
Abstract
In vivo imaging of the tau protein has the potential to aid in quantitative diagnosis of Alzheimer’s disease, corroborate or dispute the amyloid hypothesis, and demonstrate biomarker engagement in clinical drug trials. A host of tau positron emission tomography agents have been designed, validated, and tested in humans. Several agents have characteristics approaching the ideal imaging tracer with some limitations, primarily regarding off-target binding. Dozens of clinical trials evaluating imaging techniques and several pharmaceutical trials have begun to integrate tau imaging into their protocols.
Similar content being viewed by others

Introduction
In 1991, Braak and Braak [1] published a proposed neuropathological staging for Alzheimer’s disease (AD). This work detailed the progression of both extraneuronal amyloid plaques and intraneuronal neurofibrillary tau inclusions throughout the cortex. To date, research has focused primarily on the amyloid cascade hypothesis and had an amyloid-centric approach to AD [2, 3]. Amyloid therapeutic approaches, particularly monoclonal antibodies against both soluble and fibrillary forms of amyloid beta, have failed in preventing clinical progression of AD, despite a reduction in the amyloid plaque burden [4]. However, there is some hope that these drugs modify disease progression if given early enough in the disease course, but long-term efficacy data in patients with prodromal AD are only starting to emerge [5, 6].
A recurring argument against the amyloid hypothesis is that despite the prominence of amyloid plaques, tau-associated pathologic changes are more closely correlated with disease severity [7]. Thus, tau may be an important protein in early detection and staging of AD. However, tau pathology is nonspecific and is a marker of neurodegeneration beyond AD in diseases such as frontotemporal lobar degeneration (FTD), traumatic brain injury, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and chronic traumatic encephalopathy.
Regarding AD, one potential advantage to evaluating tau in vivo is the superior identification of a clinically relevant neurodegenerative process over amyloid positron emission tomography (PET) imaging. Amyloid has been shown to accumulate in the normal aging population with the percentage of cognitively normal adults who are amyloid PET-positive being 24 %, 35 %, and 49 % at the ages of 70 years, 80 years, and 90 years, respectively [8]. Therefore, older individuals are more likely to have amyloid PET positivity when they may not actually have AD dementia. Tau imaging, used alone or in conjunction with amyloid imaging, may therefore improve diagnostic certainty of AD by documenting the presence of neurodegeneration. Tau imaging may also be beneficial in evaluating the rate of disease progression, which has been unattainable with amyloid imaging alone, and measuring the efficacy of disease interventions, particularly in AD clinical trials.
Tau Biology
Microtubule-associated protein tau is an intracellular protein that plays a major role in cell structure. Tau is critical to the formation of microtubules that transport nutrients, waste, and chemical transmitters throughout neurons. The tau protein has 6 isoforms that have either 3 or 4 binding domain repeats (3R or 4R). These isoforms are hypothesized to have potentially distinct roles within the neuron, as well as differential distribution and different roles in various regions of the brain [9].
In disease, the equilibrium of bound and unbound tau proteins changes with an increase in the amount of unbound tau protein. The detachment of tau from microtubules is likely secondary to abnormal phosphorylation that is modulated by a homeostasis between phosphatase and kinase enzymatic activity. Unbound tau, particularly the hyperphosphorylated unbound tau seen in AD, then often forms insoluble fibers. The insoluble fibers form morphologically different aggregates, typically the paired helical filaments (PHFs) characteristically found in AD, straight filaments, or irregularly twisted filaments [10]. All 6 isoforms of tau are found to be abnormally phosphorylated in AD [11]. There is also evidence that a shift from 3R to 4R isoforms of the tau protein in the basal forebrain and hippocampus may be associated with formation of neurofibrillary tangles (NFTs) [12]. PHFs are typically composed of both 3R and 4R isoforms of tau, while other tauopathies are more associated with one specific isoform [13].
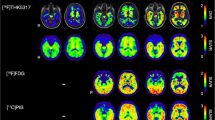
The pattern of PHF deposition in the cortex has been described and is hypothesized to spread in a sterotyped fashion in AD [1, 14]. In general, the transentorhinal and entorhinal areas are affected first, followed by the hippocampus, with an orderly progression through the subiculum and CA1, followed by CA2, CA3, and CA4, and then the inferior temporal lobe [1, 15–17] (see Fig. 1). After this, the middle and superior temporal lobes are next affected, followed by the inferior parietal and anterior frontal lobes (see Fig. 1). Finally, the PHFs spread to the remainder of the frontal and parietal cortices [1]. Both cognitively normal and impaired elderly individuals may have some degree of PHFs in the transentorhinal, entorhinal, and hippocampal areas [14]. However, if PHFs are found beyond these regions, there is a higher likelihood of having clinical symptoms.

Tau positron emission tomography images from cognitively normal controls, patients with mild cognitive impairment (MCI) with either virtually normal appearing tau imaging or those with high amount of tau, and patients with Alzheimer’s disease. Note the very low binding of tau ligand in the cortex of both patients with cognitively normal and low-tau mild cognitive impairment. In the high-tau MCI, stage I commonly observes binding in the anterior, inferior, and lateral temporal lobes. In the dementia stages the tau deposition in the previously involved areas becomes denser and tau binding is also observed in the other associations fields like the parietal and frontal lobes as seen in the figure
Tau Imaging Approaches
Historically, visualization of the neurodegenerative changes thought to be inflicted, in large part, by tau hyperphosphorylation and deposition was achieved through advanced magnetic resonance imaging methodologies that proved capable of depicting the earliest AD-associated changes in hippocampal structure. Many of these approaches modeled the hippocampal shape using computational anatomy deformation techniques [18, 19]. This enabled researchers to map the progression of AD pathology through the hippocampal structure in vivo [18, 20–24]. Yet, these approaches provided only inferential knowledge about tau-related pathology, and newer methods have since evolved.
The first agent designed for imaging tau, [18F]FDDNP, has been shown to be relatively nonselective as it binds to both intracellular NFTs and extracellular amyloid β [13]. Another nonselective tau tracer, [11C]PBB3 binds both NFTs and amyloid plaques in patients with AD and in patients with other tauopathies [25]. Thus, [18F]FDDNP and [11C]PBB3 have far inferior selectivity in binding to tau than more modern tracers. Additional disadvantages of [11C]PBB3 also include the short half-life of the C-11 radioisotope (approximately 20 min), as well as the presence of a radiometabolite than could potentially cross the blood–brain barrier and confound results [26].
Characteristics of successful tau imaging agents have been proposed and include the following: high binding affinity to PHF-tau with low nonspecific binding, high level of selectivity for PHF-tau (perhaps > 20-fold) over amyloid β, long half-life, good blood–brain barrier permeability, and very low metabolism [27, 28]. More recently, selective tau tracers that approach the theoretical ideal have been designed. The most used tracer to date is [18F]T807, which was renamed [18F]AV-1451 (also known as flortaucipir) after Avid Radiopharmaceuticals (a subsidiary of Eli Lilly and Company) acquired the tracer from Siemens Medical Solutions in April 2013 [29]. This benzimidazole-pyrimidine derivative has an approximately 29 times greater selectivity for PHF-tau over amyloid β and showed uptake in a pattern similar to that predicted by Braak staging in patients with AD [30]. The tracer also does not appear to bind with high affinity to the straight tau filaments seen in other tauopathies [31]. Being an F-18 compound, its half-life is approximately 110 min.
One of the major issues raised with this tracer is the “off-target” binding. The tracer has been reported to bind to neuromelanin and melanin containing cells (substantia nigra, pigmented cells in the eye, leptomeninges), as well as to areas of acute and subacute hemorrhage [31, 32]. There have also been reports of the tracer binding to the choroid plexus leading to a linear signal intensity along the perimeter of the hippocampus which could confound interpretation in this crucial region [33]. Choroid plexus binding of tau tracers may be related to normal pathologic changes in this area and may simply be a characteristic that should be accounted for in image interpretation [34].
From a safety standpoint, [18F]AV-1451 was evaluated for the potential risk of QT prolongation (slowing of the interval between the Q wave and T wave in the electrocardiogram). The potential risk of QT prolongation was determined with an in vitro hERG (human ether-à-gogo-related gene) assay, though in vivo canine studies failed to show any risk of QT interval changes at clinical doses [35]. Patients with known risk factors for QT prolongation or who are taking medications that are known to prolong the QT interval are excluded from receiving [18F]AV-1451 until more human data are available.
Another series of tau tracers has been developed by Tohoku University in Japan and has been given the abbreviation THK. Several quinolone and benzimidazole derivatives have been created, the first of which was [18F-THK523] which had several limitations, including high white matter retention and only 12-fold selectivity of tau over amyloid, preventing its clinical utility [36]. The next-generation derivatives ([18F]THK5105, [18F]THK5117, [18F]THK5351) have shown superior binding and improved signal to noise ratio than [18F]THK523 [36, 37]. Several novel tau tracers ([11C]RO6924963, [11C]RO6931643, and [18F]RO6958948) have been used successfully in a small Phase I study from F. Hoffman-La Roche Ltd (Roche) [38].
To date, tau radiotracers have been designed to bind with high affinity to PHF-tau. This is potentially useful in identifying patterns associated with AD and other conditions associated with both 3R and 4R isoforms of tau. However, other neurodegenerative conditions which are more specific to a single isoform, or conditions specific to certain post-translational modifications to tau may not be identified with current tracers [13]. Additionally, if shifts in populations of isoforms are related to AD initiation, a variety of tracers may be necessary for earliest disease identification. Autoradiography work on autopsy data has shown that [18F]AV-1451 has a higher affinity for 3R/4R neurofibrillary tangles than pure 3R or 4R tangles as seen in other neurodegenerative diseases [32]. Additionally, it was reported that there is a much lower affinity for TDP-43 (TAR-DNA binding protein 43) pathologies and the “ghost” tangles associated with tangle predominant dementia [32].
Tau Imaging Research Findings
The first studies with tau PET imaging in research populations are emerging and findings are generally supportive of the important role of tau in AD and other dementias, as well as the Braak and Braak staging of tau pathology. On tau PET imaging, both patients with mild cognitive impairment (MCI) and AD show increased binding relative to cognitively normal (CN) older adults (see Fig. 1). Specifically, patients with AD show significant tau PET tracer uptake in the temporal, parietal, and frontal lobes, while the primary sensory/motor cortices are relatively spared [30, 39–44]. Tau in the inferior temporal lobe is associated with increased amyloid deposition on PET, as well as greater cognitive impairment and disease severity in AD [41, 44]. Tau PET can also be used for Braak staging of participants. Data suggest that most amyloid-positive patients with AD are in Braak stage VI, while the majority of amyloid-negative patients with AD had Braak stages of III or lower [41, 44]. Across AD and CN participants, tau PET measures also significantly correlate with cerebrospinal fluid (CSF) levels of amyloid and tau [39]. A comparison between CSF biomarker levels and [18F]AV-1451 imaging has shown a direct correlation between phosphorylated tau levels in CSF and uptake in temporal limbic and neocortical regions [45]. A longitudinal tau PET study showed an increase in tracer signal of approximately 5 % in the inferior temporal and fusiform gyri in mild AD, and an increase of 5 % to 8.6 % in tracer retention in the fusiform, parahippocampal, and inferior temporal gyri in moderately impaired patients with AD [46]. Patients with MCI also show greater uptake than CN adults in the inferior temporal lobe, posterior cingulate, and fusiform, entorhinal, and parahippocampal gyri [30, 40, 41, 44]. As the MCI stage is quite heterogeneous, patients can demonstrate a full range of Braak stages, from I to VI [44]. Notably, patients with MCI who are amyloid positive on PET showed much greater tau binding than those who were amyloid negative on PET, who rarely showed a Braak stage > 0 [44]. Finally, in CN adults only, amyloid positivity on PET is associated with increased tau tracer uptake in the medial and lateral temporal and parietal lobes [39, 43]. A potential limiting factor for tau PET imaging is that Braak staging does not correlate perfectly with clinical symptomatology. In the oldest old NTFs in the hippocampus and neocortical regions are often found in the absence of amyloid deposition [47]. Future studies are needed to determine whether tau PET measurements will be predictive of future cognitive decline in MCI and CN.
Other neurodegenerative diseases and dementias have also been investigated with tau PET imaging. A case study of a symptomatic carrier of an autosomal dominant AD mutation in presenilin 1 (PSEN1) showed significant tau deposition in the posterior cingulate, precuneus, and parietal and occipital cortices [48]. Patients with posterior cortical atrophy, an atypical form of AD, showed increased tau in the primary visual cortex, the medial and lateral parietal, the occipital, and the posterior temporal lobes (temporo-parieto-occipital regions) [42, 49]. Another atypical form of AD, logopenic aphasia, demonstrated as-expected asymmetric tau deposition (left greater than right) in the majority of patients, in temporoparietal, occipital, and anterior temporal lobe regions, with more severely affected patients showing the more uptake in anterior temporal regions than less affected individuals [42, 49]. Studies have also investigated various forms of frontotemporal dementia with tau PET techniques. Recent tau studies have shown increased tau binding in frontal and temporal cortices in behavioral variant FTD that is correlated with increased clinical symptoms [50, 51]. Studies in forms of FTD with motor symptoms, including CBD and PSP, have shown deposition of tau in the supplementary motor area, midbrain, subthalamus, perirolandic area, basal ganglia, and cerebral and cerebellar white matter regions in the former and significant uptake in the brainstem, basal ganglia, globus pallidum, thalamus, subthalamic nucleus, midbrain, perirolandic areas, cerebellum, and frontal cortex in the latter [40, 50–52].
Overall, to date, the expected distribution of tau deposition across tauopathies has generally been observed using tau PET techniques. Future studies investigating the timing of initial tau deposition, the sensitivity of the available tau tracers to the earliest tau deposits, the variables associated with tau spread, and the implications of increased tau deposition on clinical outcomes and symptoms, as well as other AD biomarkers, are needed to understand fully tau in AD and other neurodegenerative diseases.
Tau Imaging in AD Clinical Trials
With the more widespread availability of tau tracers for PET imaging, clinical trials are beginning to include this modality as a secondary outcome. Tau PET imaging in AD trials could have several important implications. First, in trials of the currently used amyloid monoclonal antibodies, showing a downstream indirect effect of reduced amyloid β on the rate of deposition of PHFs would further bolster the amyloid hypothesis and lend credence to the presumptive disease-modifying impact of these drugs. Certainly as tau monoclonal antibodies are designed and trialed, tau PET imaging will be helpful in demonstrating and quantifying desired target engagement. Many current trials use CSF biomarkers of tau and phospho-tau to detect target engagement with little existing data on how spinal fluid biomarkers and tau PET imaging correlate. Further, tau PET imaging may help to confirm that changes in tau deposition are correlated with clinical disease progression.
Owing to tau PET tracers being labeled radiopharmaceuticals a complete listing of all active trials can be found on ClinicalTrials.gov. Observational studies make up the majority of the listed AD clinical trials using tau imaging. Many studies use [18F]T807 (AV-1451), including studies from Washington University [53, 54], Mayo Clinic [55], University of Pennsylvania [56], University of Southern California [57], Alzheimer’s Disease Cooperative Study [58], Molecular Neuroimaging (division in Invicro LLC) [59], Avid Radiopharamaceuticals [60–63], and St. Joseph’s Hospital [64], and others. A single study using [18F]THK-5351 from Asan Medical Center is being used to investigate findings in many patients ranging from normal subjects to those with AD [65]. A collaboration between Molecular Neuroimaging and Genentech (division of Roche) is underway to investigate the novel tau agent [18F]MNI-798 in normal controls and patients with AD [66]. Two other novel agents ([18F]MNI-777, [18F]MNI-815) are being investigated by Molecular Neuroimaging in a host of tauopathies including AD [67, 68].
There are relatively few AD clinical trials involving active treatment arms that are using tau imaging, though many are expected to use this modality in the near future. The Dominantly Inherited Alzheimer’s Network (DIAN) is an international clinical trial with observation and treatment arms. The treatment arm of the DIAN trial uses either solanezumab (Eli Lilly and Co.) or gantenerumab (Roche) monoclonal antibodies targeting amyloid β. The trial is an early adopter of tau imaging using [18F]T807 (AV-1451), which is available to enrolled participants [69, 70]. The goals of adding tau imaging to the DIAN trial are hypothesized to include the following: demonstrate binding in presymptomatic stages of AD, correlate with CSF tau biomarkers, correlate with symptoms, and predict the conversion from presymptomatic to symptomatic patients [69]. ExpeditionPRO, a subsequent phase III trial of Eli Lilly’s antiamyloid monoclonal antibody solanezumab, will evaluate tau imaging as a secondary outcome measure. This trial, which began enrolling in June 2016, will enroll > 2000 patients with prodromal AD and use [18F]T807 (AV-1451) at baseline and at 24 months to look at the change in neocortical tau deposits [71]. NAVIGATE-AD, another trial using an agent named LY3202626, will be using [18F]T807 (AV-1451) as a primary outcome measure of change in standardized uptake value ratio from baseline to week 52 [72].
Conclusions
Tau imaging agents have evolved from nonspecific agents to ligands with very high selectivity for PHF-tau over the last 5 years. Several promising agents are currently available and will help broaden the understanding of AD progression/screening, help support/disprove the amyloid hypothesis, and aid in our understanding of downstream biomarker engagement using existing and future treatments for AD. Further development in understanding off-target binding, radiotracer metabolites, and clinicopathologic correlations of tau imaging agents will clarify their utility in research and potential clinical utility, as well as lead to even the development of more useful agents in the future.
References
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239-259.
Hardy J, Higgins G. Alzheimer's disease: the amyloid cacade hypothesis. Science 1992;256:184-185.
Hardy J, Selkoe D. The amyloid hypothesis of alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353-356.
Giacobini E, Gold G. Alzheimer disease therapy—moving from amyloid-(beta) to tau. Nat Rev Neurol 2013;9:677-686.
Liu-Seifert H, Siemers E, Holdridge K, et al. Delayed-start analysis: mild alzheimer's disease patients in solanezumab trials, 3.5 years. Alzheimers Dement 2015;1:111-121.
Siemers E, Sundell K, Carlson C, et al. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer's disease patients. Alzheimers Dement 2016;12:110-120.
Arriagada P, Growdon J, Hedley-Whyte E, Hyman B. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 1992;42:631-639.
Ossenkoppele R, Jansen W, Rabinovici G, et al. Prevalence of amyloid PET positivity in dementia syndromes. JAMA 2015;313:1939-1950.
Ballatore C, Lee V-Y, Trojanowski J. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 2007;8:663-672.
Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof P. Tau protein isoforms, phosphorylation and role in neurodgenerative disorders. Brain Res Rev 2000;33:95-130.
Goedert M, Spillantini M, Cairns N, Crowther R. Tau proteins of alzheimer paird helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 1992;8:159-168.
Ginsberg S, Che S, Counts S, Mufson E. Shift in the ratio of three-repeat tau and four-repeat tau mRNAs in individual cholingeric basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. J Neurochem 2006;96:1401-1408.
Villemagne V, Furomoto S, Fodero-Tavoletti M, et al. The challenges of tau imaging. Future Neurol 2012;7:409-421.
Delacourte A, David J, Sergeant N, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease. Neurology 1999;52:1158-1165.
Schonheit B, Zarski R, Ohm T. Spatial and temporal relationships between plaques and tangles in Alzehimer-pathology. Neurobiol Aging 2004;25:697-711.
West M, Gundersen H. Unbiased sterological estimation of the number of neurons in the human hippocampus. J Comp Neurol 1990;296:1-22.
West M, Kawas C, Stewart W, Rudow G, Troncoso J. Hippocampal neurons in pre-clinical Alzheimer's disease. Neurobiol Aging 2004;25:1205-1212.
Csernansky JG, Wang L, Joshi S, et al. Early DAT is distinguished from aging by high-dimensional mapping of the hippocampus. Dementia of the Alzheimer type. Neurology 2000;55:1636-1643.
Thompson PM, Hayashi KM, De Zubicaray GI, et al. Mapping hippocampal and ventricular change in Alzheimer disease. Neuroimage 2004;22:1754-1766.
Apostolova LG, Dutton RA, Dinov ID, et al. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Arch Neurol 2006;63:693-699.
Csernansky JG, Wang L, Swank J, et al. Preclinical detection of Alzheimer's disease: hippocampal shape and volume predict dementia onset in the elderly. Neuroimage 2005;25:783-792.
Apostolova LG, Thompson PM, Green AE, et al. 3D comparison of low, intermediate, and advanced hippocampal atrophy in MCI. Hum Brain Mapp 2010;31:786-797.
Apostolova LG, Dinov ID, Dutton RA, et al. 3D comparison of hippocampal atrophy in amnestic mild cognitive impairment and Alzheimer's disease. Brain 2006;129:2867-2873.
Apostolova L, Mosconi L, Thompson P, et al. Subregional hippocampal atrophy predicts alzheimer's dementia in the cognitively normal. Neurobiol Aging 2010;31:1077-1088.
Maruyama M, Shimada H, Suhara T, et al. Imaging to tau pathology in a tuaopathy mouse model and in alzheimer patients compared to normal controls. Neuron 2013;79:1094-1108.
Hashimoto H, Kawamura K, Takei M, et al. Identification of a major radiometabolite of [11C]PBB3. Nuclear Med Biol 2015;42:905-910.
Villemagne V, Fodero-Tavoletti M, Masters C, Rowe C. Tau imaging: early progress and future directions. Lancet Neurol 2015;14:114-124.
Okamura N, Harada R, Furomoto S, Arai H, Yanai K, Kudo Y. Tau PET imaging in alzheimer's disease. Curr Neurol Neurosci Rep 2014;14:500.
Eli Lilly. Lilly acquires novel tau tangle diagnostic program to bolster alzheimer's disease research and development 2013. Available at: https://investor.lilly.com/releasedetail.cfm?ReleaseID=757053. Accessed June 3, 2016.
Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis 2013;34:457-468.
Marquie M, Normandin M, Vandergurg C, et al. Validating novel tau positron emission tomography trace [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 2015;78:787-800.
Lowe V, Curran G, Fang P, et al. An autoradiographic evaluation of AV-1451 tau PET in dementia. Acta Neuropathol Commun 2016;4:1-19.
Villemagne V, Okamura N. Tau imaging in the study of ageing, alzheimer's disease, and other neurodegenerative conditions. Curr Opin Neurobiol 2016;36:43-51.
Ikonomovic M, Abrahamson E, Price J, Mathis C, Klunk W. [F-18]AV-1451 positron emission tomography retention int he choroid plexus: more than “off-target” binding. Ann Neurol 2016;80:307-308.
Avid Radiopharmaceuticals. Investigator's brochure, 18F-AV-1451 ([F-18]T807) injection for brain tau imaging. 22 February 2016.
Dani M, Brooks DJ, Edison P. Tau imaging in neurodegenerative diseases. Eur J Nuclear Med Mol Imaging 2016;43:1139-1150.
Okamura N, Furumoto S, Fodero-Tavoletti M, et al. Non-invasive assessment of alzheimer's disease neurofibrillary pathology using 18F-THK5105 PET. Brain 2014;137:1762-1771.
Wong D, Borroni E, Kuwabara H, et al. First in-human PET study of 3 novel tau radiopharmaceticals: 11CRO6924963, 11CRO6931643, and 18FRO6958948. Alzheimers Dement 2015;11(7 Suppl.):850-851.
Brier MR, Gordon B, Friedrichsen K, et al. Tau and Abeta imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med 2016;8:338ra66.
Chiotis K, Saint-Aubert L, Savitcheva I, et al. Imaging in-vivo tau pathology in Alzheimer's disease with THK5317 PET in a multimodal paradigm. Eur J Nuclear Med Mol Imaging 2016;43:1686-1699.
Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 2016;79:110-119.
Ossenkoppele R, Schonhaut DR, Scholl M, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain 2016;139:1551-1567.
Scholl M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron 2016;89:971-982.
Schwarz AJ, Yu P, Miller BB, et al. Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016;139:1539-1550.
Chhatwal J, Schultz A, Marshall G, et al. Temporal T807 binding correlates with CSF tau and phospho-tau in normal elderly. Neurology 2016;87:920-926.
Ishiki A, Okamura N, Furukawa K, et al. Longitudinal assessment of tau pathology in patients with alzheimer's disease using [18F]THK-5117 positron emission tomography. PLOS ONE 2015;10:e0140311.
Saava G, Warton S, Ince P, et al. Age, neuropathology, and dementia. N Engl J Med 2009;360:2302-2309.
Smith R, Wibom M, Olsson T, et al. Posterior accumulation of tau and concordant hypometabolism in an early-onset Alzheimer's disease patient with presenilin-1 mutation. J Alzheimers Dis 2016;51:339-343.
Ossenkoppele R, Schonhaut DR, Baker SL, et al. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol 2015;77:338-342.
Dickerson B, Domoto-Reilly K, Daisy S, Brickhouse M, Stepanovic M, Johnson KA. Imaging tau pathology in vivo in FTLD: initial experience with [18F]T807 PET. Alzheimers Dement 2014;10:P131.
Suhara T, Shimada H, Shinotoh H, et al. In vivo tau PET imaging using [11C]PBB3 in Alzheimer's disease and non-Alzheimer's disease tauopathies. J Nucl Med 2014;55(Suppl. 1):1824.
Shinotoh H, Shimada H, Hirano S, et al. Imaging of tau pathology in patients with non-Alzheimer's disease tauopathies by [11C]PBB3 PET. Alzheimers Dement 2014;10:6-7.
Washington University School of Medicine. F 18 T807 Tau PET imaging of alzehimer's disease (T807IND) Bethesda (MD): National Library of Medicine (US). Available at: https://clinicaltrials.gov/ct2/show/NCT02414347. Accessed June 2, 2016.
Washington University School of Medicine. F 18 T807 Tau PET imaging of progressive posterior cortical dysfunction (IND 123119, Protocol E) Bethesda (MD): National Library of Medicine (US). Available at: https://clinicaltrials.gov/ct2/show/NCT02414282. Accessed June 2, 2016.
Clinic M. Imaging of brain amyloid plaques in the aging population. Available at: https://clinicaltrials.gov/ct2/show/NCT00950430. Accessed June 6, 2016.
University of Pennsylvania. Tau imaging in young onset dementia. Available at: https://clinicaltrials.gov/ct2/show/NCT02289118. Accessed June 6, 2016.
University of Southern California, Northern California Institute of Research and Education, National Institute on Aging (NIA), Alzheimer's Therapeutic Research Institute. Alzheimer's disease neuroimaging intiative 2 (ADNI2).Available at: https://clinicaltrials.gov/ct2/show/NCT01231971. Accessed June 6, 2016.
Alzheimer's Disease Cooperative Study, Janssen Research and Development LLC. Down syndrome biomarker initiative (DSBI). Available at: https://clinicaltrials.gov/ct2/show/NCT02141971. Accessed June 6, 2016.
Molecular Neuroimaging, Institue for Neurodegenerative Disorders, AG RP. Quantitative evaluation of [18f]t807 as a potential PET radioligant for imaging tau in patients with alzheimer's disease. Available at: https://clinicaltrials.gov/ct2/show/NCT02370524. Accessed June 6, 2016.
Avid Radiopharmaceuticals. 18F-AV-1451 Autopsy study Bethesda (MD): National Library of Medicine (US). Available at: https://clinicaltrials.gov/ct2/show/study/NCT02516046. Accessed June 2, 2016.
Avid Radiopharmaceuticals. 18F-AV-1451 High Resolution Autopsy study Bethesda (MD): National Library of Medicine (US). Available at: https://clinicaltrials.gov/ct2/show/NCT02350634. Accessed June 2, 2016.
Avid Radiopharmaceuticals. Clinical Evaluation of 18F-AV-1451 Bethesda (MD): National Library of Medicine (US). Available at: https://clinicaltrials.gov/ct2/show/NCT02278367. Accessed June 2, 2016.
Avid Radiopharmaceuticals. Analysis of 18F-AV-1451 PET imaging in cognitively healthy, MCI and AD subjects. Bethesda (MD): National Library of Medicine (US). Available at: https://clinicaltrials.gov/ct2/show/NCT02016560. Accessed June 2, 2016.
St. Joseph's Hospital and Medical Center Phoenix, Banner Alzheimer's Insitutue Phoenix, Translational Genomics Research Institute Phoenix. Relationship between down syndrome (dx) and alzehimer's disease (ad) Bethesda (MD): National Library of Medicine (US). Available at: https://clinicaltrials.gov/ct2/show/NCT02759887. Accessed June 2, 2016.
Jae Seung Kim, Asan Medical Center. [18f]THK-5351 positron emission computed tomorgraphy study of normal, mild cognitive impairment, alzheimer's disease and other neurodegenerative disease Bethesda (MD): National Library of Medicine (US). Available at: https://clinicaltrials.gov/ct2/show/NCT02656498. Accessed June 2, 2016.
Molecular Neuroimaging. Longitudinal evaluation of [18F]MNI-798 as a PET radioligand for imaging tau in the brain of patients with alzheimer's disease Bethesda (MD): National Library of Medicine (US). Available at: https://clinicaltrials.gov/ct2/show/NCT02640092. Accessed June 2, 2016.
Molecular Neuroimaging, Institue for Neurodegenerative Disorders. Evaluation of [18F]MNI-777 PET as a marker of tau pathology in subjects with taupathies compared to healthy subjects.. Available at: https://clinicaltrials.gov/ct2/show/NCT02103894. Accessed June 6, 2016.
Molecular Neuroimaging, Piramal Imaging SA. Evaluation of [18F]MNI-815 as a potential PET radioligand for imaging tau protein in the brain of patients with tauopathies. Available at: https://clinicaltrials.gov/ct2/show/NCT02531360. Accessed June 6, 2016.
Benzinger T. F 18 T807 Tau PET imaging in dominantly inherited alzheimer's network (DIAN Project). Available at: https://clinicaltrials.gov/show/NCT02414178. Accessed May 11, 2016.
Washington University School of Medicine, Eli Lilly and Company, Roche H-L, Alzheimer's Association, National Institute on Aging (NIA), Avid Radiopharmaceuticals, et al. Dominantely inherited alzheimer network trial: an opportunity to prevent dementia. A study of potential disease modifying treatments in individuals at risk for or with a type of early onset alzheimer's disease caused bya genetic mutation (DIAN-TU). Bethesda (MD): National Library of Medicine (US). Available at: https://clinicaltrials.gov/ct2/show/NCT01760005. Accessed June 2, 2016.
Eli Lilly. A study of solanezumab (LY2062430) in participants with prodromal alzheimer's disease (ExpeditionPRO). Available at: https://clinicaltrials.gov/show/NCT02760602. Accessed May 11, 2016.
Eli Lilly. A study of LY3202626 on disease progression in participants with mild alzheimer's disease dementia (NAVIGATE-AD). Available at: https://clinicaltrials.gov/ct2/show/NCT02791191. Accessed June 6, 2016.
Acknowledgments
This work was supported by NIA P30 AG010133, NIA K01 AG049050, NIA R01 AG040770, NIA K02 AG048240, the Easton Consortium for Alzheimer Drug Discovery and Biomarker Development, the Alzheimer’s Association, the Indiana University Health-Indiana University School of Medicine Strategic Research Initiative, and the Indiana Clinical and Translational Science Institute.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Brosch, J.R., Farlow, M.R., Risacher, S.L. et al. Tau Imaging in Alzheimer’s Disease Diagnosis and Clinical Trials. Neurotherapeutics 14, 62–68 (2017). https://doi.org/10.1007/s13311-016-0490-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0490-y