
Abstract
Introduction
Empagliflozin, a highly selective sodium-glucose cotransporter 2 (SGLT2) inhibitor, improves glycaemic control in patients with type 2 diabetes mellitus (T2DM) by inducing urinary glucose excretion. Combination therapy with empagliflozin and glucagon-like peptide-1 (GLP-1) receptor agonists had not previously been assessed, so we investigated the safety, tolerability and efficacy of empagliflozin as an add-on therapy to liraglutide, a GLP-1 receptor agonist.
Methods
This was a randomised, double-blind, parallel-group phase 4 trial of empagliflozin (10 mg or 25 mg) for 52 weeks as an add-on therapy to liraglutide (0.9 mg/day) in Japanese patients with T2DM insufficiently controlled by liraglutide alone.
Results
59.4% (19/32) and 66.7% (22/33) of patients in the empagliflozin 10 mg and 25 mg groups, respectively, reported at least one adverse event (AE). 9.4% (3/32) and 21.2% (7/33) of patients, respectively, reported drug-related AEs (primary endpoint). From baseline to week 52, adjusted mean changes with empagliflozin 10 mg and 25 mg, respectively, were: − 0.55 (standard error: 0.15) and − 0.77 (0.14)% for glycated haemoglobin; − 32.5 (4.6) and − 36.0 (4.5) mg/dL for fasting plasma glucose; − 2.6 (0.4) and −3.1 (0.3) kg for body weight; − 6.7 (2.2) and − 8.4 (2.1) mmHg for systolic blood pressure; and − 3.0 (1.2) and − 4.7 (1.1) mmHg for diastolic blood pressure.
Conclusion
Empagliflozin as an add-on to liraglutide for 52 weeks was well tolerated and led to clinically meaningful and sustained improvements in glycaemic control, body weight and blood pressure in Japanese patients with T2DM.
Trial Registration
ClinicalTrials.gov with the identifier NCT02589626.
Funding
Nippon Boehringer Ingelheim Co. Ltd.
Similar content being viewed by others


Introduction
Non-insulin glucose-lowering agents play a central role in the management of type 2 diabetes mellitus (T2DM) and its complications. The 2016 revision of the Japanese Clinical Practice Guideline for diabetes recommends the use of glucose-lowering agents for patients with T2DM who fail to achieve favourable glycaemic control following lifestyle intervention(s). In patients receiving glucose-lowering agent monotherapy who fail to achieve their glycaemic target, the guideline recommends increasing the dose of the first-line agent, changing to a more potent agent, or adding another glucose-lowering agent with a different mechanism of action [1].
Glucose-lowering agents available in Japan for T2DM management are sulfonylureas (SU), biguanides, α-glucosidase inhibitors, thiazolidinediones (TZD), glinides, dipeptidyl peptidase (DPP)-4 inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists and sodium-glucose cotransporter-2 (SGLT2) inhibitors [1].
SGLT2 inhibitors decrease renal glucose reabsorption, which leads to urinary glucose excretion and, consequently, reduce blood glucose levels in patients with T2DM [2]. Empagliflozin is a highly selective SGLT2 inhibitor [3]. In a phase 3 trial in Japanese patients with T2DM, empagliflozin (10 mg or 25 mg) as an add-on therapy to oral antidiabetic agents (SUs, biguanides, α-glucosidase inhibitors, TZDs, glinides or DPP-4 inhibitors) for 52 weeks significantly reduced glycated haemoglobin (HbA1c), fasting plasma glucose (FPG) and body weight, and was well tolerated [4]. Moreover, in the EMPA-REG OUTCOME® trial of patients with T2DM and established cardiovascular disease, empagliflozin given in addition to standard of care reduced the composite major adverse cardiovascular events (MACE) of cardiovascular death, nonfatal myocardial infarction or nonfatal stroke as well as the risks of cardiovascular death, all-cause mortality, hospitalization for heart failure and incident or worsening nephropathy in comparison to placebo, and led to significant reductions in the urinary albumin-to-creatinine ratio [5,6,7]. Currently, empagliflozin is the only SGLT2 inhibitor with proven cardiovascular and all-cause mortality benefit in patients with T2DM and established cardiovascular disease [8]. The risk reductions for cardiovascular outcomes and mortality in the EMPA-REG OUTCOME® trial were consistent for the overall population and the Asian/East Asian subpopulation [9].
GLP-1 receptor agonists increase glucose-dependent insulin secretion, lower postprandial glucagon levels, slow gastric emptying, and produce satiety and a reduced calorie intake [10]. In the LEADER trial, liraglutide, a GLP-1 receptor agonist, significantly reduced MACE, death from cardiovascular causes, death from any cause and the development and progression of diabetic kidney disease compared with placebo [11, 12].
Based on the evidence for empagliflozin and liraglutide, the American Diabetes Association stated in its Standards of Medical Care in Diabetes 2018 that, “In patients with type 2 diabetes and established atherosclerotic cardiovascular disease, antihyperglycaemic therapy should begin with lifestyle management and metformin and subsequently incorporate an agent proven to reduce major adverse cardiovascular events and cardiovascular mortality (currently empagliflozin and liraglutide) after considering drug-specific and patient factors” [13].
The effects of SGLT2 inhibitors and GLP-1 receptor agonists are favourable in patients with T2DM and various cardiovascular risk factors. As these agents exert their antihyperglycaemic and cardiovascular protective effects via different mechanisms, combination therapy is expected to provide additive effects over those achieved with either agent as monotherapy or in combination with other glucose-lowering agents [14,15,16].
To date, combination therapy with empagliflozin and GLP-1 receptor agonists has not been reported. In this trial, we investigated the safety, tolerability and efficacy of empagliflozin as 52-week add-on therapy to liraglutide in Japanese patients with T2DM insufficiently controlled by liraglutide alone.
Methods
Compliance with Ethics Guidelines
All procedures performed in studies involving human participants were approved by the respective institutional review boards (IRBs) according to Japanese regulations and were conducted in compliance with the Japanese Ethical Guidelines for Clinical Studies and the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Study Design and Inclusion/Exclusion Criteria
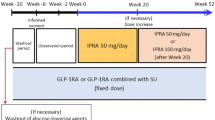
This was a multicentre, randomised, double-blind, parallel-group phase 4 trial of empagliflozin taken once daily for 52 weeks as add-on therapy to the GLP-1 receptor agonist liraglutide in Japanese patients with T2DM. The trial is registered at ClinicalTrials.gov with the identifier NCT02589626 [17]. The study design is shown schematically in Fig. 1. Eligible patients were male and female adults (≥ 20 years) with T2DM and a body mass index (BMI) of ≤ 40.0 kg/m2 at the screening visit who had been pretreated with: (A) liraglutide 0.9 mg/day for at least 10 weeks prior to informed consent and with a HbA1c level of 7.0–10.0% at screening; (B) liraglutide 0.9 mg/day in combination with one oral antidiabetic (OAD) for at least 10 weeks prior to informed consent and with a HbA1c level of 7.0–9.0% at screening and 7.0–10.0% at the beginning of the placebo run-in period; (C) one OAD for at least 10 weeks prior to informed consent and with a HbA1c level of 7.0–10.0% at screening and at the beginning of the placebo run-in period. SUs were permitted only if the dose was ≤ 50% of the daily maximum approved dose (160 mg, 10 mg and 6 mg for gliclazide, glibenclamide and glimepiride, respectively). Insulin, TZDs and SGLT-2 inhibitors were not allowed.

Study design. OAD oral antidiabetic drug, R randomisation
Patients were excluded if they had uncontrolled hyperglycaemia with a glucose value > 270 mg/dL (15 mmol/L) after an overnight fast; were drug-naïve at screening or were treated with insulin, TZD or SGLT-2 inhibitor within 10 weeks prior to informed consent; had acute coronary syndrome, stroke, transient ischaemic attack or indication of liver disease; had impaired renal function, defined as an estimated glomerular filtration rate (eGFR) < 45 mL/min/1.73 m2; were treated with antiobesity drugs within 12 weeks prior to informed consent.
Patients who met the inclusion criteria after screening entered a switch/washout period based on their pretreatment history. Patients who had been pretreated with liraglutide 0.9 mg/day for at least 10 weeks prior to informed consent skipped this step. Patients who had been pretreated with liraglutide 0.9 mg/day in combination with an OAD for at least 10 weeks prior to informed consent had a washout of OAD for 10 weeks. Patients who had been pretreated with an OAD only for at least 10 weeks prior to informed consent were switched to liraglutide 0.9 mg/day for 14 weeks including a maximum 4 weeks’ uptitration of liraglutide in accordance with the Japanese label. Patients then entered a 2-week, open-label, placebo run-in phase prior to randomisation. At the start of double-blind treatment, patients were randomised 1:1 to receive empagliflozin 10 mg or 25 mg as add-on therapy to liraglutide. Randomisation was conducted via interactive response technology. Treatment continued for 52 weeks, with follow-up performed 1 week after the end of treatment. All study drugs were taken orally once daily in the morning. Patients, investigators and study personnel remained blinded to the randomised treatment assignments until database lock.
Primary, Secondary and Additional Endpoints
The primary endpoint of the trial was the proportion of patients with drug-related adverse events (AE) during 52 weeks of treatment with empagliflozin as add-on therapy to liraglutide. Additional safety endpoints were AEs of special interest which included: metabolic acidosis, ketoacidosis and diabetic ketoacidosis; decreased renal function or hepatic injury; and events involving lower limb amputation. In addition, any AEs of ‘hypoglycaemic events’, defined as glucose concentrations < 54 mg/dL (< 3.0 mmol/L), and symptomatic and severe hypoglycaemic episodes were evaluated and recorded.
The secondary endpoint was the change from baseline in HbA1c level after 52 weeks of treatment with empagliflozin as add-on therapy to liraglutide.
Additional efficacy endpoints were: change from baseline to week 52 in FPG; change from baseline to week 52 in body weight; change from baseline to week 52 in systolic blood pressure (SBP) and diastolic blood pressure (DBP); proportion of patients with > 5% decrease in body weight from baseline to week 52; change from baseline to week 52 in fasting plasma insulin.
Statistical Analysis
The sample size for this trial was determined based on consultation with the Japanese Pharmaceuticals and Medical Devices Agency (PMDA), which, for safety evaluations but not for testing the statistical significance of efficacy, requires at least 50 patients who receive over 52 weeks of exposure to 2 doses of the study drug. Assuming a discontinuation/dropout rate of approximately 20% during the trial, the sample size was calculated at 32 patients each for the empagliflozin 10 mg and 25 mg arms.
Safety analyses were assessed descriptively. No confirmatory statistical analysis was planned. The statistical model for the secondary endpoint used a restricted maximum likelihood-based mixed model with repeated measures (MMRM) which included treatment, baseline renal function, visit, treatment-by-visit interaction and baseline HbA1c-by-visit interaction as fixed effects and baseline HbA1c as covariate. Continuous (additional) efficacy endpoints were summarised using descriptive statistics by treatment group. A MMRM model similar to that described above for the secondary endpoint analysis was used. The model included baseline value for the corresponding endpoint and its interaction with visit as additional covariates. Binary efficacy endpoints were described using frequency statistics.
Results
Patient Disposition
The trial was conducted at 16 study sites in Japan from November 2015 to June 2017. Patient disposition is shown in Fig. 2. Of 84 patients screened in the trial, 17 were deemed screening failures for failing to meet inclusion criteria (n = 12) or for other reasons (n = 5). Of 67 patients who entered the placebo run-in phase, 61 had been pretreated with liraglutide alone (skipped washout/switch period), 3 with liraglutide plus 1 OAD (underwent washout of OAD), and 3 with one OAD alone (switched to liraglutide). Of 65 patients who entered the double-blind treatment phase, 32 and 33 patients were randomised into the empagliflozin 10 mg and 25 mg groups, respectively, with stratification based on baseline HbA1c value (< 8.0%, ≥ 8.0%) and eGFR value (≥ 90, 60 to < 90, 45 to < 60 mL/min/1.73 m2); the remaining 2 patients were not randomised as they failed to meet the inclusion criteria. Five patients in total (3 patients allocated to empagliflozin 10 mg and 2 patients allocated to empagliflozin 25 mg) discontinued study medication prematurely before week 52 due to AEs (1 from each group), refusal to continue the study (1 from each group), or for other reason (n = 1 in the 10-mg group). Thus, 29 patients randomised to empagliflozin 10 mg and 31 patients randomised to empagliflozin 25 mg completed the study.

Patient disposition
The demographic and clinical characteristics of the treatment groups at baseline are summarised in Table 1. The patient population was predominantly male (73.8%). Mean [standard deviation (SD)] age was 57.3 (9.6) years, and 23.1% were elderly (≥ 65 years). Most patients (84.6%) had a T2DM disease history of more than 5 years. As a group, the study population was overweight [mean BMI 27.8 (5.2) kg/m2 and 69.2% with BMI ≥ 25], had poor glycaemic control [mean HbA1c 8.76 (0.83)% and 83.1% with HbA1c ≥ 8.0%], and showed only mild loss of renal function [eGFR 85.6 (22.8) mL/min/1.73 m2]. Baseline characteristics were generally well balanced between treatment groups except for the proportion of females (6 (18.8%) and 11 (33.3%) in the empagliflozin 10 mg and 25 mg groups, respectively) due to the small sample size.
Safety
An overall summary of AEs is provided in Table 2. A total of 19 patients (59.4%) in the empagliflozin 10 mg group and 22 patients (66.7%) in the empagliflozin 25 mg group reported at least one AE during the course of the study. All AEs except for one were mild or moderate in intensity. There were no deaths, episodes of diabetic ketoacidosis, or lower leg amputations reported during the study.
Drug-related AEs (primary endpoint) were reported in 3 (9.4%) and 7 (21.2%) patients in the empagliflozin 10 mg and 25 mg groups, respectively. Except for vulvocandidiasis, which was reported in 2 patients, all other drug-related AEs were reported in a single patient each.
Serious AEs were reported in 2 patients (6.3%) in the empagliflozin 10 mg group (loss of consciousness; large intestine polyp) and 1 patient (3.0%) in the empagliflozin 25 mg group (unstable angina). The episodes of loss of consciousness (considered to be related to empagliflozin 10 mg) and unstable angina (considered to be unrelated to empagliflozin 25 mg) led to treatment discontinuation. The large intestine polyp was considered to be unrelated to the empagliflozin 10 mg. Loss of consciousness was reported as a severe AE.
During the study, a hypoglycaemic AE was reported for 1 patient (3.0%) in the empagliflozin 25 mg group. The patient experienced an episode of symptomatic hypoglycaemia, which was mild in intensity and required no assistance. The blood glucose level was not measured. Events consistent with urinary tract infections were identified in 1 patient (3.1%) in the empagliflozin 10 mg group (asymptomatic bacteriuria) and 2 patients (6.1%) in the empagliflozin 25 mg group (funguria; urinary tract infection). All events were mild in intensity and recovered. Only urinary tract infection in the empagliflozin 25 mg group was considered by the investigator to be drug-related. Events consistent with genital tract infections were identified in 2 patients (6.1%) in the empagliflozin 25 mg group (vulvovaginal candidiasis). Both events were mild in intensity and recovered. Both events were considered by the investigator to be drug-related. AE related to bone fracture (coccyx fracture caused by a fall) was reported in 1 patient (3.1%) in the empagliflozin 10 group. Both the fall and the coccyx fracture were mild in intensity and not considered to be related to the drug by the investigator. There were no reports of events consistent with volume depletion, diabetic ketoacidosis, decreased renal function or lower limb amputation in either treatment group.
Changes from baseline to week 52 in vital signs and laboratory values are summarised in Table S1 of the Electronic supplementary material (ESM). The haematocrit was increased in both treatment groups. Total ketone bodies, acetoacetic acid and β-hydroxybutyrate were increased from baseline to week 52 in both treatment groups. No clinically meaningful changes were observed in total, HDL or LDL cholesterol, free fatty acid, triglyceride, serum electrolytes, liver enzymes [aspartate aminotransferase and alanine aminotransferase] and bone markers [alkaline phosphatase, urinary N-terminal telopeptide (NTx)/creatinine ratio and parathyroid hormone] in either treatment group. There were no clinically meaningful changes in pulse rate in the empagliflozin 10 mg and 25 mg groups.
Efficacy
Empagliflozin reduced HbA1c levels from baseline, with changes observed at all time points from week 8 to 52 (Fig. S1 in the ESM). Adjusted mean change [standard error (SE)] in HbA1c from baseline to week 52 was − 0.55 (0.15) and − 0.77 (0.14)% in the empagliflozin 10 mg and 25 mg groups, respectively (Fig. 3a).

Changes from baseline to week 52 in a HbA1c, b FPG, c body weight, d SBP and e DBP. Data are shown as the mean ± standard error (SE). For HbA1c, the model includes baseline HbA1c as a linear covariate and baseline eGFR, treatment, visit, visit by treatment interaction and baseline HbA1c by visit interaction as fixed effects. A similar model was used for FPG, body weight, SBP and DBP. The model included baseline value for the corresponding endpoint and its interaction with visit as additional covariates. DBP diastolic blood pressure, FPG fasting plasma glucose, HbA1c glycated haemoglobin, SBP systolic blood pressure
Empagliflozin decreased FPG from baseline to week 52. Adjusted mean change (SE) in FPG at week 52 was − 32.5 (4.6) and − 36.0 (4.5) mg/dL in the empagliflozin 10 mg and 25 mg groups, respectively (Fig. 3b).
Empagliflozin reduced body weight from baseline to week 52, with changes observed at all time points from week 8 to week 52 (Fig. S2 in the ESM). Adjusted mean change (SE) in body weight at week 52 was − 2.6 (0.4) and − 3.1 (0.3) kg in the empagliflozin 10 mg and 25 mg groups, respectively (Fig. 3c).
The proportion of patients with > 5% reduction in body weight from baseline to week 52 was 28.1% and 36.4% in the empagliflozin 10 mg and 25 mg groups, respectively (Fig. S3 in the ESM). Reductions in body weight were consistent across all HbA1c and BMI subgroups (Table 3).
Waist circumference was decreased with empagliflozin at week 52. Adjusted mean (SE) change from baseline in waist circumference was − 2.7 (0.7) and − 2.1 (0.7) cm in the empagliflozin 10 mg and 25 mg groups at 52 weeks, respectively (Fig. S4 in the ESM).
Empagliflozin reduced SBP and DBP levels from baseline to week 52. Adjusted mean change (SE) in SBP at week 52 was − 6.7 (2.2) and − 8.4 (2.1) mmHg in the empagliflozin 10 mg and 25 mg groups, respectively (Fig. 3d). Adjusted mean change (SE) in DBP at week 52 was −3.0 (1.2) and − 4.7 (1.1) mmHg in the empagliflozin 10 mg and 25 mg groups, respectively (Fig. 3e).
Fasting plasma insulin levels were decreased with empagliflozin from baseline to week 52. Mean change (SD) in fasting plasma insulin at week 52 was − 18.22 (20.02) and − 19.09 (45.00) pmol/L in the empagliflozin 10 mg and 25 mg groups, respectively (Table S1 in the ESM).
Discussion
This is the first study to assess the long-term safety, tolerability and clinical effectiveness of empagliflozin in combination with a GLP-1 receptor agonist, liraglutide. In the EMPA-REG OUTCOME® trial, empagliflozin significantly decreased the risk of cardiovascular death, all-cause mortality and hospitalization for heart failure compared with placebo in patients with T2DM and established cardiovascular disease [5]. In the LEADER trial, compared with placebo, liraglutide was also shown to significantly reduce cardiovascular death and all-cause mortality in patients with T2DM and a high cardiovascular risk [11]. These results sparked considerable interest in using these agents in combination to address cardiovascular risk in patients with T2DM.
Compared with a previous study which investigated empagliflozin as monotherapy for 52 weeks in Japanese patients with T2DM [18], our study population was younger and had greater adiposity (higher BMI) and poorer glycaemic control (higher HbA1c and FPG), which is likely reflected in the background use of liraglutide in 95.5% of patients at the time of screening. Although in our study there was no placebo comparison to accurately measure the clinical effect of add-on therapy with empagliflozin, compared with baseline values, empagliflozin 10 or 25 mg added to liraglutide 0.9 mg/day produced clinically meaningful and sustained reductions in HbA1c, FPG, body weight and blood pressure in agreement with the results reported with empagliflozin monotherapy [18].
Empagliflozin as an add-on therapy to liraglutide was generally well tolerated over the 52-week double-blind treatment period. Reported AEs and analyses of vital signs and laboratory values were consistent with the known experience of empagliflozin from previous clinical trials [19, 20], including studies conducted specifically in Japanese patients [4, 18]. The proportion of patients with drug-related AEs, which was the primary endpoint of the study, was 9.4% with empagliflozin 10 mg and 21.2% with empagliflozin 25 mg. These values were within the range expected from previous studies of empagliflozin, and the types of drug-related AEs reported were consistent with the known safety profile of empagliflozin. The incidence of drug-related genital infection (vulvovaginal candidiasis) was 2 (6.1%) in the empagliflozin 25 mg group and 0 in the empaglilfozin 10 mg group. The 2 genital infection cases in the empagliflozin 25 mg group were females with no history of genital infection. In a previous pooled safety analysis, females were found to have a higher incidence of genital infection than males in East Asian patients [20]. The higher rate of drug-related AEs in the empagliflozin 25 mg group might be in part due to the difference in the proportion of females between the groups (6 females (18.8%) and 11 females (33.3%) in the empagliflozin 10 mg and 25 mg groups, respectively).
Incidences of hypoglycaemia and of events consistent with UTIs, genital infection or volume depletion were similar with both doses of empagliflozin/liraglutide combination therapy and were comparable to those in a previous study of empagliflozin monotherapy [18]. There were no reports in any patient with diabetic ketoacidosis, decreased renal function or events involving lower limb amputation. Also, no drug-related bone fracture was observed in this trial. One notable event which involved loss of consciousness in a patient treated with empagliflozin 10 mg was deemed severe by the investigator and led to hospitalisation. Treatment was discontinued and the event resolved in a few days. No reason for the loss of consciousness could be identified, although a relationship with empagliflozin could not be ruled out.
The efficacy and safety of using a SGLT2 inhibitor as an add-on to a GLP-1 receptor agonist has been investigated in other trials. In the multinational, randomised, controlled DURATION-8 trial in patients with T2DM inadequately controlled by metformin, exenatide plus dapagliflozin over 28 weeks was significantly superior to either drug alone for glycaemic measures and cardiovascular risk factors, with no differences in the safety profile observed compared with single-agent treatment [21]. Other studies have reported a benefit of using SGLT2 inhibitor/GLP-1 receptor agonist combinations in Japanese patients with T2DM. An early study showed that dapagliflozin as monotherapy or in combination with existing antidiabetic drugs, including liraglutide (10.4% of patients), was an effective way to improve glycaemic control and was well tolerated over 52 weeks [22]. In an open-label single-arm study, luseogliflozin added to liraglutide significantly reduced HbA1c, FPG and body weight (all p < 0.001 vs baseline) over 52 weeks and was well tolerated [23]. The study revealed that a reduction in body weight was attributable to a reduction in fat mass with luseogliflozin. In this study, empagliflozin reduced waist circumference, indicating that fat mass would be reduced with an add-on of empagliflozin. In this trial, treatment with empagliflozin led to clinically meaningful reductions in HbA1c, FPG, body weight, SBP and DBP without an increase in pulse rate, as also seen in previous studies [4, 18]. Collectively, these studies confirm that SGLT2 inhibitor plus GLP-1 receptor agonist combinations provide additional benefits to patients with T2DM in terms of improving not only glycaemic control but also other cardiovascular risk factors such as body weight and blood pressure without compromising safety.
The current study is limited by the absence of a placebo control group and the lack of a confirmatory statistical analysis of efficacy or safety, although these are not required by the PMDA for safety evaluations. Despite these limitations, the study adds to the evidence indicating the safety and efficacy of SGLT2 inhibitor plus GLP-1 receptor agonist combinations in Japanese patients with T2DM, and provides the first important information about the tolerability and clinical effectiveness of the previously unexplored combination of empagliflozin and liraglutide, which were shown to reduce the risks of MACE, cardiovascular death and renal disease progression. This study also showed that the reduction in body weight achieved with an add-on of empagliflozin was consistent across all HbA1c and BMI subgroups.
Conclusions
To conclude, empagliflozin as an add-on therapy to liraglutide for 52 weeks was well tolerated, and the safety profile of the combination was consistent with the established safety profiles of empagliflozin and liraglutide. Combined treatment with empagliflozin and a GLP-1 receptor agonist provided clinical benefit in terms of glycaemic control and reductions in body weight, SBP and DBP in Japanese patients with T2DM. Further study is needed to investigate additive or synergistic cardiovascular and renal benefits with the combination therapy of empagliflozin and liraglutide.
References
Haneda M, Noda M, Origasa H, et al. Japanese clinical practice guideline for diabetes 2016. J Diabetes Investig. 2018;9:657–97.
Chao EC, Henry RR. SGLT2 inhibition—a novel strategy for diabetes treatment. Nat Rev Drug Discov. 2010;9:551–9.
Grempler R, Thomas L, Eckhardt M, et al. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes Metab. 2012;14:83–90.
Araki E, Tanizawa Y, Tanaka Y, et al. Long-term treatment with empagliflozin as add-on to oral antidiabetes therapy in Japanese patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2015;17:665–74.
Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–28.
Wanner C, Inzucchi SE, Lachin JM, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med. 2016;375:323–34.
Cherney DZI, Zinman B, Inzucchi SE, et al. Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: an exploratory analysis from the EMPAGLIFLOZIN-REG OUTCOME randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017;5:610–21.
Zelniker TA, Wiviott SD, Raz I, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393:31–9.
Kaku K, Lee J, Mattheus M, et al. Empagliflozin and cardiovascular outcomes in Asian patients with type 2 diabetes and established cardiovascular disease®—results from EMPAGLIFLOZIN-REG OUTCOME®. Circ J. 2017;81:227–34.
Meier JJ. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8:728–42.
Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311–22.
Mann JFE, Ørsted DD, Brown-Frandsen K, et al. Liraglutide and renal outcomes in type 2 diabetes. N Engl J Med. 2017;377:839–48.
American Diabetes Association. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes—2018. Diabetes Care. 2018;41(Suppl. 1):S73–85.
Nauck MA, Meier JJ. GLP-1 receptor agonists and SGLT2 inhibitors: a couple at last? Lancet Diabetes Endocrinol. 2016;4:963–4.
DeFronzo RA. Combination therapy with GLP-1 receptor agonist and SGLT2 inhibitor. Diabetes Obes Metab. 2017;19:1353–62.
Goncalves E, Bell DSH. Combination treatment of SGLT2 inhibitors and GLP-1 receptor agonists: symbiotic effects on metabolism and cardiorenal risk. Diabetes Ther. 2018;9:919–26.
National Institutes of Health. ClinicalTrials.gov. Study record: A 52-week randomised, double-blind, parallel group, safety and efficacy study of empagliflozin once daily as add-on therapy to glucagon-like peptide-1 receptor agonist in Japanese type 2 diabetes mellitus patients with insufficient glycaemic control. https://clinicaltrials.gov/ct2/show/NCT02589626. Accessed 6 Mar 2019.
Kadowaki T, Haneda M, Inagaki N, et al. Efficacy and safety of empagliflozin monotherapy for 52 weeks in Japanese patients with type 2 diabetes: a randomized, double-blind, parallel-group study. Adv Ther. 2015;32:306–18.
Kohler S, Zeller C, Iliev H, Kaspers S. Safety and tolerability of empagliflozin in patients with type 2 diabetes: pooled analysis of phase I–III clinical trials. Adv Ther. 2017;34:1707–26.
Yabe D, Yasui A, Ji L, et al. Safety and tolerability of empagliflozin in East Asian patients with type 2 diabetes: pooled analysis of phase I–III clinical trials. J Diabetes Investig. 2019;10:418–28. https://doi.org/10.1111/jdi.12910 (Epub ahead of print).
Frías JP, Guja C, Hardy E, et al. Exenatide once weekly plus dapagliflozin once daily versus exenatide or dapagliflozin alone in patients with type 2 diabetes inadequately controlled with metformin monotherapy (DURATION-8): a 28 week, multicentre, double-blind, phase 3, randomised controlled trial. Lancet Diabetes Endocrinol. 2016;4:1004–16.
Kaku K, Maegawa H, Tanizawa Y, et al. Dapagliflozin as monotherapy or combination therapy in Japanese patients with type 2 diabetes: an open-label study. Diabetes Ther. 2014;5:415–33.
Seino Y, Yabe D, Sasaki T, et al. Sodium-glucose cotransporter-2 inhibitor luseogliflozin added to glucagon-like peptide 1 receptor agonist liraglutide improves glycemic control with bodyweight and fat mass reductions in Japanese patients with type 2 diabetes: a 52-week, open-label, single-arm study. J Diabetes Investig. 2018;9:332–40.
Acknowledgements
The authors would like to thank all study investigators and participants.
Funding
The study, including article processing charges, was funded by Nippon Boehringer Ingelheim Co. Ltd. All authors had full access to all the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their final approval for this version to be published.
Medical Writing and Editorial Assistance
Editorial assistance in the preparation of this manuscript was provided by Robert Furlong and Kerry Dechant of Content Ed Net, with funding from Nippon Boehringer Ingelheim Co. Ltd., Tokyo, Japan.
Disclosures
Yasuo Terauchi has received honoraria for speaker bureaus from Astellas Pharma Inc., AstraZeneca K.K., Bayer Yakuhin, Ltd., Daiichi Sankyo Company Limited, Sumitomo Dainippon Pharma Co., Ltd., Eli Lilly Japan K.K., Kowa Pharmaceutical Company Ltd., Merck Sharp & Dohme (MSD) K.K., Mitsubishi Tanabe Pharma Corporation, Nippon Boehringer Ingelheim Co., Ltd., Novo Nordisk Pharma Ltd., Ono Pharmaceutical Co., Ltd., Sanwa Kagaku Kenkyusho Co., Ltd., Sanofi K.K., Shionogi & Co., Ltd., Taisho Toyama Pharmaceutical Co., Ltd., and Takeda Pharmaceutical Company Limited; and grants from Astellas Pharma Inc., Daiichi Sankyo Company Limited, Sumitomo Dainippon Pharma Co., Ltd., Eli Lilly Japan K.K., Kowa Pharmaceutical Company Ltd., MSD K.K., Mitsubishi Tanabe Pharma Corporation, Nippon Boehringer Ingelheim Co., Ltd., Novo Nordisk Pharma Ltd., Ono Pharmaceutical Co., Ltd., Sanwa Kagaku Kenkyusho Co., Ltd., Sanofi K.K., Shionogi & Co., Ltd, and Takeda Pharmaceutical Company Limited. Kazunori Utsunomiya has received honoraria for speaker bureaus/advisory panels from Astellas, Astra Zeneca, Eli Lilly, Kowa, MSD, Novo Nordisk, Sanofi, and Taisho; and research support from Arkray, Astellas, Boehringer Ingelheim, Kowa, Kyowa Hakko Kirin, Kissei, MSD, Novo Nordisk, Ono, Taisho, Tanabe-Mitsubishi, and Terumo. Atsutaka Yasui is an employee of Nippon Boehringer Ingelheim Co. Ltd. Tetsuo Seki was an employee of Nippon Boehringer Ingelheim Co. Ltd. and is currently an employee of the Foundation for Biomedical Research and Innovation at Kobe. Gang Cheng is an employee of Boehringer Ingelheim (China) Investment Co. Ltd. Kosuke Shiki is an employee of Nippon Boehringer Ingelheim Co. Ltd. Jisoo Lee is an employee of Boehringer Ingelheim Pharma GmbH & Co. KG.
Compliance with Ethics Guidelines
All procedures performed in studies involving human participants were approved by the respective institutional review boards (IRBs) at each study site according to Japan regulations and were conducted in compliance with the Japanese Ethical Guidelines for Clinical Studies and the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Data Availability
The datasets obtained and/or analysed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.7844348.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Terauchi, Y., Utsunomiya, K., Yasui, A. et al. Safety and Efficacy of Empagliflozin as Add-On Therapy to GLP-1 Receptor Agonist (Liraglutide) in Japanese Patients with Type 2 Diabetes Mellitus: A Randomised, Double-Blind, Parallel-Group Phase 4 Study. Diabetes Ther 10, 951–963 (2019). https://doi.org/10.1007/s13300-019-0604-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-019-0604-8