
Abstract
Phosphoinositide 3-kinase γ (PI3Kγ) expressed in immune cells is linked to neuroinflammation in several neurological diseases. However, the expression and role of PI3Kγ in preclinical traumatic brain injury (TBI) have not been investigated. In WT mice, we found that TBI induced rapid and extensive expression of PI3Kγ in neurons within the perilesional cortex and the ipsilateral hippocampal subfields (CA1, CA3), which peaked between 1 and 3 days and declined significantly 7 days after TBI. Intriguingly, the induction of neuronal PI3Kγ in these subregions of the brain spatiotemporally coincided with both the TBI-induced activation of the neuronal ER stress pathway (p-eIF2α, ATF4, and CHOP) and neuronal cell death (marked by TUNEL-positive neurons) 3 days after TBI. Further, we show that the absence of PI3Kγ in knockout mice profoundly reduced the TBI-induced activation of the ER stress pathway and neuronal cell death. White matter disruption is a better predictor of long-term clinical outcomes than focal lesion size. We show that PI3Kγ deficiency not only reduced brain tissue loss but also alleviated white matter injury (determined by axonal injury and demyelination) up to 28 days after TBI. Importantly, PI3Kγ-knockout mice exhibited greater functional recovery including forepaw use, sensorimotor balance and coordination, and spatial learning and memory up to 28 days after TBI. These results unveil a previously unappreciated role for neuronal PI3Kγ in the regulation of ER stress associated with neuronal cell death, white matter damage, and long-term functional impairment after TBI.
Similar content being viewed by others


Introduction
Traumatic brain injury (TBI) is a major cause of death and disability among young adults [1]. The pathophysiology of TBI involves a primary, direct mechanical insult to the brain tissue and the secondary, delayed progressive gray and white matter damage [2]. The hippocampus, a structure in the temporal lobe, is critical for learning and memory [3]. The neurons in the hippocampus are known to be vulnerable to insults such as ischemia, hypoxia, hypoglycemia, and inflammation [4, 5]. Experimental and clinical studies have shown that hippocampal neurons are particularly vulnerable to TBI, and damage to these neurons can continue to evolve over a period of days, weeks, or even months after moderate-to-severe TBI [6, 7]. TBI patients are often faced with persistent cognitive deficits, including spatial learning and memory impairment that are caused by secondary damage to the hippocampus [8,9,10,11,12]. Persistent cognitive deficits can interfere with post-TBI recovery, rehabilitation, and living an independent life. However, to date, no clinically proven neuroprotective therapy has been identified to reduce neuronal damage and improve cognitive function [13, 14]. Thus, there is an urgent need to better understand the mechanisms of secondary brain injury caused by TBI and to identify novel therapeutic targets for TBI.
The endoplasmic reticulum (ER) is an important organelle that plays a major role in the synthesis, folding, and quality control of secreted and membrane proteins. In response to various stimuli such as ischemia or cytoplasmic Ca2+ elevation, impairment of ER function causes the accumulation of unfolded and misfolded proteins in the ER lumen, resulting in ER stress [15]. When ER stress occurs, the unfolded protein response (UPR) is activated which offers the cell a chance to restore normal ER function. The activation of UPR can be initiated by 3 proximal sensors of ER stress, namely PERK (PKR-like ER kinase), ATF6 (activating transcription factor 6), and IRE-1 (inositol requiring enzyme 1). These proximal sensors regulate the UPR through their distinct signaling cascades [15]. Activated PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), thereby reducing the rate of translation and the protein load on the ER. Phosphorylation of eIF2α paradoxically increases translation of ATF4 mRNA to produce a transcription factor that activates expression of several UPR target genes. Activation of the ER protein kinase IRE1 induces cleavage of X box-binding protein 1 (XBP1) mRNA and produces spliced XBP1 protein. XBP-1 is a key transcriptional regulator of the UPR that activates genes involved in protein folding, secretion, and degradation to restore ER function. Concurrently, ATF6 is released from GRP78 and transits to the Golgi body where it is cleaved to release a transcriptionally active fragment. Cleaved ATF6 acts in concert with spliced XBP1 protein to induce expression of genes encoding protein chaperones and components of the ER-associated degradation machinery. Among the 3 proximal sensors, the PERK–eIF2α–ATF4 pathway is the most extensively studied in TBI and is critically involved in TBI-induced neuronal ER stress and neuronal apoptosis via the upregulation of the C/EBP homologous protein (CHOP), therefore representing a potential therapeutic target for TBI [16, 17]. Over the past decade, neuronal ER stress has emerged as an important mechanism involved in the pathogenesis of neurodegenerative disorders [16]. More recently, neuronal ER stress has been proposed as a mechanistic link between acute neurotrauma and progressive neurodegeneration after TBI [17]. Therefore, manipulation of the neuronal ER stress pathway might represent a promising strategy to battle secondary neuronal injury and neurodegeneration after TBI.
PI3Kγ (phosphatidylinositol 3-kinase gamma) is preferentially expressed in immune cells and has been well recognized as a key regulator of immune and inflammatory responses in autoimmune and cardiovascular diseases [18]. Previous experimental studies have shown that increased expression of PI3Kγ in brain immune cells (including infiltrated leukocytes and activated microglia) is linked to neuroinflammation in preclinical models of Alzheimer’s disease, ischemic brain injury, and surgical brain injury [19,20,21,22]. However, the role of PI3Kγ in secondary brain injury after TBI has not been investigated. In this study, we aimed at investigating the expression and role of neuronal PI3Kγ in ER stress-induced neuronal and white matter damage and long-term functional deficits in a murine model of traumatic brain injury.
Materials and Methods
Animals
C57BL/6J mice (wild type [WT]) were purchased from Jackson Laboratories (Bar Harbor, ME). PI3Kγ−/− (KO) mice were kindly provided by Dr. P.J.M. (the Institute for Molecular Biotechnology of the Austrian Academy of Sciences, Vienna, Austria) [23]. PI3Kγ KO mice (backcrossed onto a C57BL/6 background for > 10 generations) exhibit normal size, weight, viability, fertility, blood cell count, and chemistries as described previously [21, 23]. Male mice (8 to 10 weeks old, 22 to 25 g) were used in this study. All animal protocols were approved by the Institutional Animal Care and Use Committee.
Experimental Design
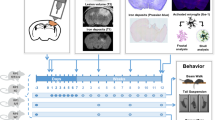
Figure 1a shows a timeline of experimental assays including behavioral tests, biochemistry, and immunohistochemistry for TBI and sham control groups up to 28 days after TBI. Suppl. Fig. 1 shows a schematic diagram of the regions of interests (ROIs) selected for image acquisition and quantitative analysis.

TBI induces PI3Kγ expression in cortical and hippocampal neurons. (a) A timeline of experimental design. WB = Western blotting; IHC = immunohistochemistry; TBI = traumatic brain injury. (b) Representative images of Western blotting (left) and quantitative analysis (right) of PI3K p110γ protein levels using protein extracts from the perilesional brain tissues of WT and KO (PI3Kγ−/−) mice subjected to TBI or sham surgery at 1 day after TBI. Protein extracts of blood neutrophils (PMN) from WT and KO mice served as a positive and a negative control, respectively. Semiquantification of immunoblots was analyzed by densitometry. Data are expressed as mean ± S.D. from 3 independent experiments. *p < 0.05 versus sham control. (c) Representative immunofluorescence images showing that PI3K p110γ was constitutively expressed in the vascular-like structures in sham control, but its expression was increased in neuron-like cells within 1 to 7 days after TBI in the perilesional cortex region of WT mice, and no expression of PI3K p110γ was detected in KO-sham and KO-TBI mice. Scale bar = 100 μm. (d) Representative immunofluorescence images (left) showing that PI3K p110γ was constitutively expressed in the vascular-like structures in sham control, but its expression was increased and colocalized with NeuN (a neuronal marker) (quantitative analysis shown in the right panel) at 3 days after TBI. White dotted boxes indicate the regions selected for higher magnification. Images were acquired from the perilesional cortex region, ipsilateral CA1 and CA3 subfields of the dorsal hippocampus at − 1.5 mm posterior to the bregma. Scale bar = 100 μm. n = 5 per group, *p < 0.05 versus sham control
Controlled Cortical Impact Injury
TBI was induced using a computer-controlled cortical impact (CCI) device (PinPoint Precision Cortical Impactor, Hatteras, PCI 3000, Cary, NC), as we described previously [24]. Mice were anesthetized with intraperitoneal injection of ketamine (80 mg/kg)/xylazine (5 mg/kg). Depth of anesthesia was assessed by monitoring respiration rate and pedal withdrawal reflexes. The anesthetized mice were placed on a stereotaxic apparatus with a heating pad to maintain body temperature at 37 ± 0.5 °C. The head was shaved and disinfected, and then the skull was exposed by a midline scalp incision. A 4-mm craniotomy was made by an electric drill on the right lateral side of the skull and centered 2.7 mm lateral from the midline and 2.0 mm posterior to the bregma. Then, a 3-mm rounded impactor tip was placed in the center of the craniotomy site, and the impact was delivered to the right parietal cortex at 2 mm depth with 2 m/s velocity and 100 ms dwell time. These parameters produce moderate-to-severe TBI injury in mice as documented previously [25, 26]. After impact, Surgicel (Johnson & Johnson, Dallas, TX) was applied to the dura. The skullcap was replaced and affixed with dental adhesive and the incision was sutured. Sham animals underwent the same procedure as CCI mice except impact.
Neurobehavioral Assessments
A battery of behavioral tests, including modified neurological severity score (mNSS: motor, sensory, reflex, and balance), foot-fault test (forelimb placement), Morris water maze test (spatial learning and memory), and novel object recognition (recognition memory) [27, 28], were performed before and at the indicated time after surgery by an investigator who was blinded to the experimental groups. Detailed methods for each test are available in the Supplemental file.
Contusion Volume Measurement
The animals were euthanized at 3 or 28 days after CCI in deep anesthesia with sodium pentobarbital (150 mg/kg) and transcardially perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA). Brains were removed, postfixed overnight in 4% FPA, cryoprotected in 30% sucrose solution, embedded in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA), then stored at − 80 °C before sectioning. Each brain was serially sectioned with a cryostat from caudal to rostral. Coronal sections (40 μm thick) were sliced every 500 μm, dry fixed, stained with Nissl staining, and photographed with a digital camera system connected to a microscope. The contused brain tissue was then measured on the digital photographs using NIH Image J software by an investigator blinded to animal groups. Contusion volume (V, mm3) was calculated based on the contused area (A, mm2) on 15 coronal sections 500 μm distance (d = 0.5 mm) apart using the following formula: V = d × (A1 × 0.5 + A2 + A3 + … + A14 + A15 × 0.5), as described previously [29].
Immunohistochemistry
The animals were euthanized at 1, 3, 7, or 28 days after CCI. After transcardial perfusion with 30 mL of cold phosphate-buffered saline (PBS, 10 mM, pH 7.4), followed by 50 mL of 4% paraformaldehyde (PFA), the brains were removed, postfixed overnight in 4% PFA, and cryoprotected for 48 h in 30% sucrose at 4 °C. Serial 10-μm-thick coronal sections were cut using a cryostat (Leica CM 1950), and stored at − 80 °C until used. Immunofluorescence staining was performed as described previously [20, 21]. The primary antibodies anti-PI3K p110γ (1:200, Abcam, Cambridge, MA), anti-NeuN (1:500, Millipore, Burlington, MA), anti-CHOP (1:100, Abcam), anti-ATF4 (1:200, Abcam), anti-phosphorylated eukaryotic translation initiation factor 2a (p-eIF2a) (1:50, Cell Signaling Technology, Danvers, MA), anti-Iba1 (1:200, Wako, Richmond, VA), anti-myelin basic protein (MBP) (1:200, Abcam), and anti-neurofilament H non-phosphorylated mouse mAb (SMI-32) (1:500, Millipore) were used. Irrelevant class- and species-matched immunoglobulins as well as incubations without the primary antibody were used as controls. After primary antibody incubation, sections were incubated with Alexa fluorochrome-conjugated secondary antibodies (1:1000 in PBS, Invitrogen, Carlsbad, CA). Images were captured using a Nikon Eclipse Ti-E Microscope connected to a computer. Five consecutive coronal sections 0.5 mm apart from the area of maximal cortical injury (from − 1.5 to − 3.5 mm relative to the bregma) were used for each different stain. The PI3K p110γ-positive neurons (marked by NeuN) were measured in 4 randomly selected microscopic fields in the perilesional cortex and hippocampal CA1 and CA3 regions; data are expressed as the number of cells per square millimeter (mm2) of the perilesional cortex and per 250 μm length of medial CA1 or CA3 as described previously [30]. The intensity of the immunoreactivity for SMI-32 and MBP was measured in the predefined regions of interest (Suppl. Fig. 1) including the perilesional cortex, corpus callosum (CC), and external capsule (EC) using Image Pro plus software (version 5.1, Media Cybernetics), and data are presented as fold change relative to sham controls. All measurements were performed in a blinded manner.
Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Staining
Early neuronal apoptosis induced by TBI was examined at 3 days after TBI. Double labeling staining of TUNEL and NeuN was performed to colocalize apoptotic neurons as described previously [31]. Serial coronal sections (10 μm thick) at the level of the dorsal hippocampus (− 1.4 to − 2.1 mm from the bregma) at 100-μm intervals were selected for staining. Sections were stained with anti-NeuN antibody (1:500; Millipore), then the TUNEL staining was performed using a commercial kit (In Situ Cell Death Detection Kit, Roche, Basel, Switzerland) according to the manufacturer’s protocol. For negative control TUNEL stain, slides were incubated with the label solution without terminal transferase for TUNEL. The data are expressed as the number of total TUNEL-positive cells or apoptotic neurons (i.e., TUNEL- and NeuN-double-positive cells) per square millimeter (mm2) of the perilesional cortex and per 250 μm length of medial CA1 or CA3 as described previously [30]. All measurements were performed in a blinded manner.
Hippocampal Neuronal Loss and White Matter Damage Measurement
Hippocampal neuronal loss induced by TBI in the chronic phase was assessed at 28 days after injury. Nissl staining was performed on serial paraffin sections (10 μm thick) at the level of the dorsal hippocampus (− 1.4 to − 2.1 mm from the bregma) at 100-μm intervals. To maintain consistency across animals, 4 rectangular boxes (1.0 × 0.25 mm) were centered over the CA1 and CA3 layers. CA1 and CA3 pyramidal neurons showing intact round nuclei were counted as surviving cells using a Nikon Ti-E light microscope at × 400 magnification. The data are expressed as the number of surviving neurons per millimeter of medial CA1 or CA3 as described previously [32].
White matter damage induced by TBI in the chronic phase was assessed at 28 days after injury. The modified Bielschowsky’s silver (KT499, American MasterTech, Lodi, CA) and luxol fast blue (IW-3005, IHCWORLD, Woodstock, MD) stain kits were used to examine the density of axons and myelin, respectively. The intensity of silver staining was measured by densitometry [33]. To determine the intensity of luxol fast blue staining, color deconvolution was used to separate an image into 3 channels corresponding to the actual colors of stains for quantification [34, 35]. The intensity of each staining was measured in the predefined regions of interest (Suppl. Fig. 1) including the corpus callosum (CC) and external capsule (EC) using Image J (National Institutes of Health, Bethesda, MD). The data collected from 5 sections and 4 fields within each section were averaged to obtain a single value for 1 animal and were presented as percentage change relative to sham controls. All measurements were performed in a blinded manner.
Western Blot Analysis
Protein extraction and Western blot analysis were performed as we described previously [21]. The animals were euthanized at 24 h or 72 h after surgery. After transcardial perfusion with normal saline, brains were removed and divided into 2 hemispheres and whole-cell lysates were prepared from the pericontused brain tissue (between − 1.5 and − 3.5 mm relative to the bregma) and homogenized in ice-cold isotonic mitochondrial buffer supplemented with protease inhibitor cocktail (1 mg/mL each of aprotinin, pepstatin, and leupeptin; 100 mg/mL phenylmethylsulfonyl fluoride and 2 mm sodium orthovanadate). The homogenized samples were centrifuged at 1000 × g at 4 °C for 10 min, and the supernatants were collected. The cytosolic and mitochondrial fractions were further prepared from the supernatants using a commercially available cytosol/mitochondrial fractionation kit according to the manufacturer’s protocol (Millipore). The protein content was determined by a BCA protein assay kit using a 96-well microplate reader (Spectra Max 340, Molecular Devices, San Jose, CA). Antibodies were used as follows: monoclonal rabbit antibody against PI3K p110γ (1:1000), Bcl-2 (1:1000), Bax (1:1000), COX-IV (1:1000) (all purchased from Cell Signaling), and mouse antibody against β-actin (1:4000, Sigma). β-Actin and COX-IV were used as loading controls for cytosolic and mitochondrial fractions, respectively. Immunopositive bands of horseradish peroxidase-conjugated secondary antibodies were detected with an ECL system (GE Healthcare, Chicago, IL) and exposure to ECL Hyperfilm.
Statistical Analysis
Data were expressed as mean ± S.D. GraphPad Prism 5 software package was used for statistical analysis. The normality of data distribution was assessed with the D’Agostino–Pearson omnibus test. For normally distributed variables, 1-way ANOVA followed by the Bonferroni post hoc tests was used to assess differences among multiple groups. The Kruskal–Wallis test followed by the Mann–Whitney U test was used to explore differences among groups in non-normally distributed variables. If only 2 groups were compared, an unpaired, 2-tailed Student t test was used. For evaluation of behavioral data, repeated-measures analyses of variance (2-way or 1-way as appropriate) were utilized to determine statistical differences. The numbers of animals used in each experiment were based on sample size calculation (power = 0.8, α = 0.05) using an online calculator (http://www.lasec.cuhk.edu.hk/sample-size-calculation.html). An estimated 5 animals per group would be required to detect ≥ 30% difference in brain tissue loss between a 2-group comparison with the 25% standard deviation of the mean value. Differences with p < 0.05 were considered statistically significant.
Results
TBI Induces PI3Kγ Expression Primarily in Cortical and Hippocampal Neurons
Western blot analysis showed that the PI3K p110γ protein expression was significantly increased in the perilesional brain tissue (between − 1.5 and − 3.5 mm relative to the bregma) 1 day after TBI in WT mice (Fig. 1b). Immunostaining showed that PI3K p110γ was expressed in vascular-like structures in WT-sham control (Fig. 1c), which is consistent with our previous data [20]. However, in WT mice, TBI robustly induced PI3K p110γ expression in neuron-like cells within the perilesional cortex, which peaked between 1 and 3 days and declined significantly by 7 days after TBI (Fig. 1c). Double immunofluorescence staining confirmed that the TBI-induced PI3K p110γ expression was located primarily in NeuN-positive neurons in the perilesional cortex and hippocampal CA1 and CA3 regions at 3 days after the insult (Fig. 1d), but rarely in reactive microglia/microphages (arrows in Fig. 1d and Suppl. Fig. 2). It is noteworthy that the absence of p110γ expression in PI3Kγ KO-sham and especially in KO-TBI mice was confirmed by both Western blot (Fig. 1b) and immunostaining (Fig. 1c). Collectively, these data indicate that PI3K p110γ is a TBI-responsive gene in brain cortical and hippocampal neurons.
Induction of Neuronal PI3Kγ Correlates Positively with Neuronal ER Stress After TBI
Endoplasmic reticulum (ER) stress contributes substantially to secondary neuronal injury following TBI [36,37,38,39]. Using immunohistochemistry, ER stress markers CHOP (C/EBP homologous protein), ATF4 (activating transcription factor 4), and p-eIF2α (phosphorylated eIF2α) were examined. In WT-TBI mice, CHOP was robustly activated in the ipsilateral cortex (Fig. 2a) and hippocampal CA1 (Fig. 2b) and CA3 (Fig. 2c) subfields at 3 days after TBI, and colocalized almost exclusively with NeuN, a neuronal cell marker. Moreover, persistent CHOP activation was detected even at 28 days after injury in WT-TBI mice (Fig. 2e). Such robust and persistent CHOP activation was significantly alleviated in KO-TBI mice in the corresponding areas (Fig. 2a–d). Further, activation of ATF4 (Fig. 2f) and p-eIF2α (Suppl. Fig. 3) in the ipsilateral cortical and hippocampal neurons suggests that TBI triggered neuronal ER stress through the PERK–eIF2a–ATF4–CHOP pathway. Of note, TBI-induced neuronal PI3Kγ was colocalized with CHOP in neurons within the ipsilateral cortex and hippocampus (Suppl. Fig. 4). Taken together, these findings suggest that TBI-induced neuronal PI3Kγ was implicated in post-traumatic ER stress.

TBI-induced neuronal PI3Kγ positively correlates with neuronal ER stress activation. (a, b, c, and d) Representative fluorescence images of CHOP (green) and NeuN (red) double immunostaining (a, b, and c) and quantitative analysis of TUNEL/CHOP-stained cells (d) in the ipsilateral perilesional cortex, CA1 and CA3 subfields of the dorsal hippocampus at 3 days after TBI. Scale bar = 100 μm; n = 5 per group, *p < 0.05 versus WT. (e) Representative fluorescence images of CHOP (green) and NeuN (red) double immunostaining in the ipsilateral perilesional cortex, CA1 and CA3 subfields of the dorsal hippocampus at 28 days after TBI. Scale bar = 100 μm. (e) Representative fluorescence images of ATF4 (green) and NeuN (red) double immunostaining in the ipsilateral perilesional cortex, CA1 and CA3 subfields of the dorsal hippocampus at 3 days after TBI. Scale bar = 100 μm
Neuronal ER Stress Contributes to Secondary Neuronal Injury Through the Mitochondrial Apoptotic Pathway
To examine the effects of neuronal PI3Kγ induction after TBI, we assessed secondary neuronal injury in both the acute and chronic phases of TBI. At 3 days after TBI, the number of TUNEL-positive apoptotic cells, which were mostly also NeuN-positive, significantly increased in the perilesional cortex and the ipsilateral hippocampal subfields of CA1 and CA3 in WT mice (Fig. 3a); however, the number of TUNEL-positive apoptotic cells was dramatically decreased in the corresponding regions of PI3Kγ KO mice (Fig. 3a). At 28 days after TBI, Nissl staining showed substantial neuronal loss in the ipsilateral CA1 and CA3 subfields of the dorsal hippocampus in WT mice, whereas the TBI-induced neuronal loss in the hippocampus was profoundly attenuated in PI3Kγ KO mice (Fig. 3b, c). As expected, the numbers of neurons in the contralateral CA1 and CA3 regions of PI3Kγ KO and WT mice were almost identical (Fig. 3b, c). These findings show that TBI exacerbated, whereas PI3Kγ deficiency alleviated, progressive neuronal apoptosis, suggesting that TBI-induced neuronal PI3Kγ was associated with secondary neuronal injury in the traumatic brain.

TBI-induced neuronal PI3Kγ is associated with secondary neuronal injury in the traumatic brain. (a) Representative fluorescence images of TUNEL (green) and NeuN (red) double staining (left) and quantitative analysis of TUNEL/NeuN-stained cells (right) in the ipsilateral perilesional cortex, CA1 and CA3 subfields of the dorsal hippocampus at 3 days after TBI. Scale bar = 100 μm; n = 5 per group, *p < 0.05 versus WT-TBI. (b) Representative immunohistochemical photomicrographs of crystal violet-stained brain tissue showing hippocampal neuronal loss at 28 days after TBI. Four rectangular boxes (1.0 × 0.25 mm) were centered over the CA1 and CA3 layer to maintain consistency across animal. Ips = ipsilateral; Ctrl = contralateral. Images correspond to brain sections about − 1.4 mm posterior to the bregma. (c) Quantitative analysis of live neurons per millimeter of medial CA1 or CA3 hippocampal subfields at 28 days after TBI. Scale bar = 250 μm; n = 5 per group, *p < 0.05 versus WT contralateral region, #p < 0.05 versus WT ipsilateral region
CHOP activation is critical for ER stress-triggered apoptotic cell death. CHOP activation downregulates anti-apoptotic Bcl-2 and translocates pro-apoptotic Bax from the cytosol to the mitochondria [40]. The mitochondrial and cytosolic fractions of the perilesional brain tissue were prepared from PI3Kγ KO and WT mice subjected to TBI or sham at 3 days after surgery, and the fractions were analyzed by Western blotting to examine the subcellular expression levels of Bcl-2 and Bax. The expression levels of both Bcl-2 and Bax in all the fractions were comparable at baseline between the WT-sham and KO-sham groups (Fig. 4a–c). However, the expression of Bcl-2 in the mitochondria was markedly reduced in the WT-TBI group, but this reduction was largely reversed in the KO-TBI group (Fig. 4a). Bax is mostly cytosolic and translocates to mitochondria following an apoptotic stimulus. We found that the expression of Bax was significantly increased in the mitochondria (Fig. 4b) with a concomitant decrease in the cytosol (Fig. 4c) in the WT-TBI group, and these changes were reversed in the KO-TBI group. Taking all the above findings together, we propose that TBI-induced neuronal PI3Kγ is implicated in neuronal ER stress which contributes to secondary neuronal injury through the mitochondrial apoptotic pathway.

Neuronal ER stress contributes to secondary neuronal injury through the mitochondrial apoptotic pathway. (a, b, and c) Representative immunoblots (left) and quantitative analysis (right) showing the mitochondrial (Mito) Bcl-2 and mitochondrial and cytosolic (Cyto) Bax protein levels using protein extracts from perilesional brain tissue at 3 days after TBI. β-Actin and COX-IV were used as loading controls for cytosolic and mitochondrial fractions, respectively. Quantitative analysis of Western blot was performed by densitometry. Data are expressed as mean ± S.D. from 3 independent experiments. *p < 0.05 versus respective sham control, #p < 0.05 versus WT-TBI
PI3Kγ Deficiency Mitigates White Matter Injury After TBI
White matter injury is a critical component of the progression of traumatic brain injury [41]. Therefore, we further examined white matter injury 28 days after TBI. In comparison with WT mice, TBI-induced loss of axons and myelin was significantly attenuated in PI3Kγ KO mice in the corpus callosum (CC) and external capsule (EC) in the ipsilateral hemisphere, as determined respectively by Bielschowsky’s silver (Fig. 5a, c) and Luxol Fast Blue staining (Fig. 5b, d). Moreover, at 28 days after TBI, argyrophilic axonal degeneration [42] was quite evident in the EC area of WT-TBI mice (arrows, Fig. 5a), whereas such argyrophilic structures were absent in other groups (WT-sham, KO-sham, KO-TBI). TBI-induced white matter injury was further confirmed using immunofluorescent double labeling for MBP, a marker of myelin integrity, and SMI32, a marker for axonal damage. The ratio of SMI32 to MBP was calculated as an indicator of white matter injury (axonal injury and demyelination) as described previously [43]. We found that PI3Kγ deficiency significantly alleviated post-traumatic white matter injury, as indicated by the decreased ratio of SMI32 to MBP staining intensity in the perilesional cortex (Fig. 6a), CC (Fig. 6b), and EC (Fig. 6c) at 28 days after TBI.

PI3Kγ deficiency mitigates white matter injury after TBI. (a and b) Representative immunohistochemical images showing Bielschowsky’s silver staining (a) and Luxol fast blue/hematoxylin–eosin (LFB/HE) staining (b) in the ipsilateral regions of the corpus callosum (CC) and external capsule (EC) at 28 days after TBI. All coronal brain sections are about − 0.5 mm posterior to the bregma. Scale bar = 200 μm. Higher magnification of immunohistochemical images of Bielschowsky’s silver staining in the ipsilateral region of EC is also shown (scale bar = 25 μm). (c and d) Quantitative analysis of axon density (c) and myelin density (d) by Bielschowsky’s silver and LFB/HE staining, respectively. Data are expressed as percentage of the immunostaining intensity relative to WT-sham control (set at 100%). n = 6 per group, *p < 0.05 versus respective sham control, #p < 0.05 versus WT-TBI

PI3Kγ deficiency attenuates axonal injury and demyelination after TBI. (a, b, and c) Representative immunofluorescence images and quantitative analysis for dephosphorylated neurofilament protein (SMI-32) and myelin basic protein (MBP) in the ipsilateral regions of the perilesional cortex (a, CTX), corpus callosum (b, CC) and external capsule (c, EC) at 28 days after TBI. The ratio of SMI32 to MBP was calculated as an indicator of white matter injury (axonal injury and demyelination). Scale bar = 25 μm; n = 6 per group, *p < 0.05 versus respective sham control, #p < 0.05 versus WT-TBI
PI3Kγ Deficiency Reduces Brain Contusion and Improves Functional Outcomes After TBI
The mNSS (Fig. 7a) and foot-fault test (Fig. 7b) were conducted to assess sensorimotor deficits after TBI. Although both PI3Kγ KO and WT mice subjected to TBI showed significant sensorimotor deficits from 1 day to 28 days after TBI, PI3Kγ KO mice exhibited significant improvements in sensorimotor functions compared to WT mice with time extension (Fig. 7a, b). Further, the Morris water maze test was conducted to assess cognitive deficits 24 days after TBI. PI3Kγ KO mice exhibited significant improvements in spatial learning (Fig. 7c) and memory functions (Fig. 7d) compared to WT mice, as demonstrated by decreased swim latency to find the platform (Fig. 7e) and increased swim time in the target quadrant when the platform was removed after the learning period (Fig. 7f). In addition, the novel object recognition test was conducted to assess recognition memory on day 28 after TBI. PI3Kγ KO mice exhibited better recognition memory as determined by the discrimination index (i.e., a percentage of time spent exploring a novel object relative to the total time exploring both novel and familiar object) compared to WT mice (Fig. 7g).

PI3Kγ deficiency improves neurological outcome and reduces brain contusion after TBI. (a and b) Two behavioral tests were performed to assess sensorimotor deficits including (a) the modified neurological severity score (mNSS) and (b) foot-fault test before (day 0) and up to 28 days after TBI. n = 12 per group. *p < 0.05 versus WT-TBI mice at respective time points. (c and d) Representative images of the swim paths of mice during the cued learning phase (c) when the submerged platform was present and the spatial memory phase (d) when the platform was removed. (e) Latency to find the hidden platform in the cued learning test from 24 to 28 days after surgery. n = 12 per group. *p < 0.05 versus respective sham control, #p < 0.05 versus WT-TBI. (f) Time spent in the target quadrant (where the platform was previously located) at 28 days after TBI. n = 12 per group. *p < 0.05 versus WT-sham control, #p < 0.05 versus WT-TBI. (g) Novel objective recognition (NOR) was measured on day 28 after surgery and expressed by percent time spent with a novel object compared with the familiar object (discrimination index). *p < 0.05 versus WT-sham control, #p < 0.05 versus WT-TBI. (h) Quantitative analysis of contusion volumes in wild-type (WT) and PI3Kγ knockout (KO) mice at 3 and 28 days after TBI. n = 12 per group. *p < 0.05 versus WT-TBI mice at respective time points
Contusion volume was measured at 3 days and 28 days after TBI in brain sections with Nissl staining. The contusion volume was 8.42 ± 0.53 mm3 in the WT group and decreased to 4.45 ± 0.56 mm3 in the PI3Kγ KO group at 3 days after TBI (Fig. 7h). At 28 days after TBI, the PI3Kγ KO mice showed significantly less cortical and hippocampal tissue loss compared with the WT mice (Fig. 7h and Suppl. Fig. 5a), suggesting that a smaller contusion volume corresponds to better preservation of neurobehavioral function. In addition, KO-TBI mice showed an earlier regain of body weight compared to WT-TBI mice (Suppl. Fig. 5b), suggesting alleviated general stress in KO-TBI mice. There was no mortality in this moderate-to-severe CCI model.
Discussion
The present study, for the first time, demonstrates that induction of neuronal PI3Kγ expression plays an important role in mediating TBI-induced ER stress and secondary neuronal injury. Three important findings contribute to our understanding of neuronal PI3Kγ in TBI pathophysiology. First, TBI induces rapid and extensive expression of PI3Kγ in neurons in the perilesional cortex and hippocampus. Second, induction of PI3Kγ in these neurons is functionally relevant as it is critically involved in post-traumatic neuronal ER stress and secondary neuronal injury. Third, PI3Kγ deficiency alleviates ER stress-mediated secondary neuronal injury, preserves white matter integrity, and promotes long-term recovery of neurological function following TBI.
The present study demonstrates that TBI induced rapid and extensive expression of PI3Kγ in neurons within the perilesional cortex and the ipsilateral hippocampus (CA1, CA3 subfields) after TBI. This finding is novel as it is different from the previous findings observed in other experimental models of neurological diseases, including cerebral ischemia [20, 21], surgical brain injury [22], and Alzheimer’s disease [19], in which PI3Kγ expression was found primarily in infiltrated leukocytes and reactive microglia that were critically involved in the development of neuroinflammation, but rarely in neurons. In the normal murine brain, PI3Kγ expression is observed in vascular-like structures [23] and in neurons within specific brain regions, such as the periaqueductal gray matter (PAG) [44], locus coeruleus (LC) [45], and dorsal root ganglion (DRG) [46], but not in the cortex and hippocampus. Taken together, these results suggest that PI3Kγ gene expression in different brain cell types can be differentially induced by distinct pathological stimuli, thereby PI3Kγ exerts its cell-type-specific effects in different neurological disease models.
Furthermore, the present study demonstrates that induction of neuronal PI3Kγ is functionally important as it is involved in TBI-induced neuronal ER stress and secondary neuronal injury. ER stress has emerged as an important mechanism of secondary neuronal injury after TBI through mitochondrial-mediated neuronal apoptosis [16, 17]. Previous studies have shown that moderate-to-severe TBI can cause persistent ER stress activation up to 21 days after the insult [37, 38]. In the present study, we show that TBI induced early (3 days) and persistent (28 days) activation of the ER stress pathway (p-eIF2α, ATF4, CHOP) in neurons in the perilesional cortex and underlying hippocampus (CA1, CA3 subfields). Notably, the induction of neuronal PI3Kγ in these subregions of the brain spatiotemporally coincided with the TBI-induced activation of the neuronal ER stress pathway (p-eIF2α, ATF4, CHOP) and neuronal cell death (marked by TUNEL-positive neurons). Also importantly, the results of this study show that the absence of PI3Kγ in knockout mice profoundly reduced the TBI-induced activation of the ER stress pathway and neuronal cell death. The phosphoinositide 3-kinase (PI3K) family comprises multiple isoforms and is involved in a variety of cellular functions in an isoform-specific manner. Although the PI3K signaling pathway has been implicated in TBI pathology [47], the role of specific PI3K isoforms including PI3Kγ in TBI has not been investigated. To our knowledge, no prior studies have examined the role of PI3Kγ in ER stress, although the PI3K pathway is implicated in ER stress in neurons [48,49,50]. The results of this study reveal a previously unappreciated role of PI3Kγ in the regulation of neuronal ER stress associated with neuronal cell death after TBI, as illustrated in Fig. 8.

Illustration showing that TBI-induced neuronal PI3Kγ contributes to ER stress-associated secondary neuronal injury. Induction of PI3Kγ in neurons by TBI exacerbates, whereas PI3Kγ deficiency alleviates, ER stress-associated secondary neuronal injury through the mitochondrial apoptotic pathway. Neuronal ER stress leads to PERK autophosphorylation, which, in turn, phosphorylates elF2α and attenuates translation of 5′-capped mRNAs. However, translation of selected mRNAs is favored under ER stress, such as ATF4, which then induces expression of CHOP. CHOP downregulates the expression of anti-apoptotic Bcl-2 and increases mitochondrial Bax, thereby carrying out neuronal apoptosis, leading to secondary neuronal injury
The present study further demonstrates that induction of neuronal PI3Kγ contributes to long-term sensorimotor and cognitive impairment after TBI. PI3Kγ has been implicated in hippocampus-dependent cognitive impairment in several neurological disease models. Choi et al. [51] have shown that overexpression of PI3Kγ in the hippocampus by local injection of flag-p110γ-expressing adeno-associated viruses (AAVs) into the hippocampal CA1 region impairs synaptic plasticity and hippocampus-dependent spatial learning. In a murine model of Alzheimer’s disease induced by intracerebroventricular injection of beta-amyloid 1-40 peptide, Passos et al. [19] have shown that pharmacological inhibition of PI3Kγ with its selective inhibitor AS605240 prevents Abeta(1-40)-induced cognitive deficits and synaptic dysfunction by attenuating the Abeta(1-40)-induced neuroinflammation as evidenced by reduced accumulation of activated astrocytes and microglia in the hippocampus. Neuronal ER stress not only contributes to acute neuronal cell death but also plays an important role in chronic axonal injury and neurodegeneration after TBI [16]. The results of the present study demonstrate the importance of induction of neuronal PI3Kγ expression in long-term functional deficits after TBI. Prolonged and diffuse white matter injury has been observed in humans after TBI and can serve as a better predictor of long-term clinical outcomes than focal lesion size [41]. Here, we show that the present moderate-to-severe TBI model not only caused significant loss of hippocampal neurons but also induced persistent and diffuse white matter injury in wild-type mice 28 days after the insult, as determined by axonal damage and demyelination within the corpus callosum (CC) and external capsule (EC), whereas the absence of PI3Kγ in knockout mice significantly reduced the loss of hippocampal neurons and preserved long-term white matter integrity. The preserved white matter integrity along with the reduced secondary neuronal injury by PI3Kγ deficiency most likely contributed to the improved sensorimotor and cognitive outcomes observed in this study. These results unveil a previously unappreciated role for neuronal PI3Kγ in the regulation of ER stress associated with neuronal cell death, white matter damage, and long-term functional impairment after TBI.
The limitations of the study are as follows: many different mechanisms may contribute to the PI3Kγ-mediated effects on secondary brain injury after TBI. Secondary brain injury after TBI involves complex mechanisms, including excitotoxicity, oxidative stress, mitochondrial dysfunction, blood–brain barrier (BBB) disruption, and neuroinflammation [52, 53]. PI3Kγ is predominantly expressed in leukocytes and endothelial cells and participated in regulating leukocyte activation and migration into inflamed tissues [54, 55]. Numerous studies have established the importance of PI3Kγ in peripheral inflammation [54], but relatively less attention has been paid to its role in neuroinflammatory response after brain injury. Recently, several studies from our laboratory and other groups have shown that genetic and pharmacological inhibition of PI3Kγ reduced neuroinflammation and brain damage in preclinical models of ischemic brain injury, surgical brain injury, and Alzheimer’s disease [19,20,21,22, 56]. However, to our knowledge, the role of PI3Kγ in neuroinflammation after TBI has not been investigated yet. In the present study, we specifically addressed the role of the induction of PI3Kγ in cortical and hippocampal neurons in TBI-induced neuronal ER stress and secondary neuronal injury. Nevertheless, other mechanisms, especially neuroinflammation, could also be involved in the PI3Kγ-mediated effects on TBI pathology. Indeed, we observed that TBI induced a significant increase in the accumulation of reactive microglia in the injured brain 3 days after TBI in WT mice and this increase was dramatically decreased in PI3Kγ knockout mice (Suppl. Fig. 2). Microglial interactions with neurons have been shown to play an important role in mediating neuroinflammation, neuronal death, and eventual functional deficits in neurological disorders including TBI [57,58,59]. The detailed mechanism by which PI3Kγ mediates the microglia–neuronal cross-talk after TBI warrants further investigation.
In conclusion, this study demonstrates that 1) PI3Kγ is a previously unrecognized TBI-responsive gene in brain neurons, and induction of neuronal PI3Kγ expression contributes to neuronal ER stress and secondary neuronal injury after TBI, and 2) the absence of PI3Kγ in knockout mice reduces neuronal ER stress and confers potent neuroprotection in both acute and chronic stages of TBI, suggesting that PI3Kγ might be a novel and promising therapeutic target for TBI. Therapeutic targeting of PI3Kγ with its selective inhibitors for TBI is under investigation in our lab.
References
Dewan MC, Rattani A, Gupta S, et al. Estimating the global incidence of traumatic brain injury. J Neurosurg 2018:1–18.
Stoica BA, Faden AI. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 2010;7(1):3–12.
Leuner B, Gould E. Structural plasticity and hippocampal function. Annu Rev Psychol 2010;61:111–140, C111–113.
Bartsch T, Schönfeld R, Müller FJ, et al. Focal lesions of human hippocampal CA1 neurons in transient global amnesia impair place memory. Science 2010;328(5984):1412–1415.
Kadar T, Dachir S, Shukitt-Hale B, Levy A. Sub-regional hippocampal vulnerability in various animal models leading to cognitive dysfunction. J Neural Transm (Vienna) 1998;105(8–9):987–1004.
Colicos MA, Dixon CE, Dash PK. Delayed, selective neuronal death following experimental cortical impact injury in rats: possible role in memory deficits. Brain Res 1996;739(1–2):111–119.
Titus DJ, Sakurai A, Kang Y, et al. Phosphodiesterase inhibition rescues chronic cognitive deficits induced by traumatic brain injury. J Neurosci 2013;33(12):5216–5226.
Lew HL, Poole JH, Guillory SB, Salerno RM, Leskin G, Sigford B. Persistent problems after traumatic brain injury: the need for long-term follow-up and coordinated care. J Rehabil Res Dev 2006;43(2):vii–x.
Reeves TM, Lyeth BG, Phillips LL, Hamm RJ, Povlishock JT. The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Res 1997;757(1):119–132.
Prins ML, Hovda DA. Traumatic brain injury in the developing rat: effects of maturation on Morris water maze acquisition. J Neurotrauma 1998;15(10):799–811.
Atkins CM, Falo MC, Alonso OF, Bramlett HM, Dietrich WD. Deficits in ERK and CREB activation in the hippocampus after traumatic brain injury. Neurosci Lett 2009;459(2):52–56.
Hamm RJ, Dixon CE, Gbadebo DM, et al. Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J Neurotrauma 1992;9(1):11–20.
Stoica B, Byrnes K, Faden AI. Multifunctional drug treatment in neurotrauma. Neurotherapeutics 2009;6(1):14–27.
Diaz-Arrastia R, Kochanek PM, Bergold P, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma 2014;31(2):135–158.
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007;8(7):519–529.
Li S, Yang L, Selzer ME, Hu Y. Neuronal endoplasmic reticulum stress in axon injury and neurodegeneration. Ann Neurol 2013;74(6):768–777.
Yin Y, Sun G, Li E, Kiselyov K, Sun D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res Rev 2017;34:3–14.
Costa C, Martin-Conte EL, Hirsch E. Phosphoinositide 3-kinase p110γ in immunity. IUBMB Life 2011;63(9):707–713.
Passos GF, Figueiredo CP, Prediger RD, et al. Involvement of phosphoinositide 3-kinase gamma in the neuro-inflammatory response and cognitive impairments induced by beta-amyloid 1-40 peptide in mice. Brain Behav Immun 2010;24(3):493–501.
Jin R, Yu S, Song Z, et al. Phosphoinositide 3-kinase-gamma expression is upregulated in brain microglia and contributes to ischemia-induced microglial activation in acute experimental stroke. Biochem Biophys Res Commun 2010;399(3):458–464.
Jin R, Song Z, Yu S, et al. Phosphatidylinositol-3-kinase gamma plays a central role in blood-brain barrier dysfunction in acute experimental stroke. Stroke 2011;42(7):2033–2044.
Huang L, Sherchan P, Wang Y, et al. Phosphoinositide 3-kinase gamma contributes to neuroinflammation in a rat model of surgical brain injury. J Neurosci 2015;35(29):10390–10401.
Sasaki T, Irie-Sasaki J, Jones RG, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 2000;287(5455):1040–1046.
Anthony Jalin AMA, Jin R, Wang M, Li G. EPPS treatment attenuates traumatic brain injury in mice by reducing Aβ burden and ameliorating neuronal autophagic flux. Exp Neurol 2019;314:20–33.
Siebold L, Obenaus A, Goyal R. Criteria to define mild, moderate, and severe traumatic brain injury in the mouse controlled cortical impact model. Exp Neurol 2018;310:48–57.
Romine J, Gao X, Chen J. Controlled cortical impact model for traumatic brain injury. J Vis Exp 2014;90:e51781.
Grayson B, Leger M, Piercy C, Adamson L, Harte M, Neill JC. Assessment of disease-related cognitive impairments using the novel object recognition (NOR) task in rodents. Behav Brain Res 2015;285:176–193.
Schaar KL, Brenneman MM, Savitz SI. Functional assessments in the rodent stroke model. Exp Transl Stroke Med 2010;2(1):13.
Dash PK, Orsi SA, Zhang M, et al. Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS One 2010;5(6):e11383.
Jover-Mengual T, Miyawaki T, Latuszek A, Alborch E, Zukin RS, Etgen AM. Acute estradiol protects CA1 neurons from ischemia-induced apoptotic cell death via the PI3K/Akt pathway. Brain Res 2010;1321:1–12.
Liang F, Shi L, Zheng J, Chen S, Wang Y, Zhang J. Neuroprotective effects of CGP3466B on apoptosis are modulated by protein-L-isoaspartate (D-aspartate) O-methyltransferase/Mst1 pathways after traumatic brain injury in rats. Sci Rep 2017;7(1):9201.
Choi S-HH, Lee DY, Kim SU, Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci 2005;25(16):4082–4090.
Shitaka Y, Tran HT, Bennett RE, et al. Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol 2011;70(7):551–567.
Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 2001;23(4):291–299.
Chen Z, Shin D, Chen S, et al. Histological quantitation of brain injury using whole slide imaging: a pilot validation study in mice. PLoS One 2014;9(3):e92133.
Hood KN, Zhao J, Redell JB, et al. Endoplasmic reticulum stress contributes to the loss of newborn hippocampal neurons after traumatic brain injury. J Neurosci 2018;38(9):2372–2384.
Truettner JS, Hu B, Alonso OF, Bramlett HM, Kokame K, Dietrich WD. Subcellular stress response after traumatic brain injury. J Neurotrauma 2007;24(4):599–612.
Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci 2014;34(10):3743–3755.
Chou A, Krukowski K, Jopson T, et al. Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury. Proc Natl Acad Sci U S A 2017;114(31):E6420-E6426.
Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004;11(4):381–389.
Kinnunen KM, Greenwood R, Powell JH, et al. White matter damage and cognitive impairment after traumatic brain injury. Brain J Neurol 2011;134(Pt 2):449–463.
Armstrong RC, Mierzwa AJ, Marion CM, Sullivan GM. White matter involvement after TBI: clues to axon and myelin repair capacity. Exp Neurol 2016;275(Pt 3):328–333.
Han L, Cai W, Mao L, et al. Rosiglitazone promotes white matter integrity and long-term functional recovery after focal cerebral ischemia. Stroke 2015;46(9):2628–2636.
Narita M, Imai S, Kasukawa A, Yajima Y, Suzuki T. Increased level of neuronal phosphoinositide 3-kinase gamma by the activation of mu-opioid receptor in the mouse periaqueductal gray matter: further evidence for the implication in morphine-induced antinociception. Neuroscience 2004;124(3):515–521.
D'Andrea I, Fardella V, Fardella S, et al. Lack of kinase-independent activity of PI3Kγ in locus coeruleus induces ADHD symptoms through increased CREB signaling. EMBO Mol Med 2015;7(7):904–917.
Cunha TM, Roman-Campos D, Lotufo CM, et al. Morphine peripheral analgesia depends on activation of the PI3Kgamma/AKT/nNOS/NO/KATP signaling pathway. Proc Natl Acad Sci U S A 2010;107(9):4442–4447.
Sun D, Gu G, Wang J, et al. Administration of tauroursodeoxycholic acid attenuates early brain injury via Akt pathway activation. Front Cell Neurosci 2017;11:193.
Lee DY, Hong SH, Kim B, Lee DS, Yu K, Lee KS. Neuropeptide Y mitigates ER stress-induced neuronal cell death by activating the PI3K-XBP1 pathway. Eur J Cell Biol 2018;97(5):339–348.
Li H, Zhang X, Qi X, Zhu X, Cheng L. Icariin inhibits endoplasmic reticulum stress-induced neuronal apoptosis after spinal cord injury through modulating the PI3K/AKT signaling pathway. Int J Biol Sci 2019;15(2):277–286.
Zhou MF, Feng ZP, Ou YC, et al. Endoplasmic reticulum stress induces apoptosis of arginine vasopressin neurons in central diabetes insipidus via PI3K/Akt pathway. CNS Neurosci Ther 2019;25(5):562–574.
Choi JH, Park P, Baek GC, et al. Effects of PI3Kγ overexpression in the hippocampus on synaptic plasticity and spatial learning. Mol Brain 2014;7:78.
Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res 2011;2(4):492–516.
Schimmel SJ, Acosta S, Lozano D. Neuroinflammation in traumatic brain injury: a chronic response to an acute injury. Brain Circ 2017;3(3):135–142.
Ruckle T, Schwarz MK, Rommel C. PI3Kgamma inhibition: towards an ‘aspirin of the 21st century’? Nat Rev Drug Discov 2006;5(11):903–918.
Puri KD, Doggett TA, Huang CY, et al. The role of endothelial PI3Kgamma activity in neutrophil trafficking. Blood 2005;106(1):150–157.
Jin R, Xiao AY, Li J, Wang M, Li G. PI3Kγ (phosphoinositide 3-kinase-γ) inhibition attenuates tissue-type plasminogen activator-induced brain hemorrhage and improves microvascular patency after embolic stroke. Hypertension 2019;73(1):206–216.
Sajja VS, Hlavac N, VandeVord PJ. Role of glia in memory deficits following traumatic brain injury: biomarkers of glia dysfunction. Front Integr Neurosci 2016;10:7.
Kempuraj D, Selvakumar GP, Zaheer S, et al. Cross-talk between glia, neurons and mast cells in neuroinflammation associated with Parkinson’s disease. J NeuroImmune Pharmacol 2018;13(1):100–112.
Gorse KM, Lafrenaye AD. The importance of inter-species variation in traumatic brain injury-induced alterations of microglial-axonal interactions. Front Neurol 2018;9:778.
Acknowledgments
This work was supported in part by National Institutes of Health grants NS089991 and NS088719 (Dr. Li) and the Louisiana State University Health Sciences Foundation Fund for Schumpert Endowed Chair (Guohong Li).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Contributions
SL, RJ, WZ, and AYX performed the experiments and data analysis. SL contributed to manuscript writing. GL designed and supervised the study and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Suppl. Fig. 1
Schematic diagram showing the region of interests (ROIs) selected for image acquisition and quantitative analysis. CC: corpus callosum; EC: external capsule (PNG 222 kb)
Suppl. Fig. 2
Representative immunofluorescence images of PI3K p110γ (green) and Iba1 (a cell marker for microglia and macrophages, red) double staining and quantitative analysis of Iba1-positive cells in the ipsilateral perilesional cortex, CA1 and CA3 subfields of the dorsal hippocampus at 3 days after TBI. Scale bar = 50 μm; n = 5 per group, *p < 0.05 vs WT-Sham; #p < 0.05 vs WT-TBI (PNG 1.97 mb)
Suppl. Fig. 3
Representative immunofluorescence images of P-eIF2α (green) and NeuN (red) double staining in the ipsilateral perilesional cortex, hippocampal CA1 and CA3 subfields at 3 days after TBI. Scale bars = 100 μm (PNG 645 kb)
Suppl. Fig. 4
Representative immunofluorescence images showing the co-localization of PI3K p110γ (green) and CHOP (red) in neurons within the ipsilateral perilesional cortex, hippocampal CA1 and CA3 subfields at 3 days after TBI. Scale bars = 50 μm (PNG 1.79 mb)
Suppl. Fig. 5
A, Nissl staining shows brain sections (from bregma −0.2 to −3.8 mm with 0.4 mm interval) from a WT mouse and a KO mouse at 28 days after TBI. Red dotted area indicates tissue loss. B, The changes in body weight were measured before (day 0) and up to 28 days after TBI. n = 12 per group, *p < 0.05 vs WT-TBI (PNG 1.01 mb)
ESM 1
(PDF 58 kb)
ESM 2
(DOCX 41 kb)
Rights and permissions
About this article

Cite this article
Liu, S., Jin, R., Xiao, A.Y. et al. Induction of Neuronal PI3Kγ Contributes to Endoplasmic Reticulum Stress and Long-Term Functional Impairment in a Murine Model of Traumatic Brain Injury. Neurotherapeutics 16, 1320–1334 (2019). https://doi.org/10.1007/s13311-019-00748-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00748-x