Ruotolo, B.T.: Collisional and coulombic unfolding of gas-phase proteins: high correlation to their domain structures in solution. Angew. Chem. (Int. ed. Engl.). 53, 9209–9212 (2014)
Morrow, J.A., Segall, M.L., Lund-Katz, S., Phillips, M.C., Knapp, M., Rupp, B., Weisgraber, K.H.: Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry. 39, 11657–11666 (2000)
Eschweiler, J.D., Farrugia, M.A., Dixit, S.M., Hausinger, R.P., Ruotolo, B.T.: A structural model of the urease activation complex derived from ion mobility-mass spectrometry and integrative modeling. Structure (London, England : 1993). 26. e593, 599–606 (2018)
Eschweiler, J.D., Frank, A.T., Ruotolo, B.T.: Coming to grips with ambiguity: ion mobility-mass spectrometry for protein quaternary structure assignment. J. Am. Soc. Mass Spectrom. 28, 1991–2000 (2017)
Degiacomi, M.T.: On the effect of sphere-overlap on super coarse-grained models of protein assemblies. J. Am. Soc. Mass Spectrom. 30, 113–117 (2019)
Hsieh, Y.H., Chou, C.Y.: Structural and functional characterization of human apolipoprotein E 72-166 peptides in both aqueous and lipid environments. J. Biomed. Sci. 18, 4 (2011)
Jurchen, J.C., Williams, E.R.: Origin of asymmetric charge partitioning in the dissociation of gas-phase protein homodimers. J. Am. Chem. Soc. 125, 2817–2826 (2003)
Acknowledgements
The mass spectrometry research was supported by the National Institutes of Health, National Institute of General Medical Science Grant 2P41GM103422 to MLG, the modeling by NIH National Institute of General Medical Science Grant 5R01GM095832 to BR, and the biochemistry by NIH RF1AG044331 to CF.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 908 kb)
Rights and permissions
About this article

Cite this article
Wang, H., Eschweiler, J., Cui, W. et al. Native Mass Spectrometry, Ion Mobility, Electron-Capture Dissociation, and Modeling Provide Structural Information for Gas-Phase Apolipoprotein E Oligomers. J. Am. Soc. Mass Spectrom. 30, 876–885 (2019). https://doi.org/10.1007/s13361-019-02148-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-019-02148-z