
Abstract
Friedreich’s ataxia (FRDA) is a neurodegenerative disorder caused by an unstable GAA repeat expansion within intron 1 of the FXN gene and characterized by peripheral neuropathy. A major feature of FRDA is frataxin deficiency with the loss of large sensory neurons of the dorsal root ganglia (DRG), namely proprioceptive neurons, undergoing dying-back neurodegeneration with progression to posterior columns of the spinal cord and cerebellar ataxia. We used isolated DRGs from a YG8R FRDA mouse model and C57BL/6J control mice for a proteomic study and a primary culture of sensory neurons from DRG to test novel pharmacological strategies. We found a decreased expression of electron transport chain (ETC) proteins, the oxidative phosphorylation (OXPHOS) system and antioxidant enzymes, confirming a clear impairment in mitochondrial function and an oxidative stress-prone phenotype. The proteomic profile also showed a decreased expression in Ca2+ signaling related proteins and G protein-coupled receptors (GPCRs). These receptors modulate intracellular cAMP/cGMP and Ca2+ levels. Treatment of frataxin-deficient sensory neurons with phosphodiesterase (PDE) inhibitors was able to restore improper cytosolic Ca2+ levels and revert the axonal dystrophy found in DRG neurons of YG8R mice. In conclusion, the present study shows the effectiveness of PDE inhibitors against axonal degeneration of sensory neurons in YG8R mice. Our findings indicate that PDE inhibitors may become a future FRDA pharmacological treatment.
Similar content being viewed by others

Introduction
Friedreich’s ataxia (FRDA) (OMIM # 229300, ORPHA:95) is a rare inherited disease. It is classified as a hereditary sensory neuropathy (with autosomal recessive inheritance) involving axonal loss that affects large neuronal fibers [1]. The first pathological changes appear in the dorsal root ganglia (DRG) and the peripheral nerves, with the loss of the proprioceptive neurons, followed by atrophy of the spinal posterior columns and the spinocerebellar and corticospinal tracts of the spinal cord [2]. These changes are accompanied by progressive distal loss of large myelinated fibers in the peripheral nerves responsible for deep sensitivity, which causes ataxia [1]. In addition, patients can develop hypertrophic cardiomyopathy and diabetes; therefore, FRDA has been described as a systemic disorder by some authors [3].
This disease is caused by deficiency of a mitochondrial protein called frataxin (FXN) [4]. Frataxin expression is seriously compromised in patients due to a hyperexpansion of GAA-TTC repeats in intron 1 of the FXN gene that decreases the transcription of the gene [5]. Frataxin is responsible for iron sulfur cluster (ISC) biosynthesis and iron homeostasis [6, 7], participating in cellular energy production [8] and the oxidative stress response [9]. In FRDA, the lack of frataxin is related to defects in mitochondrial respiration [10] with increased oxidative stress [11,12,13], abnormal Ca2+ homeostasis [14], and overload of cellular iron [15]. In FRDA, deficient ISC synthesis is the most accepted early initiating event that alters activities of ISC-dependent enzymes and those of ETC complexes which contain ISC subunits [6]. In this respect, endomyocardial biopsies of two FRDA patients showed decreased activities of aconitase and complexes I, II, and III [16], fibroblast of FRDA patients have been shown to present defects in the activities of complexes I and II [17], and more recently, downregulated expression of NDUFAI subunit of complex I has also been described in the blood of FRDA patients [18]. Besides showing a defective ETC activity, the oxidative phosphorylation is uncoupled and ATP production is decreased in skeletal muscle of FRDA patients [10]. Thus, FRDA is considered an OXPHOS deficient mitochondrial disease [19]. These early defects in ISC biosynthesis and mitochondrial respiration precede other mitochondrial alterations such as oxidative stress, mitochondrial iron accumulation, and iron-mediated oxidative stress as a common underlying mechanism present in several neurodegenerative disorders [20].
Current pharmacological treatments and therapeutic strategies in FRDA can be classified into five categories: palliative and symptomatic treatments, iron chelators, antioxidants, FXN level modifiers, and gene therapy (for review, see [21,22,23,24,25]). Despite the fact that treatments directly target the main pathophysiological key points such as oxidative stress or iron accumulation, FRDA has no treatment that can alter its natural history. For this reason, our interest focused on discovering what other signaling pathways are involved in the pathophysiological mechanisms of neurodegeneration in FRDA, as well as testing novel and effective related treatments, using the YG8R mouse model.
The YG8R mouse is a transgenic animal that contains the entire FRDA locus from a Friedreich’s ataxia patient with GAA expansions in a null mouse Fxn background [26]. These “humanized” mice exhibit progressive neurological symptoms resembling those of FRDA patients, such as degeneration of the large sensory neurons of the DRG [26]. Cellular studies performed in primary culture of DRG from YG8R mice have determined that the frataxin deficiency in sensory neurons involves global mitochondrial dysfunction with depolarized mitochondria, increased reactive oxygen production (ROS) production, and improper Ca2+ handling which together cause axonal dystrophy in the neurodegenerative process [27]. The multiple axonal spheroids, formed mainly due to Ca2+ imbalance, can be reverted by prolonged treatments with Ca2+ chelators or metalloprotease inhibitors [27].
Calcium is strongly connected with two other cellular second messengers, cyclic guanosine monophosphate (cGMP) and cyclic adenosine monophosphate (cAMP). These second messenger pathways have reciprocal regulation, for instance Ca2+ waves (which increase cytosolic Ca2+) cause a cytosolic increase of cAMP and cGMP that decreases cytosolic levels of Ca2+ and restores basal levels [28,29,30]. In neurons, Ca2+ and cAMP transduce extracellular signals through G protein-coupled receptors (GPCRs) to regulate essential neuronal processes such as differentiation [31], axonal growth [32] and guidance [33], excitability and synaptic transmission [34], and gene expression [35]. In fact, pharmacological strategies promoting cyclic nucleotide signaling have been shown to improve axonal health [36,37,38].
Cellular cAMP and cGMP levels are regulated by adenylate cyclase (AC) and guanylate cyclase (GC), in charge of their synthesis, and by phosphodiesterases (PDEs), responsible for their degradation. For their synthesis, AC is able to integrate positive or negative signals directly from GPCRs or indirectly via intracellular signals mediated by protein kinase A (PKA), protein kinase C (PKC), and calcium/calmodulin-dependent protein kinase (CaMK) [39]. Of these, the most important in activating AC and raising cAMP levels is the G protein alpha subunit (Gαs) liberated after GPCR activation.
PDE enzymes belong to a superfamily comprising 11 subtypes based on their subcellular distribution, their regulatory mechanisms, and, especially, their affinity to each of the cyclic nucleotides. Because cAMP and cGMP are involved in a wide variety of neuronal functions, alterations in the levels of these nucleotides can be related to neurodegenerative processes in time and space [30, 40]. In particular, an alteration in calcium levels may lead to improper cAMP or cGMP signaling causing pathogenic effects on cells [29].
In the current investigation, we performed a proteomic study of the DRG of YG8R mice. The proteomic profile showed that frataxin deficiency in this tissue is associated with defects in the proteins related to GPCR signal transduction. Because GPCRs regulate the synthesis of intracellular second messengers such as cAMP and Ca2+, then we should expect reduced cAMP levels in the DRG of YG8R mice with a defective intracellular response mediated by GPCRs. For this reason, we chose a pharmacological strategy based on PDE inhibitors to act on cyclic nucleotide signaling, avoiding the axonal dystrophy seen in the DRG neurons of YG8R mice. We have confirmed that PDE inhibitors, as promoters of GPCR signaling, recover Ca2+ overload and abnormal mitochondrial network morphology and reverse the formation of axonal spheroids in frataxin-deficient sensory neurons. Therefore, we propose PDE inhibitors as a potential therapeutic treatment for Friedreich’s ataxia.
Methods
Animals, Primary Culture, and Cell Lines
The experiments were performed using the YG8R FRDA mouse model purchased from The Jackson Laboratory Repository (Stock no. 008398). The YG8R mouse model has FXN gene targeted alleles and carries two human FXN genes with GAA triplet sequences of 82 and 190 repeats. Previous publications have demonstrated that both C57BL6/J or wild-type littermates are the correct controls for YG8R mice [26, 41]. Thus, the C57BL/6J mouse was used as control in this study. The crossing and genotyping was carried out as described by Mollá et al. [42]. Animals were group-housed under standard housing conditions with a 12-h light–dark cycle and food and water ad libitum. The local Animal Ethics Review Committee of Spanish National Research Council (CSIC) approved all mouse experiments. Primary culture of DRG was performed as previously described [27].
The lymphoblast lines were obtained from the CIBERER Biobank (www.ciberer-biobank.es). Briefly, B lymphocytes from peripheral blood mononuclear cells (PBMC) were transformed by adding EBV supernatant to the PBMC in transformation medium (RPMI 1640 + 20% FBS + 1% L-glutamine + 1% de penicillin–streptomycin + 1 μg/ml cyclosporin). The cells were incubated in 5% CO2 at 37 °C, in vented filter cap tissue culture flasks placed in an upright position. FRDA patient selection and recruitment was carried out with the approval of the Biomedical Research Ethics Committee (CEIB) of Hospital La Fe (Valencia). Informed consent was obtained from all participants. Immortalized lymphoblasts from healthy volunteers were a gift from Dr. García-Gimenez of CIBERER.
Proteomic Study by 2D-DIGE
The proteomic study was performed in DRG tissue comparing FXN-deficient YG8R mice and C57BL/6J control mice at 24 months of age. Changes in the protein expression pattern were evaluated by 2D-DIGE, performed at the Proteomics Unit (Two-Dimensional Electrophoresis) of the Central Research Unit (UCIM), Central Service for Support to Experimental Research (SCSIE) of the University of Valencia. Spots that varied in value by more than 1.3 were digested with trypsin and analyzed by MALDI-TOF (4700 Proteomics Analyzer, ABSciex) in the Proteomic Core Facility at the Príncipe Felipe Research Center (CIPF). The spots that were not well identified were reanalyzed by Liquid Mass, LC-MS/MS, (5600 TripleTOF, ABSciex) in the SCSIE (University of Valencia). ProteinPilot (ABSciex) default parameters were used to generate a peak list directly from MALDI-TOF and LC-MS/MS files. To identify peptide sequences, database searches on Swiss-Prot, NCBInr, and Expasy were used. Only the proteins for which there were individual evidence (unique peptides with enough confidence) have been listed. We based our selection on the Unused ProtScore as a measure of the protein confidence for a detected protein; the peptides (95%) as the number of distinct peptides having at least 95% confidence; the % coverage (95) as the percentage of matching amino acids identified peptides having confidence greater than or equal to 95% divided by the total number of amino acids in the sequence. For the study, we collected the DRG from three experimental biological replicates comprising 24-month-old YG8R (n = 3) mice and C57BL/6J (n = 3) control mice.
Immunodetection of Protein Expression by Western Blot
We studied protein expression by western blot in DRG tissues of 24-month-old YG8R (n = 6) and C57BL6/J control mice (n = 6). DRGs were resuspended in 200 μl of ice cold lysis buffer (50 mM Tris-HCl pH 7.4; 1% (v/v) Triton X-100; 1.5 mM MgCl, 50 mM NaF, 5 mM EDTA, 1 mM sodium orthovanadate, 0.1 mM PMSF, 1 mM DTT, protease, and phosphatase inhibitor cocktails (Sigma-Aldrich, St. Louis, MO)) and were mechanically homogenized simultaneously with TissueLyser II (QIAGEN, Hilden, Alemania) through high-speed shaking in plastic tubes with stainless steel beads (diameter 5 mm). Five cycles of 50 Hz for 30 s were applied with 30 s between each cycle. Then, protein lysates from tissues were centrifuged at 14,000 rpm for 15 min at 4 °C. The supernatant containing whole protein extracts were collected and quantified with Bradford protein assay (Bio-Rad). Electrophoresis, transference, and blocking were performed as [42]. Membranes were incubated in blocking buffer overnight at 4 °C with primary antibodies against CREB (1:1.000, Abcam, Cambridge, England, UK), p-CREB (1:1.000, Cell Signaling), PKA (1:1.000, Abcam), and p-PKA (1:1.000, Abcam). After incubation with the appropriate secondary antibodies, protein bands were detected using a Fujifilm Las-3000 after incubation with the ECL Plus Western Blotting Detection System (GE Healthcare, Chicago, Illinois, USA). Densitometry was measured using the ImageJ software (N.I.H., USA). Densities of phosphorylated protein bands for each sample were normalized to the density of the corresponding total protein bands.
cAMP Measurement by ELISA
We measured cAMP levels in DRG tissues of 24-month-old YG8R (n = 2) and C57BL6/J control mice (n = 3) using an ELISA kit (Cayman Chemical Company, Ann Arbor, MI, USA). DRG tissues were prepared following the manufacturer’s instructions and samples were measured using the Wallac Victor 2TM 1420 Multilabel Counter (Perkin Elmer, Waltham, Massachusetts, USA). Supernatants from the tissue extraction were also quantified with a BCA protein assay (Thermo-Scientific, Waltham, Massachusetts, USA). Each cAMP absorbance measure was normalized to corresponding protein quantification.
Measurement of Cytosolic Ca2+ In Vivo
Calcium measure was performed with Fluo-8 AM (Abcam) in live DRG neurons cultured at 5 days in vitro (DIV). Neurons were incubated with 200 nM of MitoTracker deep red (Molecular Probes, Eugene, Oregón, USA), 5 μM Fluo-8 AM and Pluronic acid 0.06% (Sigma-Aldrich) for 45 min at 37 °C in HHBS buffer (Hank’s buffer with 20 Mm HEPES at pH 7.0). Fluo-8 AM binds to intracellular Ca2+ and fluorescence intensity increases upon Ca2+ binding. Fluorescence of Fluo-8 AM (emission 525 nm) was monitored in live neuronal imaging using a 40× objective on a Leica TCS SP8 laser-scanning confocal microscope. Cultured neurons were identified by morphological criteria, and fluorescence intensity relative to area was measured in neuronal somas with ImageJ. At least 108 neurons were analyzed in three or more independent experiments for each treatment and genotype. For experiments using PDE inhibitors, DRG cultures were treated for 5 DIV with i) 13.5 μM nicardipin (Sigma-Aldrich), ii) 300 nM sildenafil (Sigma-Aldrich), or iii) 0.5 μM rolipram (Sigma-Aldrich). Doses were selected based on previously published data [43,44,45].
Analyses of Frataxin Levels
The lymphoblast was grown in RPMI 1640 (Gibco, Invitrogen, Carlsbad, California, USA) supplemented with 20% fetal bovine serum containing 2 mM L-glutamine and antibiotics and maintained at 37 °C in an atmosphere of 5% CO2 in air. For experiments using PDE inhibitors, lymphoblasts were treated for 24 h with i) 13.5 μM nicardipin (Sigma-Aldrich), ii) 300 nM sildenafil (Sigma-Aldrich), or iii) 0.5 μM rolipram (Sigma-Aldrich). Western blotting was performed as described by Bolinches-Amoros et al. [14]. Membranes were stained with specific antibodies: frataxin (Abcam) and actin was used as a loading control (Sigma-Aldrich).
Mitochondrial Morphology in DRG Neurons
The mitochondrial morphology analysis was performed as previously described [27]. For experiments using PDE inhibitors, DRG cultures were treated for 5 DIV with i) 13.5 μM nicardipin (Sigma-Aldrich), ii) 300 nM sildenafil (Sigma-Aldrich), or iii) 0.5 μM rolipram (Sigma-Aldrich). Doses were selected based on previously published data [43,44,45].
Statistical Analysis
The GraphPad Prism 5.00.288 software was used to generate the graphs and statistical analysis. The mean data were compared using one-way ANOVA followed by Bonferroni post hoc test to determine the significance of values between different experimental groups. Significant P values *P < 0.05, **P < 0.01, and ***P < 0.001 were considered.
Results
Reduction of Frataxin Levels Decreases Protein Expression in DRG
To investigate the molecular pathways involved in sensory neuron degeneration due to frataxin deficiency, we carried out a proteomic study of DRG in the FRDA mouse model. The proteomic expression profile was obtained from DRG samples from 24-month-old YG8R and C57BL/6J mice using two-dimensional fluorescence difference gel electrophoresis (2D-DIGE) technology. The comparative study showed 15 protein spots with significant differential expression (P < 0.05) between YG8R mice and C57BL/6J control mice. These spots were analyzed and 964 differential proteins corresponding to 495 different genes were identified. Strikingly, all identified proteins were downregulated in YG8R mice compared to C57BL/6J control mice, suggesting a protein expression defect in the DRG of FRDA (Fig. 1 A, B). The Protein Analysis Through Evolutionary Relationships (PANTHER) software based on Gene Ontology (GO) database was used to search for biological and functional features of targeted proteins. Classification by molecular function showed that the majority of altered proteins belonged to the catalytic activity and binding proteins categories (Fig. 1 C). To identify the signaling pathways in which the defective proteins were involved, we performed in silico analyses with Paintomics online tools, based on KEGG (Kyoto Encyclopedia of Genes and Genomes). Paintomics analysis revealed the implication of the 495 decreased genes in 199 KEGG pathways involved in signal transduction (PI3K-Akt, calcium, cGMP-PKG, and cAMP signaling pathways), energy metabolism (oxidative phosphorylation), cardiovascular diseases, and neurodegenerative diseases, among others (Table 1 ). Overall, the DRG of YG8R mice showed a decrease in protein expression in pathways related to cellular mechanisms, neuronal processes, and metabolic pathways, some of which, namely OXPHOS, antioxidant systems, and Ca2+ signaling, have been previously described in the pathology of the YG8R mouse model [26, 27, 33, 46].

Protein profile differential expression in DRG from frataxin-deficient mouse YG8R versus C57BL/6J control. (A) Representative 2D-DIGE blot of DRG protein extraction. The spots showing significant differences in protein levels between cases and controls are labeled. (B) 15 spots were identified as different between YG8R versus C57BL/6J, with ratio varying between − 1.33 and − 4.19. (C) Representation of molecular function of differential proteins expressed in YG8R mice versus C57BL/6J control by Gene Ontology (GO) database with PANTHER classification system. 44.30% proteins have catalytic activity (GO:0003824), 35.20% are binding proteins (GO:0005488), 22.80% proteins have structural molecule activity (GO:0005198), 8.20% proteins have enzyme regulator activity (GO:0030234), 5.70% have a receptor activity (GO:0004872), 2.70% are nucleic acid binding transcription factor activity (GO:0001071), 2.50% have translation regulator activity (GO:0045182), 0.70% have protein binding transcription factor activity (GO:0000988), and 0.20% have antioxidant activity (GO:0016209)
Frataxin Deficiency Causes Defects in OXPHOS and Antioxidant Enzymes
The proteomic study of the DRG of YG8R mice revealed a decrease in expression of proteins related to the OXPHOS system compared with the C57BL/6J control. The defect in the OXPHOS system was extensive and involved seven subunits distributed between complexes I, II, and III of the electron transport chain (ETC), two alternative ways/means of electron entry into the ETC and two subunits of complex V (Table 2 ). In complex I, the affected proteins were NDUFAF7, NDUFS3, NDUFA10, and NDUFS1 (subunits relevant to complex I function). NDUFAF7 participates in the assembly and stability of complex I, and the catalytic core subunits NDUFS3, NDUFA10, and NDUFS1 are directly involved in electron flow. In complex II, the decreased protein was the catalytic subunit SDHA that converts succinate into fumarate by transferring electrons to CoQ. We previously reported that SDHA interacts physically with frataxin [19]. In complex III, we found a reduction in core protein I and Cyt c1 subunits. Core protein I acts as a link to complex formation between Cyt c and Cyt c1, which is part of the heme group that is directly involved in electron flow. The YG8R mouse also had decreased levels of ETFα and GPD2, which transfer electrons to the ETC from the mitochondrial β-oxidation of fatty acids within mitochondria and the Krebs cycle, respectively. ETFα, involved in the initial step of the β-oxidation of fatty acids in the mitochondria, is also able to interact physically with frataxin [19]. Finally, in complex V, we found a reduction in the α and β subunits of ATP synthase, which phosphorylate ADP to generate ATP, suggesting a possible defect in ATP production.
In addition, the YG8R mouse had decreased levels of two antioxidant proteins, thioredoxin and thioredoxin domain-containing protein 5. The thioredoxin deficit, as well as several other antioxidant systems, has already been described in the DRG of YG8R mice by Shan and collaborators [47] and relates FXN deficiency with the oxidative stress suffered by sensory neurons. All these results suggest mitochondrial respiratory impairment in the DRG of YG8R mice that correlates with the mitochondrial depolarization and oxidative stress previously reported in primary culture of sensory neurons and neuronal tissues from YG8R mice [27, 42, 46].
Deficit of Frataxin Causes Defects in GPCR Signal Transduction
Comparison of proteomic profiles confirmed a relevant defect in GPCR signaling proteins in the DRG of YG8R mice with respect to the C57BL/6J control mice. FXN-deficient DRG showed a decrease in 11 proteins belonging to the G protein family and several effector molecules of the signal transduction pathways related to GPCRs: IP3/Ca2+ and cAMP pathways (Table 2 ).
Regarding the IP3/Ca2+ signaling pathway that modulates the intracellular level of Ca2+, we have found that YG8R mice have decreased levels in i) four G protein subunits, two subunits of Gαq type (GNA11, GNA14), and two subunits of Gβγ type (GNB1 and GNB2); ii) the effector molecule PLC3β; and iii) two subunits of PKC (PKCα and PKCβ). The defect in G proteins and PLC3β could explain the altered store operated calcium entry (SOCE) mechanism described in sensory neurons of YG8R mice [27].
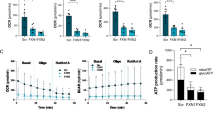
In relation to the cAMP signaling pathway, YG8R mice have decreased expression of three regulatory subunits of PKA (PKAR1A, PKAR1B, and PKAR2B) and the transcription factor CREB1. Both PKA and CREB are directly activated by cAMP. Therefore, to determine whether these protein defects could affect cAMP cellular signaling, we measured cAMP levels by ELISA and p-PKA/PKA and p-CREB/CREB ratios by western blot in DRG tissue from YG8R mice. We found a reduction in cAMP levels (Fig. 2 A), although p-PKA/PKA and p-CREB/CREB ratios were no different (Fig. 2 B–D) in YG8R compared to C57BL6/J control mice. Despite reduced cAMP levels in YG8R mice, the cellular signaling through this pathway does not seem to be affected.

cAMP measurements and PKA and CREB phosphorylation. (A) DRG tissues of YG8R mice and C57BL6/J were analyzed with cAMP enzyme immunoassay kit (Cayman Chemical Company). There was a significant variation in YG8R mice versus C57BL6/J. (B) Western blot analysis shows that the phosphorylation of PKA and CREB proteins were similar in YG8R and C57BL6J mice. Western blot results were quantified for each lane using Fujifilm’s Multi-Gauge Software. The ratio between phosphorylated and total forms was calculated and represented in (C) p-CREB/CREB and (D) p-PKA/PKA
Deficit of Frataxin Causes Defects in Ca2+ Binding Proteins
DRG of YG8R mice showed lower levels of Ca2+ binding proteins such as calmodulin, calcineurin (PP2B), and calpain compared with the C5BL/6J control mice, suggesting inefficient Ca2+-sensitive signaling (Table 2 ). The reduction of calpain activity previously reported in sensory neurons in YG8R mice [27] could be due to the decrease in calpain protein levels observed in this work.
PDE Inhibitors Rescue Degeneration in Frataxin-Deficient Sensory Neurons
The extensive defect in the GPCR signaling pathway found in the DRG of FXN-deficient mice suggests that GPCR signaling might be involved in the pathophysiology of FRDA. Therefore, a pharmacological action on this pathway could prevent neuronal degeneration. To confirm this hypothesis, we proposed a pharmacological strategy based on PDE inhibitors that inhibit cAMP/cGMP degradation and increases their levels. Evidence that intervention in cyclic nucleotide signaling improves axonal health has already been published [36,37,38].
Three different PDE inhibitors were selected as a result of their ability to increase cGMP and cAMP levels. Sildenafil is a specific inhibitor of PDE5 that increases the cytosolic levels of cGMP [37], rolipram (a PDE4 inhibitor) increases cAMP levels [48], and nicardipine (a PDE1 inhibitor) is able to augment both cAMP and cGMP levels [49]. In addition, nicardipine can act as a L-type Ca2+ channel blocker, which decreases cytosolic Ca2+ levels [49]. These drugs have been used in primary culture of sensory neurons obtained from YG8R mice, which illustrate the multifocal axonal neurodegenerative model of frataxin deficiency [27].
The intracellular Ca2+ levels were measured in vivo with Fluo-8 AM, and mitochondrial distribution was analyzed. Under basal conditions, we observed increased Ca2+ levels in YG8R mice neurons (1.00 ± 0.0) compared with C57BL/6J control mice (0.6733 ± 0.3246). After treatment with PDE inhibitors, the Ca2+ levels decreased in all cases, but only when using sildenafil a complete restoration of the control levels was achieved (Fig. 3 A). The least effective means of decreasing cytosolic Ca2+ level were both the PDE1 inhibition and the L-type Ca2+-channel blockade by nicardipine treatment. This result confirms that PDE1 inhibition is not as effective as PDE4 or PDE5 inhibition in increasing cAMP and cGMP levels and suggests that the L-type Ca2+ channels do not participate in the increase of cytosolic Ca2+ in frataxin-deficient sensitive neurons. In these frataxin-deficient neurons, oxidative stress and Ca2+ dyshomeostasis act as initiating factors of axonal focal lesion [27], with a mitochondrial pathology as an ultrastructural sign of early damage. After treatments, confocal images showed a physiological mitochondrial distribution along YG8R mice neurons (Fig. 3 B).

In vivo measurement of cytosolic Ca2+ in sensory neurons of YG8R mouse model. (A) Quantification of Fluo-8 AM fluorescence corresponding with intracellular Ca2+ levels by confocal microscopy. Final values were expressed as a ratio of the YG8R basal and the graph represents the mean ± S.E.M. of three experimental repeats (N = 3) with a total of 92, 135, 130, 131, and 108 measured neurons corresponding with C57BL/6J basal, YG8R basal, YG8R treated with nicardipine, YG8R treated with sildenafil, and YG8R treated with rolipram. One-way ANOVA (genotype); the results did not show statistically significant differences. (B) Microscopy images of Fluo-8 AM (green) and MitoTracker fluorescence (red) in primary culture of DRG of FRDA mouse model. Arrowheads show neuronal bodies and arrows show axonal spheroids with calcium and mitochondria retained. 40×, confocal microscopy. Scale 50 μm
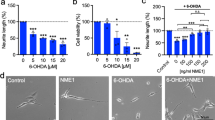
Previous studies have demonstrated how resveratrol, a PDE inhibitor, increases frataxin levels [50]. Thus, it was interesting to confirm that sildenafil, nicardipine, and rolipram had the same effect on sensory neurons. Using lymphoblasts from FRDA patients and healthy control, we showed that frataxin expression does not increase after treatment with sildenafil, rolipram, and nicardipine (Fig. 1S), suggesting that Ca2+ level modulation with PDE inhibitors may be critical to improve neuronal axonopathy observed in FXN-deficient cells.
Next, we analyzed the mitochondrial network of neurons using MitoTracker and β-tubulin III antibody. We observed important alterations in the mitochondrial morphology in YG8R mice neurons compared with C57BL/6J control mice under basal conditions. In control neurons, mitochondria were distributed homogenously in the proximal and distal axonal segments. In contrast, in frataxin-deficient neurons, mitochondria were retained in axonal spheroids forming bead chains as a clear marker of neurodegeneration (Fig. 4 A). In the proximal segments of YG8R mice neurons, mitochondria increased in number and in percentage of occupied area (Fig. 4 B, C) and were less elongated and more interconnected than in control neurons (Fig. 4 D, E). Moreover, YG8R mice mitochondria were swollen, reaching values that duplicated their sizes compared with control neurons (Fig. 4 F, G). Successfully, YG8R mice neurons treated with PDE inhibitors rescued this phenotype showing similar mitochondrial characteristics compared to controls (Fig. 4 C–G), except for the number of mitochondria. Treatment with sildenafil or rolipram did not have any effect on the number of mitochondria, whereas it increased using nicardipine (Fig. 4 B). Following PDE inhibitor treatments, YG8R mice neurons also showed higher elongation and lower interconnectivity of mitochondrial networks compared to basal conditions (Fig. 4 D, E) and decreased swelling in mitochondria (Fig. 4 F, G) leading to values similar to control neurons.

Treatment with PDE inhibitors recovers mitochondrial morphology in frataxin-deficient neurons. (A) Pattern of neuritic and mitochondrial network by immunodetection of β-tubulin III (green) and MitoTracker fluorescence (red) in primary culture of DRG from YG8R mouse. Arrowheads show neuronal bodies and arrows show axonal spheroids with mitochondria retained in YG8R mice sensory neurons that are absent in YG8R mice treated with PDE inhibitors. 40×, confocal microscopy. Scale 50 μm. (B–F) Quantification of mitochondrial network descriptors in proximal axon: number of mitochondria per 100 μm of neurite (B), percentage of axonal area occupied by mitochondria (C), mitochondrial elongation index (D), mitochondrial interconnectivity (E), and mitochondrial swelling (F) are expressed as mean ± S.E.M. of three experimental repeats (N = 3) with a total of 185, 188, 25, 213, and 193 measured neurons corresponding with C57BL/6J basal, YG8R basal, YG8R nicardipine, YG8R sildenafil, and YG8R rolipram. One-way ANOVA followed by Bonferroni post hoc test to determine the significance of values between different experimental groups. Significant P values: *P < 0.05, **P < 0.01, and ***P < 0.001 were considered. (G) Mitochondrial swelling expressed as cumulative distribution was analyzed using the Kolmogorov–Smirnov test
All these results confirm the effectiveness of PDE inhibitors against axonal degeneration in frataxin-deficient neurons in culture.
Discussion
The DRG is the primary site of neurodegeneration in FRDA; hence, it makes the ideal target tissue to investigate the pathophysiological mechanism of this disease. In this study, we found a general protein deficit in the DRG of YG8R mice. Protein depletion was observed in different pathways such as ETC, OXPHOS, and antioxidant systems, confirming their alteration in FRDA as previously reported in several studies (Table 3 ). However, in this work, we identified a newly affected biochemical pathway that so far has not been described in FRDA: the GPCR signaling pathway. The DRG of YG8R mice also showed decreased levels of four G proteins and four effectors of transduction cascades, namely PLCβ, PKC, PKA, and CREB (suggesting an impairment in the GPCR signaling pathway). The altered expression of these proteins could induce decreased levels of cAMP, as indeed we have confirmed by measuring the cAMP levels. Nevertheless, lower levels of cAMP do not seem to affect the activation of PKA and CREB, because the p-PKA/PKA and p-CREB/CREB ratios were not altered in the YG8R mice compared to C57BL/6J control mice. This fact might be explained by the possible involvement of alternative mechanisms and targets for the cellular action of cAMP. For instance, a family of novel cAMP effector proteins called EPACs (exchange proteins directly activated by cAMP) [55] has recently been related with axon specification and axonal elongation function [56]. Therefore, additional investigations through cAMP signaling pathway should be made to gain further insights into the secondary effectors involved in the cAMP defect in FRDA. Overall, defects in GPCR signaling impede gene expression and Ca2+-mediated signaling (which modulate different cellular processes, e.g., neuronal survival or synaptic activity and plasticity) [57]. Furthermore, defects in GPCR signaling generate a lower neuroprotective response to oxidative stress [58], less neurite outgrowth [59], and less synaptic plasticity of neurons [57].
In addition to GPCR signaling impairment, the DRG of YG8R mice showed reduced levels of the Ca2+-binding proteins calmodulin, calcineurin, and calpain (suggesting alterations in Ca2+-mediated signaling in FRDA). We previously reported an increase in intracellular Ca2+ levels, defective SOCE mechanism and less calpain activity in sensory neurons of YG8R mice [27]. These alterations in Ca2+ homeostasis may be the result of the GPCR signaling defects herein described. Specifically, the reduced amount of Ca2+-binding proteins reported in this work is probably the cause of lower calpain activity that has previously been reported in YG8R mice [27].
The cell surface GPCRs produce the large majority of the ubiquitous second messenger cAMP and, together with the PDE enzymes that degrade the cAMP and cGMP, maintain the appropriate amounts of both cyclic nucleotides. Promotion of cAMP and cGMP levels using PDE inhibitors is commonly used in clinical practice for treating the pathophysiological dysregulation of cyclic nucleotide signaling in several disorders including erectile dysfunction, pulmonary hypertension, and cardiac failure. Moreover, their potential therapeutic applications in neurodegenerative diseases have been described and PDE inhibitors are currently under clinical study in Alzheimer’s and Huntington’s disease [60] and also in FRDA [50]. Rolipram, a selective PDE4 inhibitor, promotes in vivo axonal regeneration of the central nervous system after spinal cord injury through CREB-dependent gene expression [44, 61] and recovers cognitive and synaptic function in Alzheimer’s disease mice models [62]. Sildenafil, which acts by inhibiting cGMP-specific PDE5, improves peripheral neuropathy in diabetic mice by stimulation of cGMP-dependent protein kinase (PKG) [63] and enhances neurogenesis and functional recovery after a stroke [37]. These PDE inhibitors provide mitochondrial bioenergetics promotion, antioxidant effects, and neuroprotective and neuroregenerative actions. Because decreased mitochondrial biogenesis has been demonstrated in mononuclear cells from peripheral blood of FRDA patients, in FRDA cells and mouse models [64, 65], as well as decreased mitochondrial potential membrane and increased ROS production in cerebellar neurons from YG8R mice [46] among other neuronal models [14, 27], it would seem that PDE inhibitors may display potential therapeutic benefits in FRDA. Resveratrol, a nonselective PDE inhibitor, increases FXN expression in cellular and mouse models of FRDA and has clinical benefits in FRDA patients by improving oxidative stress and clinical outcomes [50]. It has been suggested that the beneficial effects of resveratrol on FRDA are obtained through the activation of SIRT1 and PGC1α, which control genes involved in mitochondrial biogenesis and antioxidant defenses [50]. Another example of PDE inhibitor with therapeutic benefits in FRDA is sulmazole which has recently been demonstrated to be efficient in reducing cardiac dilatation in a Drosophila model of FRDA [66]. Lastly, forskolin treatment through the increase in the intracellular concentration of cAMP normalizes mitochondrial oxidative status and prevents apoptosis in frataxin-silenced β-cells and primary islets and neurons [67]. Therefore, increasing cAMP through different pathways seems to improve the pathological phenotype in FRDA, although much still remains unknown about the beneficial mechanisms of cAMP in the pathophysiology of FRDA.
Taking all this evidence into account, we propose the use of PDE inhibitors to treat degeneration of sensory neurons in FRDA. In this work, we tested the effectiveness of three PDE inhibitors (sildenafil, rolipram, and nicardipine) in counteracting axonal degeneration of sensory neurons of YG8R mice. FXN deficiency in these neurons causes alterations in mitochondrial networks related to intracellular Ca2+ overload [27]. Mitochondria appear spherical, swollen, and interconnected and are retained in the proximal region of neurites, forming axonal spheroids and promoting axonal degeneration [27].
The promotion of GPCR signaling, especially of those pathways involving Ca2+-cAMP-cGMP as second messengers with an important role in the regulation of neuronal functions, might be beneficial in frataxin deficiency. Therefore, we propose the modulation of the cAMP/cGMP levels as a promising intervention to ameliorate the pathophysiology of FRDA, providing novel molecular targets for therapeutic intervention preventing axonal degeneration.
We found that all three PDE inhibitors decreased intracellular Ca2+ levels and improved mitochondrial network morphology reaching reversion of axonal spheroid formation. Treatment with nicardipine was less effective in reducing Ca2+ levels, indicating that L-type Ca2+ channels are not involved in Ca2+ overload in FRDA. However, sildenafil and rolipram treatments were equally effective at reducing Ca2+ overload and recovering mitochondrial morphology. Previous studies have demonstrated that the effect of sildenafil on the calcium signaling pathway is cGMP mediated. These include the inhibition of IP3 formation by phospholipase C [68] and the activation of the sarco/endoplasmic reticulum calcium ATPases (SERCA) [69]. In any case, the consequences are the [Ca2+]i decrease. In the case of rolipram, the effect is mediated via PKA or EPAC activation that in turn mediates different cellular effects. The PKA activation decreases the intracellular Ca2+ levels [70], and EPAC regulates matrix Ca2+ entry via the mitochondrial calcium uniporter, preventing mitochondrial permeability transition (MPT) [71].
The discovery of a local cAMP/PKA signaling cascade in the mitochondrial matrix that promotes respiratory chain activity and ATP production [72, 73], and the demonstration of cross-talk between cAMP and Ca2+ signaling inside mitochondria [74] open up new questions on the molecular mechanism by which PDE inhibitors lead to mitochondrial recovery and mitochondrial Ca2+ signaling promoting neuronal survival in FRDA. Our results demonstrate axonal dystrophy reversion by PDE inhibitors through decreasing intracellular Ca2+ levels, and it is probable that the cause is by increasing mitochondrial Ca2+ uptake (Fig. 5 ). These data support the use of PDE inhibitors as promising pharmacological treatments to suppress mitochondrial dysfunction and dying-back neurodegeneration in FRDA.

Presumptive mechanism by which PDE inhibitors recover axonal dystrophy in FRDA neurons. According to previous reports (see references), frataxin-deficient neurons show (1.) a decrease in mitochondrial Fe-S proteins; (2.) a reduced mitochondrial membrane potential; (3.) a failure in mitochondrial biogenesis; (4.) a defect in Ca2+ buffering by mitochondria; (5.) high cytosolic Ca2+ levels; and (6.) axonal dystrophy. Treatment of neurons with PDE inhibitors recovers cytosolic Ca2+ to normal levels and repair axonal morphology (shown in this work). A possible recovery of Ca2+ influx activity due to restoration of mitochondrial function and mitochondrial biogenesis after PDE inhibitor treatments could explain our findings. A summary of previous reports is listed in black, data obtained in this work are shown in blue, and possible mechanisms explaining the action of PDE inhibitors are listed in red
References
Morral JA, Davis AN, Qian J, Gelman BB, Koeppen AH. Pathology and pathogenesis of sensory neuropathy in Friedreich’s ataxia. Acta neuropathologica. 2010;120(1):97–108. https://doi.org/10.1007/s00401-010-0675-0.
Koeppen AH, Mazurkiewicz JE. Friedreich ataxia: neuropathology revised. J Neuropathol Exp Neurol. 2013;72(2):78–90. https://doi.org/10.1097/NEN.0b013e31827e5762.
Koeppen AH. Friedreich’s ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci. 2011;303(1–2):1–12. https://doi.org/10.1016/j.jns.2011.01.010.
Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science (New York NY). 1996;271(5254):1423–7.
Cossee M, Campuzano V, Koutnikova H, Fischbeck K, Mandel JL, Koenig M et al. Frataxin fracas. Nat Genet. 1997;15(4):337–8.
Vaubel RA, Isaya G. Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Molecular and Cellular Neurosciences. 2012. https://doi.org/10.1016/j.mcn.2012.08.003.
Chiang S, Kovacevic Z, Sahni S, Lane DJ, Merlot AM, Kalinowski DS et al. Frataxin and the molecular mechanism of mitochondrial iron-loading in Friedreich’s ataxia. Clinical Science. 2016;130(11):853–70. https://doi.org/10.1042/CS20160072.
Ristow M, Pfister MF, Yee AJ, Schubert M, Michael L, Zhang CY et al. Frataxin activates mitochondrial energy conversion and oxidative phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(22):12239–43.
Calabrese V, Lodi R, Tonon C, D'Agata V, Sapienza M, Scapagnini G et al. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J Neurol Sci. 2005;233(1–2):145–62. https://doi.org/10.1016/j.jns.2005.03.012.
Lodi R, Cooper JM, Bradley JL, Manners D, Styles P, Taylor DJ et al. Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(20):11492–5.
Emond M, Lepage G, Vanasse M, Pandolfo M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology. 2000;55(11):1752–3.
Schulz JB, Dehmer T, Schols L, Mende H, Hardt C, Vorgerd M et al. Oxidative stress in patients with Friedreich ataxia. Neurology. 2000;55(11):1719–21.
Bradley JL, Homayoun S, Hart PE, Schapira AH, Cooper JM. Role of oxidative damage in Friedreich’s ataxia. Neurochem Res. 2004;29(3):561–7.
Bolinches-Amoros A, Molla B, Pla-Martin D, Palau F, Gonzalez-Cabo P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Frontiers in cellular neuroscience. 2014;8:124. https://doi.org/10.3389/fncel.2014.00124.
Lamarche JB, Cote M, Lemieux B. The cardiomyopathy of Friedreich’s ataxia morphological observations in 3 cases. Can J Neurol Sci. 1980;7(4):389–96.
Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D et al. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17(2):215–7.
Lobmayr L, Brooks DG, Wilson RB. Increased IRP1 activity in Friedreich ataxia. Gene. 2005;354:157–61. https://doi.org/10.1016/j.gene.2005.04.040.
Salehi MH, Kamalidehghan B, Houshmand M, Yong Meng G, Sadeghizadeh M, Aryani O et al. Gene expression profiling of mitochondrial oxidative phosphorylation (OXPHOS) complex I in Friedreich ataxia (FRDA) patients. PLoS ONE. 2014;9(4):e94069. https://doi.org/10.1371/journal.pone.0094069.
Gonzalez-Cabo P, Vazquez-Manrique RP, Garcia-Gimeno MA, Sanz P, Palau F. Frataxin interacts functionally with mitochondrial electron transport chain proteins. Hum Mol Genet. 2005;14(15):2091–8.
Isaya G. Mitochondrial iron-sulfur cluster dysfunction in neurodegenerative disease. Frontiers in Pharmacology. 2014;5:29. https://doi.org/10.3389/fphar.2014.00029.
Gonzalez-Cabo P, Llorens JV, Palau F, Molto MD. Friedreich ataxia: an update on animal models, frataxin function and therapies. Adv Exp Med Biol. 2009;652:247–61. https://doi.org/10.1007/978-90-481-2813-6_17.
Puccio H, Anheim M, Tranchant C. Pathophysiogical and therapeutic progress in Friedreich ataxia. Revue neurologique. 2014;170(5):355–65. https://doi.org/10.1016/j.neurol.2014.03.008.
Aranca TV, Jones TM, Shaw JD, Staffetti JS, Ashizawa T, Kuo SH et al. Emerging therapies in Friedreich’s ataxia. Neurodegenerative Disease Management. 2016;6(1):49–65. https://doi.org/10.2217/nmt.15.73.
Burk K. Friedreich ataxia: current status and future prospects. Cerebellum & Ataxias. 2017;4:4. https://doi.org/10.1186/s40673-017-0062-x.
Strawser C, Schadt K, Hauser L, McCormick A, Wells M, Larkindale J et al. Pharmacological therapeutics in Friedreich ataxia: the present state. Expert Review of Neurotherapeutics. 2017;17(9):895–907. https://doi.org/10.1080/14737175.2017.1356721.
Al-Mahdawi S, Pinto RM, Varshney D, Lawrence L, Lowrie MB, Hughes S et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88(5):580–90.
Molla B, Munoz-Lasso DC, Riveiro F, Bolinches-Amoros A, Pallardo FV, Fernandez-Vilata A et al. Reversible axonal dystrophy by calcium modulation in frataxin-deficient sensory neurons of YG8R mice. Frontiers in Molecular Neuroscience. 2017;10:264. https://doi.org/10.3389/fnmol.2017.00264.
Hofer AM. Interactions between calcium and cAMP signaling. Curr Med Chem. 2012;19(34):5768–73.
Di Benedetto G, Scalzotto E, Mongillo M, Pozzan T. Mitochondrial Ca(2)(+) uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab. 2013;17(6):965–75. https://doi.org/10.1016/j.cmet.2013.05.003.
Averaimo S, Nicol X. Intermingled cAMP, cGMP and calcium spatiotemporal dynamics in developing neuronal circuits. Frontiers in Cellular Neuroscience. 2014;8:376. https://doi.org/10.3389/fncel.2014.00376.
Gomez-Villafuertes R, del Puerto A, Diaz-Hernandez M, Bustillo D, Diaz-Hernandez JI, Huerta PG et al. Ca2+/calmodulin-dependent kinase II signalling cascade mediates P2X7 receptor-dependent inhibition of neuritogenesis in neuroblastoma cells. The FEBS Journal. 2009;276(18):5307–25. https://doi.org/10.1111/j.1742-4658.2009.07228.x.
del Puerto A, Diaz-Hernandez JI, Tapia M, Gomez-Villafuertes R, Benitez MJ, Zhang J et al. Adenylate cyclase 5 coordinates the action of ADP, P2Y1, P2Y13 and ATP-gated P2X7 receptors on axonal elongation. J Cell Sci. 2012;125(Pt 1):176–88. https://doi.org/10.1242/jcs.091736.
Nicol X, Hong KP, Spitzer NC. Spatial and temporal second messenger codes for growth cone turning. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(33):13776–81. https://doi.org/10.1073/pnas.1100247108.
Huang Y, Thathiah A. Regulation of neuronal communication by G protein-coupled receptors. FEBS letters. 2015;589(14):1607–19. https://doi.org/10.1016/j.febslet.2015.05.007.
Fukuchi M, Tabuchi A, Kuwana Y, Watanabe S, Inoue M, Takasaki I et al. Neuromodulatory effect of Galphas- or Galphaq-coupled G-protein-coupled receptor on NMDA receptor selectively activates the NMDA receptor/Ca2+/calcineurin/cAMP response element-binding protein-regulated transcriptional coactivator 1 pathway to effectively induce brain-derived neurotrophic factor expression in neurons. The Journal of Neuroscience: the official journal of the Society for Neuroscience. 2015;35(14):5606–24. https://doi.org/10.1523/JNEUROSCI.3650-14.2015.
Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci. 2001;21(13):4731–9.
Zhang R, Wang Y, Zhang L, Zhang Z, Tsang W, Lu M et al. Sildenafil (Viagra) induces neurogenesis and promotes functional recovery after stroke in rats. Stroke. 2002;33(11):2675–80.
Zhang L, Zhang RL, Wang Y, Zhang C, Zhang ZG, Meng H et al. Functional recovery in aged and young rats after embolic stroke: treatment with a phosphodiesterase type 5 inhibitor. Stroke. 2005;36(4):847–52. https://doi.org/10.1161/01.STR.0000158923.19956.73.
Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annual Review of Pharmacology and Toxicology. 2001;41:145–74. https://doi.org/10.1146/annurev.pharmtox.41.1.145.
Cui Q, So KF. Involvement of cAMP in neuronal survival and axonal regeneration. Anatomical Science International. 2004;79(4):209–12. https://doi.org/10.1111/j.1447-073x.2004.00089.x.
Anjomani Virmouni S, Ezzatizadeh V, Sandi C, Sandi M, Al-Mahdawi S, Chutake Y et al. A novel GAA-repeat-expansion-based mouse model of Friedreich’s ataxia. Dis Model Mech. 2015;8(3):225–35. https://doi.org/10.1242/dmm.018952.
Molla B, Riveiro F, Bolinches-Amoros A, Munoz-Lasso DC, Palau F, Gonzalez-Cabo P. Two different pathogenic mechanisms, dying-back axonal neuropathy and pancreatic senescence, are present in the YG8R mouse model of Friedreich’s ataxia. Dis Model Mech. 2016;9(6):647–57. https://doi.org/10.1242/dmm.024273.
Soeda H, Tatsumi H, Katayama Y. Neurotransmitter release from growth cones of rat dorsal root ganglion neurons in culture. Neuroscience. 1997;77(4):1187–99.
Nikulina E, Tidwell JL, Dai HN, Bregman BS, Filbin MT. The phosphodiesterase inhibitor rolipram delivered after a spinal cord lesion promotes axonal regeneration and functional recovery. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(23):8786–90. https://doi.org/10.1073/pnas.0402595101.
Jia L, Wang L, Chopp M, Zhang Y, Szalad A, Zhang ZG. MicroRNA 146a locally mediates distal axonal growth of dorsal root ganglia neurons under high glucose and sildenafil conditions. Neuroscience. 2016;329:43–53. https://doi.org/10.1016/j.neuroscience.2016.05.005.
Abeti R, Parkinson MH, Hargreaves IP, Angelova PR, Sandi C, Pook MA et al. 'Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia'. Cell Death & Disease. 2016;7:e2237. https://doi.org/10.1038/cddis.2016.111.
Shan Y, Schoenfeld RA, Hayashi G, Napoli E, Akiyama T, Iodi Carstens M et al. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxidants & Redox Signaling. 2013;19(13):1481–93. https://doi.org/10.1089/ars.2012.4537.
Boswell-Smith V, Spina D, Page CP. Phosphodiesterase inhibitors. British Journal of Pharmacology. 2006;147 Suppl 1:S252–7. https://doi.org/10.1038/sj.bjp.0706495.
Sharma RK, Wang JH, Wu Z. Mechanisms of inhibition of calmodulin-stimulated cyclic nucleotide phosphodiesterase by dihydropyridine calcium antagonists. J Neurochem. 1997;69(2):845–50.
Yiu EM, Tai G, Peverill RE, Lee KJ, Croft KD, Mori TA et al. An open-label trial in Friedreich ataxia suggests clinical benefit with high-dose resveratrol, without effect on frataxin levels. J Neurol. 2015;262(5):1344–53. https://doi.org/10.1007/s00415-015-7719-2.
Selak MA, Lyver E, Micklow E, Deutsch EC, Onder O, Selamoglu N et al. Blood cells from Friedreich ataxia patients harbor frataxin deficiency without a loss of mitochondrial function. Mitochondrion. 2011;11(2):342–50. https://doi.org/10.1016/j.mito.2010.12.003.
Sutak R, Xu X, Whitnall M, Kashem MA, Vyoral D, Richardson DR. Proteomic analysis of hearts from frataxin knockout mice: marked rearrangement of energy metabolism, a response to cellular stress and altered expression of proteins involved in cell structure, motility and metabolism. Proteomics. 2008;8(8):1731–41.
Telot L, Rousseau E, Lesuisse E, Garcia C, Morlet B, Leger T et al. Quantitative proteomics in Friedreich’s ataxia B-lymphocytes: a valuable approach to decipher the biochemical events responsible for pathogenesis. Biochimica et biophysica acta Molecular Basis of Disease. 2018;1864(4 Pt A):997–1009. https://doi.org/10.1016/j.bbadis.2018.01.010.
Swarup V, Srivastava AK, Padma MV, Rajeswari MR. Quantitative profiling and identification of differentially expressed plasma proteins in Friedreich’s ataxia. J Neurosci Res. 2013;91(11):1483–91. https://doi.org/10.1002/jnr.23262.
de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396(6710):474–7. https://doi.org/10.1038/24884.
Munoz-Llancao P, Henriquez DR, Wilson C, Bodaleo F, Boddeke EW, Lezoualc'h F et al. Exchange protein directly activated by cAMP (EPAC) regulates neuronal polarization through Rap1B. The Journal of Neuroscience: the official journal of the Society for Neuroscience. 2015;35(32):11315–29. https://doi.org/10.1523/JNEUROSCI.3645-14.2015.
Martin B, Lopez de Maturana R, Brenneman R, Walent T, Mattson MP, Maudsley S. Class II G protein-coupled receptors and their ligands in neuronal function and protection. Neuromolecular Med. 2005;7(1–2):3–36.
Espada S, Ortega F, Molina-Jijon E, Rojo AI, Perez-Sen R, Pedraza-Chaverri J et al. The purinergic P2Y(13) receptor activates the Nrf2/HO-1 axis and protects against oxidative stress-induced neuronal death. Free Radical Biology & Medicine. 2010;49(3):416–26. https://doi.org/10.1016/j.freeradbiomed.2010.04.031.
Bromberg KD, Iyengar R, He JC. Regulation of neurite outgrowth by G(i/o) signaling pathways. Frontiers in Bioscience: a journal and virtual library. 2008;13:4544–57.
Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nature Reviews Drug Discovery. 2014;13(4):290–314. https://doi.org/10.1038/nrd4228.
Hannila SS, Filbin MT. The role of cyclic AMP signaling in promoting axonal regeneration after spinal cord injury. Experimental Neurology. 2008;209(2):321–32. https://doi.org/10.1016/j.expneurol.2007.06.020.
Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114(11):1624–34. https://doi.org/10.1172/JCI22831.
Wang L, Chopp M, Szalad A, Liu Z, Bolz M, Alvarez FM et al. Phosphodiesterase-5 is a therapeutic target for peripheral neuropathy in diabetic mice. Neuroscience. 2011;193:399–410. https://doi.org/10.1016/j.neuroscience.2011.07.039.
Jasoliya MJ, McMackin MZ, Henderson CK, Perlman SL, Cortopassi GA. Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans. Human Molecular Genetics. 2017;26(14):2627–33. https://doi.org/10.1093/hmg/ddx141.
Lin H, Magrane J, Rattelle A, Stepanova A, Galkin A, Clark EM et al. Early cerebellar deficits in mitochondrial biogenesis and respiratory chain complexes in the KIKO mouse model of Friedreich ataxia. Disease Models & Mechanisms. 2017;10(11):1343–52. https://doi.org/10.1242/dmm.030502.
Palandri A, Martin E, Russi M, Rera M, Tricoire H, Monnier V. Identification of cardioprotective drugs by medium-scale in vivo pharmacological screening on a Drosophila cardiac model of Friedreich’s ataxia. Disease Models & Mechanisms. 2018;11(7). https://doi.org/10.1242/dmm.033811.
Igoillo-Esteve M, Gurgul-Convey E, Hu A, Romagueira Bichara Dos Santos L, Abdulkarim B, Chintawar S et al. Unveiling a common mechanism of apoptosis in beta-cells and neurons in Friedreich’s ataxia. Human Molecular Genetics. 2015;24(8):2274–86. https://doi.org/10.1093/hmg/ddu745.
Lincoln TM, Cornwell TL. Intracellular cyclic GMP receptor proteins. FASEB Journal: official publication of the Federation of American Societies for Experimental Biology. 1993;7(2):328–38.
Cornwell TL, Pryzwansky KB, Wyatt TA, Lincoln TM. Regulation of sarcoplasmic reticulum protein phosphorylation by localized cyclic GMP-dependent protein kinase in vascular smooth muscle cells. Molecular Pharmacology. 1991;40(6):923–31.
Xin W, Li N, Cheng Q, Petkov GV. BK channel-mediated relaxation of urinary bladder smooth muscle: a novel paradigm for phosphodiesterase type 4 regulation of bladder function. The Journal of Pharmacology and Experimental Therapeutics. 2014;349(1):56–65. https://doi.org/10.1124/jpet.113.210708.
Wang Z, Liu D, Varin A, Nicolas V, Courilleau D, Mateo P et al. A cardiac mitochondrial cAMP signaling pathway regulates calcium accumulation, permeability transition and cell death. Cell Death & Disease. 2016;7:e2198. https://doi.org/10.1038/cddis.2016.106.
Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metabolism. 2009;9(3):265–76. https://doi.org/10.1016/j.cmet.2009.01.012.
Acin-Perez R, Russwurm M, Gunnewig K, Gertz M, Zoidl G, Ramos L et al. A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J Biol Chem. 2011;286(35):30423–32. https://doi.org/10.1074/jbc.M111.266379.
Di Benedetto G, Pendin D, Greotti E, Pizzo P, Pozzan T. Ca2+ and cAMP cross-talk in mitochondria. The Journal of Physiology. 2014;592(2):305–12. https://doi.org/10.1113/jphysiol.2013.259135.
Acknowledgments:
This work was supported by grants from the Spanish Ministry of Economy and Competitiveness (Grant No. PI11/00678; SAF2015-66625-R) within the framework of the National R + D + I Plan and cofunded by the Instituto de Salud Carlos III (ISCIII)-Subdirección General de Evaluación y Fomento de la Investigación and FEDER funds, Fundación Ramón Areces (CIVP18A3899), and the Generalitat Valenciana (PROMETEOII/2014/067, PROMETEOII/2014/029, ACIF/2014/090, ACOMP/2014/058). CIBERER is an initiative developed by the Instituto de Salud Carlos III in cooperative and translational research on rare diseases. We would like to thank the staff of the CIBERER Biobank (Valencia, Spain) for their help in generating the lymphoblastoid cell lines (LCLs).
Author information
Authors and Affiliations
Contributions
BM conducted and designed experiments, analyzed the results, and wrote the manuscript. DM and PC performed experiments. MI and AF customized mito-morphology macro of ImageJ for morphometric mitochondrial analysis. FVP and MDM interpreted the data and wrote the manuscript. FP and PG designed the study, supervised the experiments, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Mollá, B., Muñoz-Lasso, D.C., Calap, P. et al. Phosphodiesterase Inhibitors Revert Axonal Dystrophy in Friedreich’s Ataxia Mouse Model. Neurotherapeutics 16, 432–449 (2019). https://doi.org/10.1007/s13311-018-00706-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-018-00706-z