
Abstract
Transcription factors recognize and bind to consensus sequence elements that are specific for each transcription factor, and the transcription factors then regulate downstream gene expression. In the bone marrow, transcription factors, such as C/EBPα, PU.1, and RUNX1, control essential genes to maintain the normal hematopoietic system. Dysregulation of transcription factors caused by gene mutations, chromosomal aberrations, or aberrant expression can lead to cancer, including acute myeloid leukemia. In the past, transcription factors were not considered “druggable” targets. However, a better understanding of the pathology of malignant tumors and mechanisms of transcriptional regulation has enabled us to develop novel therapeutic strategies that target transcription factors. In this review, we focus on transcription factors that play important roles in leukemogenesis and current efforts and prospects in the development of transcriptional therapy. We believe that such a therapeutic approach will benefit patients with cancers that involve acute myeloid leukemia in the near future.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) is characterized by clonal expansion of immature myeloid cells and suppression of normal hematopoietic cells in the bone marrow and peripheral blood, leading to anemia, leukocytopenia, and thrombocytopenia. Recent advances in our understanding of the pathogenesis and therapy of AML have dramatically improved clinical outcomes. The complete remission ratio of patients with newly diagnosed AML who receive standard chemotherapy-based treatments is 70–80%. However, the 5-year survival rate is only 30–40% [1]. Therefore, a novel therapeutic approach is required for complete cure of AML.
The conventional chemotherapy for AML inhibits DNA synthesis, cell division, and mitosis in both cancerous and normal cells, which can cause serious side effects, such as bone marrow suppression. Development of targeted therapy, which inhibits aberrant activity of oncoproteins caused by somatic genetic alterations, has benefited patients with some cancers; however, it is not the ultimate solution for cancer therapy due to some limitations, such as primary and secondary resistance.
Transcription factors contribute to stem cell maintenance, determination of differentiation, and cell maturation in hematopoietic stem cells and hematopoietic progenitor cells. Mutations, translocations, or aberrant expression of these transcription factors can cause malignant transformation of hematopoietic cells. Transcription factors had not been traditionally considered “druggable”, as they lack enzymatic activities.
Recent advances in our understanding of genetic and epigenetic mechanisms of regulation of transcription factors and the development of new technologies to manipulate their expression have contributed to the development of new strategies to target transcription factors (Table 1).
In this review, we will describe recent strategies to target transcription factors, including findings from our laboratory.
Transcription factors and therapeutics strategies
CCAAT/enhancer-binding protein alpha (C/EBPα)
C/EBPα is a transcription factor mainly expressed in hematopoietic organs, the liver, and adipose tissue [2]. In hematopoietic cells, C/EBPα upregulates its target genes, such as CSF3R, MPO, and ELANE, which are necessary for granulocyte differentiation and maturation, and concurrently suppresses genes essential for erythroid or lymphoid cell differentiation or expansion, such as EPOR [3,4,5]. Furthermore, C/EBPα competes with PU.1 to suppress monocyte differentiation and leads myeloid progenitor cell-to-granulocyte differentiation [6]. The requirement of C/EBPα for granulocyte differentiation is supported by observations, showing that a lack of C/EBPα leads to loss of mature granulocytes and eosinophils in C/EBPα-knockout mice [2].
CEBPA mRNA has two translation start sites, and these generate two isoforms of C/EBPα: p42 (full length, 42 kDa) and p30 (lacking part of the N-terminal, 30 kDa). CEBPA mutations are observed in 9% of acute myeloid leukemia patients [7]. Mutations in the N-terminus of C/EBPα cause frameshift and dominant p30 translation. The p30 isoform has less transcriptional activity, because it lacks part of the transcription activity domain. The p30 isoform inhibits the transcriptional function of the p42 isoform by forming a p42–p30 complex [8], so it acts in a dominant-negative fashion. On the other hand, mutations in the C-terminus of C/EBPα are located in the leucine zipper domain. The mutations in the C-terminus of C/EBPα make C/EBPα unable to bind DNA and function as a transcription factor. This C-terminus mutation in C/EBPα may contribute to leukemogenesis in mice [9]. In addition, accumulating evidence suggests that oncogenic fusion proteins can lead to a decrease in C/EBPα expression, which may contribute to a blocking of differentiation and leukemogenesis. BCR–ABL inhibits C/EBPα translation by upregulating heterogeneous ribonuclear protein E2 (hnRNP E2) in patients with chronic myeloid leukemia in blastic crisis (CML-BC) [10]. AML1-ETO (RUNX1–RUNX1T1) inhibits binding of C/EBPα to its own promoter and suppresses positive feedback in AML patients with t(8;21) [11].
These results prompted us to hypothesize that upregulation or reactivation of C/EBPα may lead to cell cycle arrest and granulocytic differentiation of leukemic cells, and could be a novel strategy to treat AML. RAS-rerated C3 botulinum toxin substrate 1 (RAC1), which indirectly suppresses C/EBPα expression, is upregulated in acute myeloid leukemia patients [12]. RAC1 inhibitor NSC23766 upregulates C/EBPα expression and induces apoptosis in leukemia cell lines [12]. WD repeat-containing protein 5 (Wdr5) is a histone modification factor associated with p30-dependent cell differentiation arrest. OICR-9249 is a Wdr5 inhibitor that induces differentiation in AML cells harboring CEBPA mutations [13].
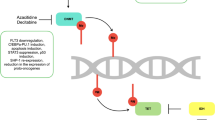
Short-activating RNAs (saRNAs) have attracted attention as a new treatment strategy (Fig. 1) [14]. saRNAs induce target gene expression. saRNAs are double-stranded RNAs consisting of a guide RNA and a passenger RNA. saRNAs are loaded onto Argonaute proteins (Ago) in the cytosol, and the active complexes translocate into the nucleus. After translocation, the passenger RNA is degraded or released from the Ago–saRNA complex. Upon releasing the passenger RNA, the Ago–guide RNA complex binds target gene promoter sequences and recruits histone modification molecules, such as histone demethylases (HDMs) or histone methyltransferases (HMTs). This histone modification induces target gene expression [14]. C/EBPA-saRNA is the saRNA designed to induce CEBPA expression. C/EBPA-saRNA induces not only CEBPA mRNA and C/EBPα expression in cell lines and mice, but also improvement of liver function in mice with liver cirrhosis and reduction of tumor volume in mice with liver cancer [15].

Mechanism of short-activating RNAs. Double-stranded RNA consisting of a passenger RNA and a guide RNA forms a complex with Ago in the cytoplasm. The Ago–guide RNA complex translocates to the nucleus and binds specific sequences, as well as recruits histone modulators
Identification of small molecules leading to C/EBPα activation is another strategy for novel therapeutics of AML. We identified ICCB280, a quinazolinone derivative, as an inducer of C/EBPα by high-throughput screening using a reporter assay (Fig. 2a). It induces morphological changes characteristic of myeloid differentiation as well as functional changes (Fig. 2b) in AML cell lines and primary AML cells isolated from patients [16]. ICCB280 induces C/EBPα and its downstream targets, such as C/EBPε and G-CSFR, at both the mRNA and protein levels (Fig. 2c). c-Myc, which is negatively regulated by C/EBPα, is downregulated by ICCB280, suggesting that ICCB280 may be effective in diseases driven by overexpression of c-Myc, such as Burkitt lymphoma. G-CSF enhances ICCB280’s ability to induce myeloid differentiation, suggesting that ICCB280 can be combined with other chemicals or cytokines as well.

ICCB280, a new small molecule, induces C/EBPα expression and granulocytic differentiation in acute myeloid leukemia cells. a Chemical structure of ICCB280. b HL-60 cells treated with either DMSO (0.1%) or ICCB280 (10.0 µM) for 7 days were subjected to Wright–Giemsa staining and NBT reduction assay. c C/EBPα and its downstream transcription factor C/EBPε were upregulated by ICCB280 treatment in HL-60 cells
PU.1
PU.1 is a transcription factor encoded by the SPI1 gene, and is specifically expressed in myeloid cells and B-lymphocytes. PU.1 upregulates many genes involved in differentiation of these cells. Examples include the CSF1R gene, which encodes macrophage colony-stimulating factor receptor (M-CSF receptor), and the IL7R gene, which encodes IL-7 receptor [17]. PU.1 also plays an important role in lineage commitment at two furcations, myeloid cells vs. erythroid cells, and granulocytes vs. monocytes. PU.1 competes with GATA-binding protein 1 (GATA-1) at the myeloid-erythroid furcation and leads to myeloid differentiation [17]. At a later stage, PU.1 competes with C/EBPα and leads to monocyte differentiation [6]. Reduction in PU.1 expression can be a trigger of AML. More than 40% of patients with AML demonstrate low PU.1 expression [18] by transcriptional and/or epigenetic mechanisms. Homozygous knockout of an enhancer located − 14 kb upstream of PU.1 (URE) causes an 80% reduction of PU.1 expression and leads to AML [19]. Paradoxically, conditional knockout of PU.1 leads to stem cell failure [20], suggesting that PU.1 is critical for stem cell maintenance. These observations also suggest that leukemic cells expressing low PU.1 may be more vulnerable to further PU.1 reduction compared to normal myeloid cells. Based on this hypothesis, Antony-Debré et al. demonstrate that inhibition of PU.1 may be a novel therapeutic strategy for AML [18]. PU.1 recognizes DNA at consensus sites harboring a 5′-GGAA/T-3′ motif in the major groove and prefers AT-rich sequences in the adjacent minor groove. These authors screened many heterocyclic diamidines that could be potent ligands for AT-rich DNA and identified three molecules (DB2313, DB2115, and DB1976) as strong PU.1 inhibitors. These small molecules decrease the tumor burden and increase survival in URE-knockout leukemic mice, possibly due to a decrease in cell growth and an increase in apoptosis.
Runt-related transcription factor 1 (RUNX1)
RUNX1 is a member of the Runt-related transcription factor (RUNX) family. The RUNX family consists of RUNX1, RUNX2, and RUNX3. Each RUNX protein forms a complex with core-binding factor beta (CBFβ) to bind its consensus sequence and regulate transcription [21]. RUNX1 regulates expression of not only hematopoiesis-associated proteins, such as C/EBPα and PU.1, but also cell cycle-associated proteins, such as p53. Deletion of floxed Runx1 causes thrombocytopenia, lymphocytopenia and reduction of these precursors [22, 23]. Genetic mutation or chromosomal aberrance of RUNX1 is commonly observed in AML patients [22]. RUNX has been regarded as a tumor suppressor [24], but the recent reports suggest that RUNX also works as a tumor promoter. These findings suggest that the tumor-promoting transcription factor RUNX1 could become a therapeutic target in cancer. Morita et al. reported a new strategy that targets RUNX, named cluster regulation of RUNX (CROX) (Fig. 3) [21]. RUNX family proteins recognize 5′-TGTGGT-3′ or, more rarely, 5′-TGCGGT-3′. Morita et al. used alkylating agent-conjugate pyrrole–imidazole (PI) polyamides that bind these specific DNA sequences, Chb-M′ for 5′-TGTGGT-3′, and Chb-50 for 5′-TGCGGT-3′. Both Chb-M′ and Chb-50 inhibit the RUNX–DNA interaction. Chb-M′ and Chb-50 then suppress expression of downstream genes regulated by RUNX, such as IL-3, CSF2RB, and CSF2. Chb-M′ has anti-leukemia effects in vitro and in mice [21].

New strategy for targeting RUNX. Alkylating agent-conjugate pyrrole-imidazole (PI) polyamide binds the RUNX consensus sequence and inhibits downstream gene transcription driven by RUNX family proteins
p53
p53 is a transcription factor that maintains genetic stability and normal cell growth by inducing cell cycle arrest and DNA repair after DNA damage by regulating downstream genes such as CDKN1A (p21) and BAX [25]. The TP53 gene is the most frequently mutated gene in human cancer: more than 50% of all human cancers harbor TP53 mutations [26]. Loss of p53 function caused by point mutations or deletions is associated with tumorigenesis [26]. Murine double minute 2 (MDM2) is an E3 ubiquitin ligase that inhibits p53 transcriptional activity and accelerates p53 degradation by the ubiquitin–proteasome system. Recently, new therapeutics that inhibit p53-MDM2 complex formation have been reported. Some therapeutics targeting p53, such as the p53-MDM2 complex formation inhibitors AMG232 and AM-8553, are being examined in clinical trials for AML, CML, and solid tumors [27,28,29,30]. PRIMA-1 and PRIMA-1MET (APR-246) upregulate p21 expression, which is downstream of p53, and induce apoptosis in the cells harboring p53 mutations [26, 30]. Moreover, PRIMA-1MET (APR-246) suppresses p63, which is the active form of mutated p53 protein [30].
c-MYC
c-MYC is a transcription factor that controls the cell cycle and cell proliferation, differentiation, and apoptosis by regulating downstream genes such as CCND1, CDKN1A, and BCL2 [31,32,33,34]. c-MYC upregulation is associated with 20% of human cancers [31]. c-MYC and its co-factors, such as MYC-associated factor X (MAX) and transformation/transcription domain-associated protein (TRRAP), form complexes that recognize and bind consensus sequences of c-MYC and regulate gene expression. The main strategies for inhibiting c-MYC are to inhibit complex formation [31, 35,36,37] and to inhibit the DNA–complex interaction [31]. Mycro3 is one of the inhibitor of c-MYC-MAX complex formation. Mycro3 decreases Ki67 expression and tumor volume in pancreatic tumor cell lines [36]. JQ-1, a bromodomain and extra-terminal motif (BET) inhibitor, downregulates c-MYC expression in AML cell lines and prolongs survival in an AML mouse model [38].
c-MYB
c-MYB is associated with cell proliferation and differentiation in hematopoiesis by regulating GATA2, STAT5A, and IKZF1 [39]. Dysregulation of c-MYB participates in proliferation and maintenance of leukemia cells [40]. Mebendazole is an anti-parasitic drug that induces c-MYB degradation. Mebendazole reduces the volume of AML cells transplanted into mice [41]. p300 is a co-factor of c-MYB that is required for its transactivation. Celastrol and plumbagin have anti-tumor activity by inhibiting c-MYB–p300 complex formation in AML mice [40, 42].
Signal transducer and activator of transcription 3 (STAT3)
STAT3 is a transcription factor that positively regulates expression of anti-apoptotic proteins, such as Bcl-2, Bcl-xL, and Mcl-1, and of cell cycle regulator proteins, such as cyclin D1 and c-MYC [43]. The JAK-STAT pathway is essential in normal hematopoiesis and hematopoietic malignancy. JAK proteins promote STAT3 and STAT5 activation [44]. STAT3 is activated in human cancers, including AML, CML, and myeloproliferative neoplasm (MPN) [44]. Galiellalactone is a fungus metabolite separated from Galiella rufa, which was found in the process of a drug screen for IL-6 production inhibitors. Galiellalactone prevents STAT3 activity and suppresses genes responsible for cell proliferation downstream of STAT3 in prostate cancer cells [43, 45].
Krüppel-like factor 4 (KLF4)
KLF4 is a zinc-finger transcription factor of the Sp/XKLF family. KLF4 regulates differentiation, migration, and inflammation in normal tissue, and serves as a tumor suppressor. KLF4 positively regulates p21 and p27 expression [46]. Downregulation of KLF4 expression is observed in several cancers, such as AML, colon cancer, and non-small-cell lung cancer [4, 46]. KLF4 is negatively regulated by metal-regulatory transcription factor 1 (MTF-1). APTO-253 induces KLF4 expression by inhibiting metal-regulatory transcription factor 1 (MTF-1), and suppresses tumor cell proliferation in AML, colon cancer, non-small-cell lung cancer, and prostate cancer [46]. APTO-253 induces p21 and BAX and suppresses Bcl-2. The tolerability of APTO-253 was confirmed by a phase 1 clinical trial for solid tumors [46].
Cyclic AMP response element-binding protein (CREB)
CREB is a leucine zipper transcription factor associated with cell proliferation, survival, differentiation, and metabolism [47]. CREB regulates downstream genes such as FOSL1, RECQL4, and GNL3 [48]. CREB is upregulated in both AML and acute lymphoid leukemia (ALL). Upregulated CREB is associated with a shorter event-free survival and a high relapse rate in AML patients [47, 49]. One report found five CREB inhibitors (STF-038533, STF-046536, STF-017794, STF-055910, and STF-046827) by high-throughput screening. These five CREB inhibitors suppress leukemia cell proliferation in KG-1 acute myeloid leukemia cells [49].
Nuclear factor erythroid 2-related factor 2 (Nrf2)
Nrf2 upregulates antioxidant-related gene expression after oxidative stress via the antioxidant response element (ARE) [50]. Upregulation of Nrf2 is caused by either mutations or epigenetic remodeling in the Nrf2-binding partner KEAP1 [51]. In AML primary cells and cell lines, Nrf2 is upregulated by chemotherapeutics such as cytarabine, daunorubicin, and arsenic trioxide. Upregulation of Nrf2 is associated with the acquisition of resistance to chemotherapy [50, 51]. The Nrf2 inhibitor brusatol renders AML cell lines susceptible to cytarabine or daunorubicin by decreasing Nrf2 expression [51].
Conclusion
Targeting transcription factors has been considered difficult and challenging compared to receptors or enzymes. However, recent progress in our understating of transcriptional control and the development of new technologies has enabled us to design novel “transcription therapies” in leukemia and other cancers. We hope that modulation of transcription factors will be an effective therapeutic option alone or in combination with other agents, eventually leading to a cure for cancer.
References
Pulte D, Gondos A, Brenner H. Expected long-term survival of patients diagnosed with acute myeloblastic leukemia during 2006–2010. Ann Oncol. 2010;21:335–41.
Lourenco AR, Coffer PJ. A tumor suppressor role for C/EBPalpha in solid tumors: more than fat and blood. Oncogene. 2017;36:5221–30.
Nuchprayoon I, Meyers S, Scott LM, Suzow J, Hiebert S, Friedman AD. PEBP2/CBF, the murine homolog of the human myeloid AML1 and PEBP2 beta/CBF beta proto-oncoproteins, regulates the murine myeloperoxidase and neutrophil elastase genes in immature myeloid cells. Mol Cell Biol. 1994;14:5558–68.
Seipel K, Marques MT, Bozzini MA, Meinken C, Mueller BU, Pabst T. Inactivation of the p53-KLF4-CEBPA axis in acute myeloid leukemia. Clin Cancer Res. 2016;22:746–56.
Suh HC, Gooya J, Renn K, Friedman AD, Johnson PF, Keller JR. C/EBPalpha determines hematopoietic cell fate in multipotential progenitor cells by inhibiting erythroid differentiation and inducing myeloid differentiation. Blood. 2006;107:4308–16.
Reddy VA, Iwama A, Iotzova G, Schulz M, Elsasser A, Vangala RK, et al. Granulocyte inducer C/EBPalpha inactivates the myeloid master regulator PU.1: possible role in lineage commitment decisions. Blood. 2002;100:483–90.
Nerlov C. C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer. 2004;4:394–400.
Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27:263–70.
Friedman AD. C/EBPalpha in normal and malignant myelopoiesis. Int J Hematol. 2015;101:330–41.
Perrotti D, Cesi V, Trotta R, Guerzoni C, Santilli G, Campbell K, et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet. 2002;30:48–58.
Pabst T, Mueller BU, Harakawa N, Schoch C, Haferlach T, Behre G, et al. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat Med. 2001;7:444–51.
Namasu CY, Katzerke C, Brauer-Hartmann D, Wurm AA, Gerloff D, Hartmann JU, et al. ABR, a novel inducer of transcription factor C/EBPalpha, contributes to myeloid differentiation and is a favorable prognostic factor in acute myeloid leukemia. Oncotarget. 2017;8:103626–39.
Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R, et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat Chem Biol. 2015;11:571–8.
Zheng L, Wang L, Gan J, Zhang H. RNA activation: promise as a new weapon against cancer. Cancer Lett. 2014;355:18–24.
Reebye V, Saetrom P, Mintz PJ, Huang KW, Swiderski P, Peng L, et al. Novel RNA oligonucleotide improves liver function and inhibits liver carcinogenesis in vivo. Hepatology. 2014;59:216–27.
Radomska HS, Jernigan F, Nakayama S, Jorge SE, Sun L, Tenen DG, et al. A cell-based high-throughput screening for inducers of myeloid differentiation. J Biomol Screen. 2015;20:1150–9.
Zhang P, Zhang X, Iwama A, Yu C, Smith KA, Mueller BU, et al. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood. 2000;96:2641–8.
Antony-Debre I, Paul A, Leite J, Mitchell K, Kim HM, Carvajal LA, et al. Pharmacological inhibition of the transcription factor PU.1 in leukemia. J Clin Invest. 2017;127:4297–313
Rosenbauer F, Wagner K, Kutok JL, Iwasaki H, Le Beau MM, Okuno Y, et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat Genet. 2004;36:624–30.
Iwasaki H, Somoza C, Shigematsu H, Duprez EA, Iwasaki-Arai J, Mizuno S, et al. Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood. 2005;106:1590–600.
Morita K, Suzuki K, Maeda S, Matsuo A, Mitsuda Y, Tokushige C, et al. Genetic regulation of the RUNX transcription factor family has antitumor effects. J Clin Invest. 2017;127:2815–28.
Guo H, Ma O, Speck NA, Friedman AD. Runx1 deletion or dominant inhibition reduces Cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood. 2012;119:4408–18.
Huang G, Zhang P, Hirai H, Elf S, Yan X, Chen Z, et al. PU.1 is a major downstream target of AML1 (RUNX1) in adult mouse hematopoiesis. Nat Genet. 2008;40:51–60.
Chuang LS, Ito K, Ito Y. RUNX family: Regulation and diversification of roles through interacting proteins. Int J Cancer. 2013;132:1260–71.
Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9:862–73.
Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol. 2010;80:724–30.
Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014;74:5855–65.
Zhao Y, Aguilar A, Bernard D, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J Med Chem. 2015;58:1038–52.
Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem. 2014;57:1454–72.
Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–36.
Chen BJ, Wu YL, Tanaka Y, Zhang W. Small molecules targeting c-Myc oncogene: promising anti-cancer therapeutics. Int J Biol Sci. 2014;10:1084–96.
Prochownik EV, Vogt PK. Therapeutic targeting of Myc. Genes Cancer. 2010;1:650–9.
Tan A, Bitterman P, Sonenberg N, Peterson M, Polunovsky V. Inhibition of Myc-dependent apoptosis by eukaryotic translation initiation factor 4E requires cyclin D1. Oncogene. 2000;19:1437–47.
Eischen CM, Packham G, Nip J, Fee BE, Hiebert SW, Zambetti GP, et al. Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene. 2001;20:6983–93.
Yin X, Giap C, Lazo JS, Prochownik EV. Low molecular weight inhibitors of Myc–Max interaction and function. Oncogene. 2003;22:6151–9.
Stellas D, Szabolcs M, Koul S, Li Z, Polyzos A, Anagnostopoulos C, et al. Therapeutic effects of an anti-Myc drug on mouse pancreatic cancer. J Natl Cancer Inst. 2014. https://doi.org/10.1093/jnci/dju320.
Savino M, Annibali D, Carucci N, Favuzzi E, Cole MD, Evan GI, et al. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS One. 2011;6:e22284.
Brondfield S, Umesh S, Corella A, Zuber J, Rappaport AR, Gaillard C, et al. Direct and indirect targeting of MYC to treat acute myeloid leukemia. Cancer Chemother Pharmacol. 2015;76:35–46.
Wang X, Angelis N, Thein SL. MYB—a regulatory factor in hematopoiesis. Gene. 2018;665:6–17.
Uttarkar S, Dasse E, Coulibaly A, Steinmann S, Jakobs A, Schomburg C, et al. Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/p300 interaction. Blood. 2016;127:1173–82.
Walf-Vorderwulbecke V, Pearce K, Brooks T, Hubank M, van den Heuvel-Eibrink MM, Zwaan CM, et al. Targeting acute myeloid leukemia by drug-induced c-MYB degradation. Leukemia. 2017;32:882–889.
Uttarkar S, Piontek T, Dukare S, Schomburg C, Schlenke P, Berdel WE, et al. Small-molecule disruption of the Myb/p300 cooperation targets acute myeloid leukemia cells. Mol Cancer Ther. 2016;15:2905–15.
Hellsten R, Johansson M, Dahlman A, Dizeyi N, Sterner O, Bjartell A. Galiellalactone is a novel therapeutic candidate against hormone-refractory prostate cancer expressing activated Stat3. Prostate. 2008;68:269–80.
Quintas-Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013;19:1933–40.
Don-Doncow N, Escobar Z, Johansson M, Kjellstrom S, Garcia V, Munoz E, et al. Galiellalactone is a direct inhibitor of the transcription factor STAT3 in prostate cancer cells. J Biol Chem. 2014;289:15969–78.
Cercek A, Wheler J, Murray PE, Zhou S, Saltz L. Phase 1 study of APTO-253 HCl, an inducer of KLF4, in patients with advanced or metastatic solid tumors. Invest New Drugs. 2015;33:1086–92.
Sakamoto KM, Grant S, Saleiro D, Crispino JD, Hijiya N, Giles F, et al. Targeting novel signaling pathways for resistant acute myeloid leukemia. Mol Genet Metab. 2015;114:397–402.
Mitton B, Chae HD, Hsu K, Dutta R, Aldana-Masangkay G, Ferrari R, et al. Small molecule inhibition of cAMP response element binding protein in human acute myeloid leukemia cells. Leukemia. 2016;30:2302–11.
Mitton B, Hsu K, Dutta R, Tiu BC, Cox N, McLure KG, et al. Small molecule screen for inhibitors of expression from canonical CREB response element-containing promoters. Oncotarget. 2016;7:8653–62.
Peng H, Wang H, Xue P, Hou Y, Dong J, Zhou T, et al. Suppression of NRF2-ARE activity sensitizes chemotherapeutic agent-induced cytotoxicity in human acute monocytic leukemia cells. Toxicol Appl Pharmacol. 2016;292:1–7.
Karathedath S, Rajamani BM, Musheer Aalam SM, Abraham A, Varatharajan S, Krishnamurthy P, et al. Role of NF-E2 related factor 2 (Nrf2) on chemotherapy resistance in acute myeloid leukemia (AML) and the effect of pharmacological inhibition of Nrf2. PLoS One. 2017;12:e0177227.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SSK receives consultant fees from Pfizer and Ono Pharmaceutical.
About this article

Cite this article
Takei, H., Kobayashi, S.S. Targeting transcription factors in acute myeloid leukemia. Int J Hematol 109, 28–34 (2019). https://doi.org/10.1007/s12185-018-2488-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-018-2488-1