
Abstract
A novel method for (ultra-)high-resolution spatial mass separation in time-of-flight mass spectrometers is presented. Ions are injected into a time-of-flight analyzer from a radio frequency (rf) trap, dispersed in time-of-flight according to their mass-to-charge ratios and then re-trapped dynamically in the same rf trap. This re-trapping technique is highly mass-selective and after sufficiently long flight times can provide even isobaric separation. A theoretical treatment of the method is presented and the conditions for optimum performance of the method are derived. The method has been implemented in a multiple-reflection time-of-flight mass spectrometer and mass separation powers (FWHM) in excess of 70,000, and re-trapping efficiencies of up to 35% have been obtained for the protonated molecular ion of caffeine. The isobars glutamine and lysine (relative mass difference of 1/4000) have been separated after a flight time of 0.2 ms only. Higher mass separation powers can be achieved using longer flight times. The method will have important applications, including isobar separation in nuclear physics and (ultra-)high-resolution precursor ion selection in multiple-stage tandem mass spectrometry.

ᅟ
Similar content being viewed by others


Introduction
One of the key techniques at the heart of mass spectrometry is the isolation of ions (i.e., the spatial separation of the ions of choice from all other ions). A clean ion population is often required to obtain well-defined information of the properties to be measured and to achieve highest sensitivity and accuracy. For example, ion isolation is an integral step in every tandem mass spectrometry experiment.
Over the years, ion isolation methods have been developed for all important types of mass analyzers [1, 2]. Sector field spectrometers are laterally dispersive and thus only require a slit for the selection of the desired ion species. Radio frequency (rf) quadrupole mass filters intrinsically rely on spatial mass separation for mass analysis as well. For trap-based mass spectrometers (i.e., Penning traps/FT-ICR mass spectrometers, rf quadrupole traps, and Orbitraps) methods have been developed for the mass-selective ion storage and ejection [3–7]. Time-of-flight mass spectrometers (TOF-MS) are longitudinally dispersive and thus require a fast deflector to convert the temporal separation of the ions into a spatial separation.
The mass separation power (i.e., the resolving power for spatial separation) achieved with these techniques is often significantly lower than the mass resolving power obtained for mass analysis. It usually lies in the range of 100 to 104. Mass separation powers in excess of 104 are typically achieved only with large and expensive double-stage sector fields and Penning traps [1, 2]. Furthermore, in the case of tandem mass spectrometers it may be a challenge to efficiently recapture the ions or even recool the fragments after the dissociation stage to achieve high resolution and mass accuracy in the mass measurement stage. The addition of a high-resolution precursor selection stage also increases the size, complexity, and cost of a mass spectrometer significantly. For this reason, most tandem mass spectrometers offer precursor separation with low or medium mass separation power only.
In nuclear physics, ion isolation with very high mass separation power is an essential stage in many high-precision experiments because the nuclides of interest must be separated from isobars, which are often produced at much higher rates. Here, mass separators capable of selecting isobars have so far been realized as large double-stage magnetic separators [8] or Penning traps [5, 9]. The maximum resolving power achieved with magnetic separators is on the order of 104 and thus not sufficient to cleanly resolve most nuclear isobars, in particular if the contaminants are produced with much higher abundances than the ions of interest. While Penning traps offer the highest resolving power, for highly resolved measurements the cycle time is on the order of 1 s and the ion capacity on the order of 1000 ions per cycle [10, 11]. This limits the accessible half-lives of the nuclei to be investigated as well as the beam intensity.
TOF mass spectrometry has a great potential for providing (ultra-)high mass separation. A TOF-MS can be converted into a mass separator by coupling it with a fast deflector or ion gate, typically a dipole deflector or a Bradbury-Nielsen gate (BNG) [12, 13]. The deflector or gate is operated such that it transmits the ions of interest but deflects all other ions. In this way, a variety of tandem TOF-MS have been developed [14–17]. At accelerator facilities, mass separation of this type was implemented by installing a deflector or BNG in a low-energy beam transport line [18–20]. Spatial isobar separation with a TOF-MS was proposed and first demonstrated by using the combination of a multiple-reflection time-of-flight mass spectrometer (MR-TOF-MS) [21, 22] with a BNG [23, 24]. Since then MR-TOF isobar separators have been installed at the accelerator facilities GSI [25, 26] and CERN [10, 27]. In addition to mass separation, MR-TOF-MS can also be used for direct mass measurements of exotic nuclei [28–30] and diagnostics purposes [10, 24, 31]. Several more devices of this type are under development and commissioning at further facilities [32–36]. Recently, even the preparation of an isomerically pure beam has been demonstrated, which further extends the applicability of this method [37]. In addition to very high mass separation powers, such TOF isobar separators have the advantage of short cycle times, which enables the access to very short-lived ions and for a given ion capacity per cycle translates to a high overall ion capacity.
A drawback of a TOF mass separator with a BNG is that it is difficult to reinject the isolated ions again into the TOF analyzer after separation. Hence these devices are used for tandem-in-space experiments (i.e., after separation the ions are transported to further experiments downstream of the TOF separator). Often this requires injecting the selected ions into a rf quadrupole or trap for recooling and accumulation [24, 38]. Recently, a mass separation method was developed for a MR-TOF-MS with nondestructive detection [39]. In this device, the ions are reinjected after TOF dispersion into the same rf trap from which they were injected into the TOF analyzer. On the way back to the trap, they are separated using an electrostatic deflector. So far, this method is limited to mass separation powers of less than 103.
In the present work, a novel method for (ultra-)high mass separation in a MR-TOF-MS is proposed and demonstrated [40–43]. Here, mass separation is performed after TOF dispersion by a dynamical re-trapping procedure in the rf trap, from which they were injected into the TOF analyzer. This re-trapping procedure is highly mass-selective. Since it is the inverse process to the ejection of the ions from the trap, it yields a relatively cool ion population even before application of additional collisional cooling. Compared with the combination of a MR-TOF-MS with a BNG, it allows for a more compact setup and enables (ultra-)high resolution tandem-in-time experiments in a MR-TOF-MS, such as the mass analysis of ions in the very same analyzer that has been used for the ion isolation in a preceding stage.
Theory
Multiple-Reflection Time-of-Flight Mass Spectrometry
In most MR-TOF-MS, ion packets are prepared prior to mass analysis in a linear rf trap (injection trap) by ion accumulation and collisional cooling in a buffer gas, typically He or N2 at pressures of a few 10− 3 mbar. Confinement of the ion bunch in the axial (z) direction is provided by applying a positive DC voltage offset to the electrodes surrounding the trap; thus an axial potential well is formed in the trap. Radial confinement is provided by the rf pseudopotential well. After cooling, the ions are injected as packets from the trap into the TOF analyzer. Typically, the analyzer of a MR-TOF-MS consists of two coaxial electrostatic reflectors. The analyzer is designed to be isochronous, i.e., such that after a certain number of reflections (typically one or two reflections) the time-of-flight of the ions only depends on their mass-to-charge ratio [44]. In most MR-TOF-MS designs, spatial confinement is ensured using an accelerating lens in each reflector. In order to inject the ions into the analyzer and to eject them again, either some voltages of the entrance and exit reflectors can be switched to a lower value, or a pulsed drift tube mounted between the reflectors can be switched when the ions fly through the drift tube in order to reduce or increase their kinetic energy [45]. The time-of-flight is measured using a TOF detector located behind the analyzer.
The mass resolving power of a TOF-MS is given by
where t is the overall time-of-flight and Δt is the spread in time-of-flight. After N a turns, the total flight time is t pt + N a t a, where t pt and t a are the time-of-flight in the pass-through from the injection trap to the detector and the time-of-flight for one turn in the analyzer, respectively. The overall spread in time-of-flight Δt is determined by the time-of-flight spreads in the pass-through Δt pt and per turn in the analyzer Δt a. These contributions to the time-of-flight spreads can be assumed to be roughly independent of each other, such that the overall time-of-flight spread is approximately given by [22]
Then, the mass resolving power of a MR-TOF-MS is given approximately by
Typically the time spread per turn Δt a is dominated by aberrations, and the time spread in the pass-through Δt pt by the turn-around time Δt ta, which is caused by the thermal motion of the ions at the start of their flight [46]. This motion gives rise to Gaussian-shaped time-of-flight peaks with a FWHM of [47]
Here, q and \( \Delta K=\frac{1}{2}{k}_{\mathrm{B}} T \) are the ion’s charge and the component of the initial kinetic energy spread in the direction of extraction, respectively, E 1 is the extraction field strength, T the temperature, and k B is Boltzmann’s constant.
Stages of an Experiment with Mass-Selective Re-Trapping
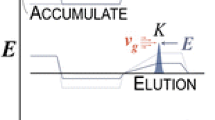
In such a MR-TOF-MS, mass separation in the framework of the re-trapping technique is performed schematically as shown in Figure 1. The ions to be separated are cooled in the injection trap, extracted by applying an accelerating voltage between the electrodes surrounding the trap, and then injected into the analyzer (Stage 1). For injection, the voltages of the entrance reflector are switched to a low value such that the ions can pass through this reflector. After the ion bunch has reached the drift section between the ion mirrors of the analyzer, the reflector voltages are switched to a high value such that they reflect the ions and the ions undergo multiple turns in the analyzer (Stage 2). After the desired flight distance, viz. mass resolving power, has been reached, the entrance reflector is opened again by switching the voltages of its electrodes to a low value (Stage 3a). The ions are thus reinjected into the injection trap. The extraction field in the injection trap is retained as it was set during ion ejection from the trap in Stage 1. The ions are retarded in this field, and at a certain re-trapping time moment t r, when the ions with the mass of interest are stopped, the retarding field is switched back to a shallow axial storage field. Ions with masses different from the mass of interest arrive back in the trap at different times. At the time moment t r they are, therefore, not stopped. Thus, only ions with the mass of interest are re-trapped, whereas all other ions escape the trap. The same MR-TOF-MS can also be used for high-resolution mass measurements if the ions are ejected from the analyzer through the exit reflector to the detector (Stage 3b). Moreover, after the ions of interest have been re-trapped in Stage 3a, a mass measurement of the re-trapped ions can be performed by repeating the Stages 1, 2, and 3b. In this way the re-trapped ions can be identified and their abundance determined.

Schematic figure showing the individual stages of an experiment with mass-selective re-trapping of ions in a MR-TOF-MS. For details see text
Quantitative Treatment
In the following, the ion motion in the Stages 1 and 3a will be considered in more detail. For simplicity, a two-stage acceleration with two homogenous fields will be assumed. The principle, however, also holds for more realistic accelerating field configurations. Ions are accelerated in the injection trap by applying an axial extraction field in the z direction with a field strength E 1 at the time moment t e. Since the ions start at different positions in the trap, they acquire somewhat different kinetic energies. After leaving the trap, the ions are further accelerated between two electrodes by the acceleration electric field E 2 and then enter the TOF analyzer. If E 1 and E 2 are approximately homogenous and if an ion initially located in the center of the trap travels through E 1 for a distance of L 1 and through E 2 for a distance of L 2 (see Figure 1) the mean kinetic energy of the ions amounts to
The primary time-focus, at which the flight time is independent of ion energy, is located at some position downstream of the injection trap. The distance of the primary time-focus from the end of the acceleration region is given by [44]
where
For a given mean kinetic energy K 0 the position of the primary time-focus is thus determined by the extraction field strength E 1.
At first, the case will be considered in which the extraction field strength is chosen such that the primary time-focus is located in the middle of the TOF analyzer. The analyzer can be tuned such that in all subsequent turns of the ions in the analyzer the intermediate time foci are also located at the same spatial position (i.e., the center of the analyzer). In the analyzer the ions perform an arbitrary odd number of reflections until the desired mass resolving power has been reached. Then the entrance reflector is opened and the ions are re-injected through this reflector back into the ion trap. Since the location of the last time-focus is the same as that of the primary time-focus (i.e., the center of the analyzer), ions of different kinetic energies but the same mass m 1 are all stopped at the same time moment t r, provided the retarding field strength is the same as the extraction field strength during initial acceleration (Figure 2). If at that moment the field in the injection trap is switched from retarding to a field creating an axial potential well, the ions of mass m 1 will be re-trapped and stored, with essential zero kinetic energy. At the same time, ions of a different mass m 2 may also be spatially located inside the injection trap, but they are stopped at a different time moment, and at t r they have non-zero energies. In the case m 2 < m 1 at the time moment t r these ions will have already been reflected by the retarding field. In the case m 2 > m 1 at the time moment t r these ions will still be moving towards the trap center. In both cases, if the re-trapping potential well is shallow enough, ions of the mass m 2 will not be trapped. This technique of efficiently stopping ions of one mass-to-charge ratio regardless of their kinetic energies is very similar to the one used for creating an almost mono-energetic ion beam in the dynamic energy buncher of the MR-TOF-MS at the FRS Ion Catcher at GSI [24, 48].

Schematic figure illustrating the concept of mass-selective re-trapping. Potential energy diagrams are shown for the phase during which ions re-enter the trap after time-of-flight in the analyzer and are retarded by an (almost) linear axial field (left panel) and the phase of re-trapping, in which an axial potential well is formed (right panel). Depending on their positions in the rf trap at the moment, at which the axial electrostatic field is switched from retarding to re-trapping, the ions have a remaining kinetic energy K. Separation of the ions is performed on the basis of differences in this kinetic energy. The re-trapping potential can only recapture those ions with an energy K < q U r and rejects those with higher kinetic energies
In Figure 1, the case is shown, for which the extraction field strength E 1 (Stage 1) and the retarding field strength E r (Stage 3a) in the injection trap are the same. Here, the positions of the primary time-focus and of the final time-focus coincide. Note that in this case the Stages 1 and 3a are symmetric to each other and that re-trapping is the time-inverse process to extraction. However, it is not a general requirement to have the same extraction and retarding field strengths. For different extraction and retarding field strengths, the primary time-focus and the final time-focus need to be located at different positions according to Equation 6. Indeed, it is not even required to perform the injection and re-trapping in the same trap. Provided the position of the final time-focus and the retarding field strength are chosen to match each other according to Equation 6, the re-trapping could also occur in a different trap (e.g., a trap located behind the exit reflector). For simplicity, the following treatment will, however, be limited to the case that re-trapping is performed in the injection trap.
It is important to emphasize that the mass separation by re-trapping is not directly based on the differences in arrival times, as it is for ion separation by a BNG. Rather, it relies on the conversion of the time differences into differences in kinetic energy followed by a separation of the ions with respect to their kinetic energies. The resulting mass separation power consequently depends on the quality of the time-of-flight separation of the ions in the TOF analyzer. Let t f = t r − t e be the time-of-flight of an ion of mass m from the extraction time moment t e to the re-trapping time moment t r. From the theory of TOF mass analyzers it is well known [44] that
At the time of re-trapping the ion velocity–time gradient is
where q is the ion charge and E r is the retarding electrostatic field strength at the position where the ions stop. Then, the velocity–mass dispersion at the time of re-trapping is
The time spread Δt at the final time-focus, which is given approximately by Equation 2, leads to a velocity spread of the ions of a single mass at the time of re-trapping of
The minimum mass difference Δm, which can be resolved by re-trapping, is determined by the relation
In Equation 12, Δv r is the velocity acceptance of the trap (i.e., the maximal velocity of recaptured ions in the trap)
where U r is the depth of the axial potential well of the trap. In Equation 12, a quadratic addition of Δv and Δv r has been assumed; the exact relation depends on the distribution functions of Δv and Δv r. Substitution of Equations 10, 11 and 13 into Equation 12 yields the mass separation power
From Equation 14 it can be seen that in case of a very shallow trap (U r → 0) or a very strong retarding field E r the mass separation power tends to the value of the mass resolving power of a TOF-MS as given in Equation 1.
Let the time window for re-trapping Δt r at the final time-focus be defined as the duration during which ions are re-trapped (i.e., during which the ions after retarding have velocities not exceeding Δv r). Assuming a homogeneous retarding field in the trap, the time window for re-trapping is related to the velocity depth of the trap Δv r as
Then, Equation 14 can be rewritten in the form
The correspondence of the parameters for the time-of-flight separation and the re-trapping mass resolution is illustrated in Figure 3. Panel a shows the envelopes of the ion packets near the position of the last time-focus. Panel b demonstrates the time-dependence of the envelopes of the ion kinetic energy in the vicinity of the moment of re-trapping. In the case of a large number of turns in the TOF analyzer, the difference t 12 between the arrival times of the ion packets of two different but close masses, m 1 and m 2, at the last time focal plane almost coincides with the difference of the flight times, t f, until the re-trapping for ions of these two masses and the time width of ion packets of one mass is given by Δt (Equation 2). Then, assuming a homogeneous axial retarding field in the trap, at the time of re-trapping, t r, (defined as the stopping time for ions of the mass m 1), the average velocity of the ions of the mass m 2 is v 2 ≈ qE r t 12/m 2. Thus the average kinetic energy K 2 of these ions is

(a) Spatial envelopes of ion packets as a function of time near the position of the last time-focus. (b) Energy envelopes of ion packets as a function of time in the vicinity of the re-trapping time. The TOF dispersion t 12 corresponds to a kinetic energy difference K 2, and the time spread of the ions of mass m 1 at the final time-focus corresponds to the kinetic energy spread Δ K 1. Note that for simplicity the figure shows the case in which the retarding field in never switched to re-trapping. (c) Figure illustrating the analogy between re-trapping and mass separation in a magnetic mass spectrometer. The TOF dispersion, the kinetic energy spread, and the energy depth of the trap of the TOF separator correspond to the spatial dispersion, the beam width, and the slit width in a magnetic separator, respectively
From Equation 4 one obtains the kinetic energy spread of the ion packet for the mass m 1
In other words, the time-of-flight dispersion at the last time-focus determines the energy difference (or velocity dispersion) at the time of re-trapping. Furthermore, the time-spread of the ion packets at the last time-focus determines the energy spread of the re-trapped ion packet. If this energy spread is smaller than the energy depth of the axial potential well of the trap (ΔK < qU r) the entire ion packet is captured in the trap. If it is larger, ions get lost during the re-trapping process. If it is significantly smaller, the mass separation power of the re-trapping technique becomes much smaller than the mass resolving power of the mass spectrometer. The optimum energy depth of the trap is, therefore, given by qU r ≈ ΔK.
From Equations 17 and 18 it can be seen that both the energy dispersion and the energy spreads of the ion bunches can be changed proportionally by varying the retarding field strength E r. This variation does not change the relative separation of the energy dispersion and the bunch energy width and, thus, does not influence the spectrometric resolving power given by Equation 1. However, it allows matching the energy spread of the bunch ΔK and the energy depth of the trap U r to optimize the mass separation power of the re-trapping technique (Equation 14), as it is illustrated in Figure 4.

Schematic figure illustrating the influence of the strength of the retarding field and the energy depth of the trap on the mass separation power of the re-trapping technique. Left-hand side: field strength as a function of the axial position in the trap; right-hand-side: energy envelopes of ion packets as a function of the time in the vicinity of the re-trapping time. Note that for simplicity, the figure shows the case in which the retarding field in never switched to re-trapping
For a packet of singly charged ions of mass 100 u, a time spread at the final time-focus of 10 ns and a retarding field strength of 100 V/mm, according to Equation 18, the kinetic energy spread amounts to 0.087 eV. It is thus on the order of the thermal energy spread. An optimized trap depth for re-trapping is therefore small compared with the typical trap depth used for conventional ion storage. Thus, care must be taken to avoid any field penetration of the acceleration field E 2 through the trap aperture during the re-trapping stage.
For some applications it may be useful to control the range of re-trapped mass-to-charge ratios by adjusting the depth of the trap as an alternative to changing the flight distance and thus the mass resolving power of the mass spectrometer. Note that since the total time-spread of the ion bunches at the final time-focus is typically proportional to \( \sqrt{m} \), the total kinetic energy spread at t r is independent of the ion mass. The mass separation power is thus also approximately independent of the ion mass.
In practice, it is usually most convenient to use the same extraction and retarding field strengths (i.e., to have the primary and final time-focus at the same position). A deep trap depth is used for the preparation of the ion packets prior to injection into the TOF analyzer, and a relatively shallow trap depth is used for re-trapping. In this way, a reduced emittance during time-of-flight analysis as well as good mass separation during the re-trapping process can be achieved simultaneously.
Analogy with Magnetic Mass Separator
There is a simple analogy between ion separation by re-trapping in TOF analyzers and the ion separation by passing an ion beam though a slit in a magnetic mass separator, as illustrated in Figure 3c. The TOF dispersion t 12 is analogous to the spatial dispersion in a magnetic separator, the energy spread of the re-trapped ion packet is analogous to the ion beam width, and the energy depth of the trap is similar to the slit width in the magnetic separator. The variation of the energy dispersion and the energy spread of the ion packets correspond to the magnification or demagnification in a magnetic separator by inserting a lens between the sector magnet and the exit slit.
Comparison with the Separation Using a BNG
It is interesting to compare the achievable mass separation power of an MR-TOF-MS using the mass-selective re-trapping technique with that of an MR-TOF-MS with a BNG. In both cases, the mass separation power is lower than the mass resolving power for mass measurements. In the case of the mass-selective re-trapping, the finite trap depth and retarding field strength lead to a reduction of the mass separation power, as explained above. This reduction can only be mitigated at the expense of losses in the efficiency of the re-trapping. In the case of an MR-TOF-MS with BNG, the mass separation power is decreased by the finite transition time of the ions through the field region of the BNG and by the finite rise time of the HV pulsers used with the BNG [26, 49]. The transition time of the ions can be made shorter by using a BNG with a smaller grid spacing, which, however, reduces the transmission efficiency. In both cases, the mass-selective re-trapping and the BNG, the additional contribution to the time distribution limiting the mass separation power is on the order of 20 ns. However, since the rise time of the HV pulsers for the BNG cannot be reduced arbitrarily, the mass-selective trapping technique may be superior for very small time-of-flight differences of mass lines, however, at the cost of efficiency. Conversely, for a large number of turns and larger time-of-flight differences, a MR-TOF-MS with a BNG may achieve a higher transmission efficiency.
Experimental
The technique of mass-selective re-trapping was implemented and tested on a mobile high-performance mass spectrometer developed for in-situ applications in analytical mass spectrometry [42, 43, 50, 51]. The system consists of an atmospheric pressure interface (API), a radio-frequency quadrupole (rfQ) ion guide, a rfQ mass filter to suppress contaminants, a rfQ cooler and a rf ion trap (injection trap) for thermalization and bunching of the ions, a TOF analyzer, and a detector. The mobile MR-TOF-MS has been developed on the basis of the MR-TOF-MS for experiments with exotic nuclei at the FRS Ion Catcher at GSI and at the Low-Energy Branch of the Super-FRS at FAIR [24, 26]. It uses a similar analyzer geometry, however, scaled to a smaller size. The analyzer consists of two reflectors with four electrodes each [48]. The innermost electrode of each reflector is operated as accelerating lens, while the another three electrodes form a repulsive potential. The analyzer can be tuned such that all time-of-flight aberrations up to the second order disappear after every full turn. A fast deflector in the center of the analyzer serves as mass range selector, which can reduce the mass range of ions in the analyzer to an unambiguous range [26]. The kinetic energy of the ions in the analyzer amounts to 1.3 keV.
In this work [43], the axial depth for ion trapping in the injection trap before injection into the analyzer was 10 V. An extraction field strength of 78 V/mm corresponding to a relative energy spread of δ = 1.4% was used. The re-trapping field strength also amounted to 78 V/mm. In the rfQ cooler and the injection trap, N2 was used as buffer gas. For each passage of the ions through the analyzer, one time-focus shift (TFS) turn [52] in the analyzer was used, which has a time-focus length that accounts also for the ion path from the injection trap the detector measurement, and several isochronous turns afterwards, which have a time-focus length of one turn each. For the exit reflector in its open state, voltage settings were used that, according to simulations, result in the same time focal length from the last intermediate time-focus in the analyzer to the detector as from the last intermediate focus in the analyzer to the position of the primary time-focus in front of the injection trap. In this way, the analyzer settings optimized experimentally for the mass measurement mode could also be applied to the re-trapping mode.
The mass separation power and the efficiency of the re-trapping technique were determined for the protonated molecular ion of caffeine (195 u). A sample of caffeine with a concentration of 10− 6 mol/L in a H2O/MeOH (1/1) solution with 0.1% formic acid was used. The measurements were performed for different turn numbers and for different re-trapping potentials. For each measurement, one TFS turn was used after injection into the analyzer, and several isochronous turns afterwards. The number of isochronous turns was set to 0.5, 7.5, 15.5, 31.5, 63.5, and 127.5, amounting to flight times of up to 3.6 ms for caffeine. The axial depth of the trap for re-trapping was varied between 1.3, 1.9, 3.2, and 6.3 V as determined from the experimentally applied voltages and the calculated electric potential on the trap axis. For each individual measurement, the re-trapping time-of-flight t f was scanned (i.e., the time from the ion extraction from the trap to the point in time, at which the trap potential was switched from retarding to re-trapping). After re-trapping, the ions were stored and cooled for a duration of about 10 ms. The ion abundance for each re-trapping cycle was then measured in a mass spectrum that was recorded with the isolated ions. The data were recorded using a ADC (FastFlight2 signal averager, Signal Recovery, Oak Ridge, TN, USA) and the custom-written data acquisition software MAc [53, 54]. For each measurement 500 spectra were averaged. Values for the mass separation power were obtained from the widths (FWHM) of the measured distributions without fitting.
The protonated molecular ions of glutamine and lysine (147 u) were chosen to demonstrate the spatial separation of ions using the re-trapping technique. The amino acids glutamine and lysine are isobars and have an absolute mass difference of 36.4 μ and a relative mass difference of 2.5 × 10− 4 only. For this measurement, the amino acids were dissolved in a H2O/MeOH (1/1) solution with 0.1% formic acid with a concentration of approximately 10− 4 mol/L. The sample was prepared to yield about the same intensity for both analytes.
Results and Discussion
Time-of-flight mass spectra of the protonated molecular ion caffeine obtained by the re-trapping technique for different flight times for a re-trapping trap depth of 1.3 V are shown in Figure 5. Every data point in a spectrum corresponds to a measurement consisting of (i) a re-trapping cycle (Stages 1, 2, and 3a in Figure 1), which served for mass separation, and (ii) a measurement cycle (Stages 1, 2, and 3b in Figure 1), which was used to determine the abundance of the re-trapped ions. For the re-trapping cycle, one TFS turn and 15.5, 63.5, or 127.5 isochronous turns were used, corresponding to flight times of 0.48, 1.8, and 3.6 ms, respectively. For the measurement cycle, one TFS turn and one isochronous turn were used. The peak width (FWHM) in all three spectra amounts to about 25 ns, corresponding to mass separation powers of 10,000, 32,000, and 71,000, respectively. Note that the complex scan sequence performed to obtain the spectra serves to demonstrate the performance of the re-trapping technique, but is not required for the selection of a particular ion species. Here, a single re-trapping cycle is sufficient.

Time-of-flight spectra of the protonated molecular ion of caffeine obtained by mass-selective re-trapping after (a) one TFS turn and 15.5 isochronous turns, (b) one TFS turn and 63.5 isochronous turns, and (c) one TFS turn and 127.5 isochronous turns. A re-trapping trap depth of 1.3 V was used
Figure 6 shows the mass separation power of the MR-TOF-MS over a large range of flight times and trap depths. The values for the mass separation power were determined using the same experimental sequence as that for the measurements shown in Figure 5. The mass separation power increases linearly with the time-of-flight. Furthermore, for smaller trap depths, a higher mass separation power is obtained, in accordance with the theoretical considerations. The experimental values are compared with the theoretical values obtained using Equation 14. In order to obtain satisfactory agreement, two experimental parameters needed to be adjusted in the calculations: the magnitude of the trap depth must be reduced from its nominal value by a constant value of 1.15 V for all measurements, and the retarding field strength must be scaled from its nominal value by a factor of 0.56 (or the trap depth scaled by the inverse square of this value) for all measurements. While it is clear that a correction such as the former should be required because of, e.g., a residual component of the rf field on the trap axis or because of imperfections in the HV switching between retarding and re-trapping, the origin of the latter correction is currently not understood. With these two adjustments, excellent agreement between the experimental results and the theoretical values is achieved for all 24 measurement points.

Mass separation power of the MR-TOF-MS in dependence of the time-of-flight for different trap depths for re-trapping. The full symbols show the results of measurements and the open symbols the corresponding theoretical values obtained using Equation 14 . For details see text
In these measurements, the maximum re-trapping efficiency amounted to 35%; however, it is likely that the efficiency was limited by space charge effects. The linear increase of mass separation power with time-of-flight indicates that even higher mass separation powers could be obtained for longer flight times, which was not done in this work for practical reasons. However, it was shown recently that with the same MR-TOF-MS, maximum mass resolving powers (in the mass measurement mode) of 300,000 could be obtained [42, 52].
If only two ion species have to be separated, the separation power of ion re-trapping is not necessarily determined by the full width of the acceptance window of the re-trapping technique, but rather by the width of its rising or falling edge. The re-trapping time can be adjusted such that one of the isobars is recaptured (i.e., is placed inside the acceptance window), but as close to the rising or falling edge as possible, whereas the other one is placed just outside the rising or falling edge of the acceptance window. Thereby, even higher separation powers than shown in Figure 6 can be obtained.
The spatial separation of close-lying mass lines is demonstrated in Figure 7. It contains three mass spectra, which were obtained after one TFS turn and 6.5 isochronous turns in the re-trapping cycle, corresponding to a flight time of 0.20 ms, and one TFS turn and 8 isochronous turns in the measurement cycle, corresponding to a flight time of 0.23 ms. In contrast to the spectra shown in Figure 5, the re-trapping time was not scanned for each spectrum; the figures show the spectra obtained in the measurement cycle. Between the three spectra, the re-trapping time was adjusted such that either both amino acids were recaptured, i.e., placed inside the time window of the re-trapping acceptance (spectrum a), or just one of them, (spectrum b and spectrum c). Clearly, glutamine as well as lysine could successfully be isolated from their respective isobaric counterpart. The two isobars have an absolute mass difference of 36.4 mu and a relative mass difference of 2.5 × 10− 4. In this measurement, the suppression of the respective contaminant is limited by ions entering the injection trap from the rfQ cooler after the re-trapping process and before re-injection of the ions into the analyzer for the measurement cycle. The relative abundance of these contaminant ions amounts to about 2%. In the future, these ions can be kept out of the injection trap by raising a potential barrier between the rfQ and the trap.

Mass spectra demonstrating the isolation of the protonated molecular ions of the amino acids glutamine and lysine. The two isobars have an absolute mass difference of 36.4 mu and a relative mass difference of 2.5 × 10–4. In spectrum (a) the re-trapping time was chosen such that both analytes were simultaneously re-trapped. In the spectra (b) and (c) either glutamine or lysine was isolated from its isobaric counterpart
Conclusions
Mass-selective re-trapping of ions is a novel method for (ultra-)high-resolution spatial mass separation in time-of-flight mass spectrometers. A theoretical treatment of the method has been developed, and the method has been implemented in a mobile multiple-reflection time-of-flight mass spectrometer. Experimental and theoretical results are in good agreement. Systematic measurements of the mass separation power have been performed for the molecular ion of caffeine. Mass separation powers in excess of 70,000 and re-trapping efficiencies of up to 35% have been obtained. With longer flight times or by separation at the edge of the acceptance window, even higher mass separation powers are feasible. The isobars glutamine and lysine with a relative mass difference of 2.5 × 10− 4 have been separated after a flight time of 0.2 ms only.
The method is very versatile, and its implementation should be feasible in most time-of-flight mass spectrometers that use an rf trap for forming ion bunches prior to time-of-flight mass analysis. An important advantage compared with the use of a pulsed deflector or ion gate in TOF isobar separators is that (1) no additional ion optical element is required, (2) a more compact setup is possible, and (3) the process of ion bunch creation, time-of-flight dispersion, and mass-selective re-trapping can be repeated for a theoretically unlimited number of times. This opens up several new and important applications: in nuclear physics experiments, an MR-TOF-MS employing the mass-selective re-trapping technique can be used as an isobar or isomer separator, which is more compact than an MR-TOF-MS with a BNG. If the suppression of abundant contaminants is not strong enough in one cycle, the isolation process can be performed several times, first with larger ion abundance and lower mass separation power and next with the preselected ion population of lower abundance and, thus, with higher mass separation power. The mass range of the isolated nuclides can be adjusted by the choice of the trap depth (e.g., to include all isobaric exotic nuclei of choice), while rejecting more abundant isobaric contaminants. After isolation of the ions of interest, a direct precision mass measurement is possible using the same TOF analyzer. Because of the preceding ion isolation, higher mass accuracies will be possible, since space charge effects due to abundant contaminants in the original ion sample can be avoided. An MR-TOF-MS employing the method of mass-selective re-trapping has been developed and has recently been installed at the TITAN facility at TRIUMF in Vancouver [32, 51, 55]. Commissioning of the device is underway.
In analytical mass spectrometry, the method of mass-selective re-trapping enables (ultra-)high resolution precursor ion selection. This paves the way for multiple-stage tandem mass spectrometry (MSN) in compact time-of-flight mass spectrometers with very high resolving power in every stage [40, 41, 43]. The implementation of this technique will be the subject of a forthcoming publication.
References
Dass, C.: Fundamentals of Contemporary Mass Spectrometry. John Wiley and Sons, New Jersey (2007)
Glish, G.L., Burinsky, D.J.: Hybrid mass spectrometers for tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 19, 161–172 (2008)
Dawson, P.H., Hedman, J.W., Whetten, N.R.: A simple mass spectrometer. Rev. Sci. Instrum. 40, 1444–1450 (1969)
Fulford, J.E., Hoa, D.N., Hughes, R.J., March, R.E., Bonner, R.F., Wong, G.J.: Radio-frequency mass selective excitation and resonant ejection of ions in a three-dimensional quadrupole ion trap. J. Vac. Sci. Technol. 17, 829–835 (1980)
Savard, G., Becker, S., Bollen, G., Kluge, H.J., Moore, R.B., Otto, T., Schweikhard, L., Stolzenberg, H., Wiess, U.: A new cooling technique for heavy ions in a Penning trap. Phys. Lett. A 158(5), 247–252 (1991)
Guan, S., Marshall, A.G.: Stored waveform inverse Fourier transform (SWIFT) ion excitation in trapped-ion mass spectrometry: theory and applications. Int. J. Mass Spectrom. Ion Processes 157/158, 5–37 (1996)
Hu, Q., Makarov, A.A., Cooks, R.G., Noll, R.J.: Resonant AC dipolar excitation for ion motion control in the Orbitrap mass analyzer. J. Phys. Chem. A 110, 2682–2689 (2006)
Wollnik, H., Becker, K.: Ion optical design for an on-line mass separator with low cross-contamination and the capability of good mass resolution. Nucl. Instrum. Methods A 238, 206–214 (1985)
Kowalska, M., Naimi, S., Agramunt, J., Algora, A., Audi, G., Beck, D., Blank, B., Blaum, K., Böhm, C., Breitenfeldt, M., Estevez, E., Fraile, L.M., George, S., Herfurth, F., Herlerth, A., Kellerbauer, A., Lunney, D., Minaya-Ramirez, E., Neidherr, D., Olaizola, B., Riisager, K., Rosenbusch, M., Rubio, B., Schwarz, S., Schweikhard, L., Warring, U.: Preparing a journey to the east of 208Pb with ISOLTRAP: Isobaric purification at A = 209 and new masses for 211–213Fr, and 211Rn. Eur. Phys. J. A 42, 351–359 (2009)
Wolf, R., Wienholtz, F., Atanasov, D., Beck, D., Blaum, K., Borgmann, C., Herfurth, F., Kowalska, M., Kreim, S., Litvinov, Y.A., Lunney, D., Manea, V., Neidherr, D., Rosenbusch, M., Schweikhard, L., Stanja, J., Zuber, K.: ISOLTRAP’s multi-reflection time-of-flight mass separator/spectrometer. Int. J. Mass Spectrom. 349/350, 123–133 (2013)
Minaya Ramirez, E., Alfaurt, P., Aouadi, M., Ascher, P., Blank, B., Blaum, K., Cam, J.F., Chaveau, P., Daudin, L., Delahaye, P., Delalee, F., Dupré, P., Abbeir, S.E., Gerbaux, M., Grévy, S., Guérin, H., Lunney, D., Metz, F., Naimi, S., Perrot, L., de Roubin, A., Serani, L., Thomas, B., Thomas, J.C.: Conception of PIPERADE: a high-capacity Penning-trap mass separator for high isobaric contamination at DESIR. Nucl. Instrum. Methods B 376, 298–301 (2016)
Cravath, A.M.: The rate of formation of negative ions by electron attachment. Phys. Rev. 33, 605–613 (1929)
Bradbury, N.E., Nielsen, R.A.: Absolute values of the electron mobility in hydrogen. Phys. Rev. 49, 388–393 (1936)
Schey, K., Cooks, R.G., Grix, R., Wollnik, H.: A tandem time-of-flight mass spectrometer for surface-induced dissociation. Int. J. Mass Spectrom. Ion Processes 77, 49–61 (1987)
Cornish, T.J., Cotter, R.J.: Tandem time-of-flight mass spectrometer. Anal. Chem. 65, 1043–1047 (1993)
Beussman, D.J., Vlasak, P.R., McLane, R.D., Seeterlin, M.A., Enke, C.G.: Tandem reflectron time-of-flight mass spectrometer utilizing photodissociation. Anal. Chem. 67, 3952–3957 (1995)
Toyoda, M., Giannakopulos, A.E., Colburn, A.W., Derrick, P.J.: Development of a tandem time-of-flight mass spectrometer “MULTUM-TOF/TOF” at Osaka University: combination of a multi-turn time-of-flight mass spectrometer and a quadratic-field ion mirror. Phys. Procedia 1, 401–411 (2008)
Schury, P., Bollen, G., Block, M., Morrissey, D., Ringle, R., Prinke, A., Savory, J., Schwarz, S., Sun, T.: Beam purification techniques for low energy rare isotope beams from a gas cell. Hyperfine Interact. 173, 165–170 (2006)
Werner, J.: [Flugzeitmassenspektrometrie und Designstudie für einen neuen rf-Quadrupol-Buncher bei SHIPTRAP.] Bachelor thesis, Justus Liebig University Gießen (2008)
Brunner, T., Mueller, A., O’Sullivan, K., Simon, M.C., Kossick, M., Ettenauer, S., Gallant, A.T., Mané, E., Bishop, D., Good, M., Gratta, G., Dilling, J.: A large Bradbury Nielsen ion gate with flexible wire spacing based on photo-etched stainless steel grids and its characterization applying symmetric and asymmetric potentials. Int. J. Mass Spectrom. 309, 97–103 (2012)
Wollnik, H., Przewloka, M.: Time-of-flight mass spectrometers with multiply reflected ion trajectories. Int. J. Mass Spectrom. Ion Processes 96, 267–274 (1990)
Plaß, W.R., Dickel, T., Scheidenberger, C.: Multiple-reflection time-of-flight mass spectrometry. Int. J. Mass Spectrom. 349, 134–144 (2013)
Plaß, W.R., Dickel, T., Petrick, M., Boutin, D., Di, Z., Fleckenstein, T., Geissel, H., Jesch, C., Scheidenberger, C., Wang, Z.: An rf quadrupole-time-of-flight system for isobar-separation and multiplexed low energy rare-isotope beam experiments. Eur. Phys. J. Special Topics 150, 367–368 (2007)
Plaß, W.R., Dickel, T., Czok, U., Geissel, H., Petrick, M., Reinheimer, K., Scheidenberger, C., Yavor, M.I.: Isobar separation by time-of-flight mass spectrometry for low-energy radioactive ion beam facilities. Nucl. Instrum. Methods B 266, 4560–4564 (2008)
Plaß, W.R., Dickel, T., Purushothaman, S., Dendooven, P., Geissel, H., Ebert, J., Haettner, E., Jesch, C., Ranjan, M., Reiter, M.P., Weick, H., Amjad, F., Ayet, S., Diwisch, M., Estrade, A., Farinon, F., Greiner, F., Kalantar-Nayestanaki, N., Knöbel, R., Kurcewicz, J., Lang, J., Moore, I., Mukha, I., Nociforo, C., Petrick, M., Pfuetzner, M., Pietri, S., Prochazka, A., Rink, A.K., Rinta-Antila, S., Schäfer, D., Scheidenberger, C., Takechi, M., Tanaka, Y.K., Winfield, J.S., Yavor, M.I.: The FRS Ion Catcher - a facility for high-precision experiments with stopped projectile and fission fragments. Nucl. Instrum. Methods B 317, 457–462 (2013)
Dickel, T., Plaß, W.R., Becker, A., Czok, U., Geissel, H., Haettner, E., Jesch, C., Kinsel, W., Petrick, M., Scheidenberger, C., Yavor, M.I.: A high-performance multiple-reflection time-of-flight mass spectrometer and isobar separator for the research with exotic nuclei. Nucl. Instrum. Methods A 777, 172–188 (2015)
Wolf, R.N., Beck, D., Blaum, K., Böhm, C., Borgmann, C., Breitenfeldt, M., Chamel, N., Goriely, S., Herfurth, F., Kowalska, M., Kreim, S., Lunney, D., Manea, V., Ramirez, E.M., Naimi, S., Neidherr, D., Rosenbusch, M., Schweikhard, L., Stanja, J., Wienholtz, F., Zuber, K.: Plumbing neutron stars to new depth with the binding energy of the exotic nuclide 82Zn. Phys. Rev. Lett. 110, 041101 (2013)
Scheidenberger, C., Attallah, F., Casares, A., Czok, U., Dodonov, A., Eliseev, S.A., Geissel, H., Hausmann, M., Kholomeev, A., Kozlovski, V., Litvinov, Y.A., Maier, M., Münzenberg, G., Nankov, N., Novikov, Y.N., Radon, T., Stadlmann, J., Weick, H., Weidenmüller, M., Wollnik, H., Zhou, Z.: A new concept for time-of-flight mass spectrometry with slowed-down short-lived isotopes. Hyperfine Interact. 132, 531–534 (2001)
Wienholtz, F., Beck, D., Blaum, K., Borgmann, C., Breitenfeldt, M., Cakirli, R.B., George, S., Herfurth, F., Holt, J.D., Kowalska, M., Kreim, S., Lunney, D., Manea, V., Menendez, J., Neidherr, D., Rosenbusch, M., Schweikhard, L., Schwenk, A., Simonis, J., Stanja, J., Wolf, R., Zuber, K.: Masses of exotic calcium isotopes pin down nuclear forces. Nature 498, 346–349 (2013)
Ito, Y., Schury, P., Wada, M., Naimi, S., Sonoda, T., Mita, H., Arai, F., Takamine, A., Okada, K., Ozawa, A., Wollnik, H.: Single-reference high-precision mass measurement with a multireflection time-of-flight mass spectrograph. Phys. Rev. C 88, 011306 (2013)
Purushothaman, S., Reiter, M.P., Haettner, E., Dendooven, P., Dickel, T., Geissel, H., Ebert, J., Jesch, C., Plaß, W.R., Ranjan, M., Weick, H., Amjad, F., Ayet, S., Diwisch, M., Estrade, A., Farinon, F., Greiner, F., Kalantar-Nayestanaki, N., Knöbel, R., Kurcewicz, J., Lang, J., Moore, I., Mukha, I., Nociforo, C., Petrick, M., Pfuetzner, M., Pietri, S., Prochazka, A., Rink, A.K., Rinta-Antila, S., Scheidenberger, C., Takechi, M., Tanaka, Y.K., Winfield, J.S., Yavor, M.I.: First online results of a cryogenic stopping cell with short-lived heavy uranium fragments produced at 1000 MeV/u. Eur. Phys. Lett 104, 42001 (2013)
Jesch, C., Dickel, T., Plaß, W.R., Short, D., Ayet, S., Dilling, J., Geissel, H., Greiner, F., Lang, J., Leach, K.G., Lippert, W., Scheidenberger, C., Yavor, M.I.: The MR-TOF-MS isobar separator for the TITAN facility at TRIUMF. Hyperfine Interact 235, 97–106 (2015)
Hirsh, T.Y., Paul, N., Burkey, M., Aprahamian, A., Buchinger, F., Caldwell, S., Clark, J.A., Levand, A.F., Ying, L.L., Marley, S.T., Morgan, G.E., Nystrom, A., Orford, R., Galván, A.P., Rohrer, J., Savard, G., Sharma, K.S., Siegl, K.: First operation and mass separation with the CARIBU MR-TOF. Nucl. Instrum. Methods B 376, 229–232 (2016)
Chauveau, P., Delahaye, P., France, G.D., Abir, S.E., Lory, J., Merrer, Y., Rosenbusch, M., Schweikhard, L., Wolf, R.N.: PILGRIM, a multi-reflection time-of-flight mass spectrometer for Spiral2-S3 at GANIL. Nucl. Instrum. Methods B 376, 211–215 (2016)
Schultz, B.E., Kelly, J., Nicoloff, C., Long, J., Ryan, S., Brodeur, M.: Construction and simulation of a multi-reflection time-of-flight mass spectrometer at the University of Notre Dame. Nucl. Instrum. Methods B 376, 251–255 (2016)
Tian, Y.L., Wang, Y.S., Wang, J.Y., Zhou, X.H., Huang, W.X.: Designing a multi-reflection time-of-flight mass analyzer for LPT. Int. J. Mass Spectrom. 408, 28–32 (2016)
Dickel, T., Plaß, W.R., Ayet San Andres, S., Ebert, J., Geissel, H., Haettner, E., Hornung, C., Miskun, I., Pietri, S., Purushothaman, S., Reiter, M.P., Rink, A.K., Scheidenberger, C., Weick, H., Dendooven, P., Diwisch, M., Greiner, F., Heiße, F., Knöbel, R., Lippert, W., Moore, I.D., Pohjalainen, I., Prochazka, A., Ranjan, M., Takechi, M., Winfield, J.S., Xu, X.: First spatial separation of a heavy ion isomeric beam with a multiple-reflection time-of-flight mass spectrometer. Phys. Lett. B 744, 137–141 (2015)
Rosenbusch, M., Atanasov, D., Blaum, K., Borgmann, C., Kreim, S., Lunney, D., Manea, V., Schweikhard, L., Wienholtz, F., Wolf, R.N.: Ion bunch stacking in a Penning trap after purification in an electrostatic mirror trap. Appl. Phys. B 114, 147–155 (2014)
Hilger, R.T., Santini, R.E., McLuckey, S.A.: Nondestructive tandem mass spectrometry using a linear quadrupole ion trap coupled to a linear electrostatic ion trap. Anal. Chem. 85, 5226–5232 (2013)
Dickel, T.: Design and commissioning of an ultra-high-resolution time-of-flight based isobar separator and mass spectrometer. PhD thesis, Justus Liebig University Gießen (2010)
Plaß, W.R., Dickel, T., Ebert, J., Lang, J., Ayet, S., Geissel, H., Haettner, E., Jesch, C., Lippert, W., Petrick, M., Scheidenberger, C., Yavor, M.I.: Multiple-reflection time-of-flight mass spectrometers for the research with exotic nuclei and for analytical mass spectrometry. In: Proceedings of the 61st ASMS Conference on Mass Spectrometry and Allied Topics, p. MP 324. Minneapolis, MN, USA (2013)
Lang, J.: PhD thesis, Justus Liebig University Gießen (submitted)
Lippert, W.: Further development and application of a mobile multiple-reflection time-of-flight mass spectrometer for analytical high-resolution tandem mass spectrometry. PhD thesis, Justus Liebig University Gießen (2016)
Yavor, M.: Optics of Charged Particle Analyzers, Advances in Imaging and Electron Physics, vol. 157. Academic Press, Elsevier, Amsterdam (2009)
Wolf, R.N., Marx, G., Rosenbusch, M., Schweikhard, L.: Static-mirror ion capture and time-focusing for electrostatic ion-beam traps and multi-reflection time-of-flight mass analyzers by use of an in-trap potential lift. Int. J. Mass Spectrom. 313, 8–14 (2012)
Wiley, W.C., McLaren, I.H.: Time-of-flight mass spectrometer with improved resolution. Rev. Sci. Instrum. 26(12), 1150–1157 (1955)
Franklin, J.L., Hierl, P.M., Whan, D.A.: Measurement of the translational energy of ions with a time-of-flight mass spectrometer. J. Chem. Phys. 47, 3148–3153 (1967)
Yavor, M.I., Plaß, W.R., Dickel, T., Geissel, H., Scheidenberger, C.: Ion-optical design of a high-performance multiple-reflection time-of-flight mass spectrometer and isobar separator. Int. J. Mass Spectrom. 381/382, 1–9 (2015)
Yoon, O.K., Zuleta, I.A., Kimmel, J.R., Robbins, M.D., Zare, R.N.: Duty cycle and modulation efficiency of two-channel Hadamard transform time-of-flight mass spectrometry. J. Am. Soc. Mass Spectrom. 16, 1888–1901 (2005)
Dickel, T., Plaß, W.R., Lang, J., Ebert, J., Geissel, H., Haettner, E., Jesch, C., Lippert, W., Petrick, M., Scheidenberger, C., Yavor, M.I.: Multiple-reflection time-of-flight mass spectrometry for in situ applications. Nucl. Instrum. Methods B 317, 779–784 (2013)
Plaß, W.R., Dickel, T., Ayet San Andres, S., Ebert, J., Greiner, F., Hornung, C., Jesch, C., Lang, J., Lippert, W., Majoros, T., Short, D., Geissel, H., Haettner, E., Reiter, M.P., Rink, A.-K., Scheidenberger, C., Yavor, M.I.: High-performance multiple-reflection time-of-flight mass spectrometers for research with exotic nuclei and for analytical mass spectrometry. Phys. Scr. T 166, 014069 (2015)
Dickel, T., Yavor, M.I., Lang, J., Plaß, W.R., Lippert, W., Geissel, H., Scheidenberger, C.: Dynamical time-focus shift in multiple-reflection time-of-flight mass spectrometers. Int. J. Mass Spectrom. 412, 1–7 (2017)
Pikhtelev, A.: MAc mass analyzer control and DAQ software (2010–2015)
Bergmann, J.: [Entwicklung und Anwendung einer integrierten Systemsteuerung und Datenaufnahme für Flugzeitmassenspektrometer.] Master thesis, Justus Liebig University Gießen (2015)
Jesch, C.: The multiple-reflection time-of-flight isobar separator for TITAN and direct mass measurements at the FRS Ion Catcher. PhD thesis, Justus Liebig University, Gießen (2016)
Acknowledgments
This work was supported by the German Federal Ministry for Education and Research (BMBF) under contracts no. 06GI185I and 06GI9114I and 05P12RGFN8, by the Hessian Ministry for Science and Art (HMWK) through the LOEWE Center HIC for FAIR and the LOEWE Focus AmbiProbe, by the Helmholtz Association (HGF) through the Nuclear Astrophysics Virtual Institute (VH-VI-417), and by Justus-Liebig-Universität Gießen and GSI under the JLU-GSI strategic Helmholtz partnership agreement.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is dedicated to R. Graham Cooks on the occasion of his election to the National Academy of Sciences. We would like to congratulate him on this fantastic recognition of his excellence in science and research. WRP would also like to thank him for being a wonderful teacher and mentor. The open-mindedness he inspires is illustrated by the multidisciplinarity of the present work with applications in analytical mass spectrometry and nuclear physics.
Rights and permissions
About this article

Cite this article
Dickel, T., Plaß, W.R., Lippert, W. et al. Isobar Separation in a Multiple-Reflection Time-of-Flight Mass Spectrometer by Mass-Selective Re-Trapping. J. Am. Soc. Mass Spectrom. 28, 1079–1090 (2017). https://doi.org/10.1007/s13361-017-1617-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1617-z