
Abstract
Mouse hepatitis virus (MHV) infection causes meningoencephalitis by disrupting the neuro-glial and glial-pial homeostasis. Recent studies suggest that MHV infection alters gap junction protein connexin 43 (Cx43)-mediated intercellular communication in brain and primary cultured astrocytes. In addition to astrocytes, meningeal fibroblasts also express high levels of Cx43. Fibroblasts in the meninges together with the basal lamina and the astrocyte endfeet forms the glial limitans superficialis as part of the blood–brain barrier (BBB). Alteration of glial-pial gap junction intercellular communication (GJIC) in MHV infection has the potential to affect the integrity of BBB. Till date, it is not known if viral infection can modulate Cx43 expression and function in cells of the brain meninges and thus affect BBB permeability. In the present study, we have investigated the effect of MHV infection on Cx43 localization and function in mouse brain meningeal cells and primary meningeal fibroblasts. Our results show that MHV infection reduces total Cx43 levels and causes its intracellular retention in the perinuclear compartments reducing its surface expression. Reduced trafficking of Cx43 to the cell surface in MHV-infected cells is associated with loss functional GJIC. Together, these data suggest that MHV infection can directly affect expression and cellular distribution of Cx43 resulting in loss of Cx43-mediated GJIC in meningeal fibroblasts, which may be associated with altered BBB function observed in acute infection.
Similar content being viewed by others


Introduction
The meninges are formed by three tissue membranes that primarily provide protective covering to the central nervous system (CNS). They comprise of the outer dura matter, middle arachnoid matter, and inner pia matter that consist of different cell populations including the fibroblasts, mast cells, endodermal cells, and smooth muscle cells [1]. The arachnoid and the pia matter, together called as leptomeninges, harbor a large population of fibroblasts [2]. Meningeal fibroblasts are known to have important role in blood–brain barrier (BBB) formation, glial scar formation during injury response, angiogenesis, and early astrocyte activation [3, 4]. In adult CNS, the meninges cover and penetrate the neural tissue deeply at every level of its organization: as large projections between the major brain structures, as stroma of the choroid plexus, and as sheaths of blood vessels forming the perivascular space. They also form the roof of the lateral ventricles, the third and the fourth ventricles. Their widespread distribution and complex organization suggest a more important role of the meninges as modulator of brain function in addition to being a mere protective covering of the brain. A further indication that the meninges are functionally linked to the neural tissue is the presence of gap junction proteins called connexins (Cxs).
Gap junctions (GJs) are intercellular channels that allow exchange of small molecules (less than 1 kDa) including small glucose derivatives [5], and second messengers such as ATP, cyclic AMP, inositol 1,4,5-trisphosphate, and Ca+2 [6,7,8] between coupled cells. GJs are composed of two hemichannels each contributed by the two opposing cells. The hemichannels or connexons are formed by six Cx protein subunits which after synthesis and oligomerization in endoplasmic reticulum/endoplasmic reticulum Golgi intermediate complex are trafficked to the cell surface where they are mostly assembled into gap junction plaques. Cx proteins, mainly Cx43, Cx30, and Cx26, have been found in the meninges and in their projections into the brain, including meningeal sheaths of blood vessels and stroma of the choroid plexus [9,10,11,12,13]. The distribution of these proteins suggests the existence of anatomical and functional interactions between meningeal cells, meningeal perivascular cells, ependymocytes, and astrocytes, thus providing a rapid means to spread signals in physiological and pathological events.
Cx43, the most abundantly expressed Cx in the CNS, is also highly expressed by the meningeal fibroblasts [13]. Recent findings suggest that Cx43-mediated functional coupling between the astrocytes and in the panglial system is altered in various neurodegenerative conditions like Alzheimer’s disease, Parkinson’s disease, ischemia, and multiple sclerosis (MS) [14,15,16,17]. Additionally, neuroinfections caused by Borna virus, bovine papillomavirus, Rous sarcoma virus, and human influenza virus can alter Cx-mediated gap junction communication [18,19,20,21,22]. Our previous studies have demonstrated that mouse hepatitis virus (MHV) infection significantly reduces Cx43 expression and functional gap junction formation in mice brain and in cultured astrocytes [23]. MHV-A59, a neurotropic strain of the coronavirus, causes meningitis and encephalitis in acute infection followed by demyelination and concurrent axonal loss in chronic stage of infection, serving as a model to study certain pathology of virus-induced meningitis and the human CNS-demyelinating disease MS [24, 25]. Although earlier reports suggest that MHV infection causes loss of Cx43 expression and negatively affects gap junction communication in the brain, particularly in astrocytes, at present, very little is known about virus-induced alterations of Cx channels in meningeal fibroblasts. This is particularly important in injury-induced brain parenchymal reaction, where GJs in the astrocyte–fibroblast interface play an important role in reforming glial limitans that forms a part of the BBB. Moreover, Cx proteins of BBB have been shown to regulate BBB permeability in inflammatory conditions [26]. In the present study, we investigated the expression and cellular distribution of Cx43 in MHV-A59-infected mouse brain meningeal cells. In parallel, we have established an in vitro model of viral meningitis by infecting fibroblast-enriched primary meningeal cultures with MHV to understand the effect of viral infection on Cx43-mediated gap junction communication in meningeal fibroblasts. Our findings provide important insights in understanding the role of glial-pial GJIC in altered barrier function of the inflamed brain.
Materials and Methods
Virus
A neurotropic demyelinating strain of mouse hepatitis virus, MHV-A59, was used from our previous studies [27], for infecting mice and primary meningeal cultures to study the effect on gap junction protein Cx43.
Inoculation of Mice
Four-week old C57BL/6 mice were intracranially inoculated with 50% LD50 dose of a neurotrophic demyelinating strain of mouse hepatitis virus, MHV-A59 (2000 pfu) as described previously [23]. Mice were monitored daily for symptoms of disease and mortality. Mock-infected controls were inoculated similarly with the vehicle containing phosphate-buffered saline (PBS) containing 0.75% bovine serum albumin (BSA) and maintained in parallel. All animals were euthanized at day 5 post-infection (p.i.) and brain tissues were harvested as described below. All experimental procedures were approved by the Institutional Animal Ethical Committee and Committee for the Purpose of Control and Supervision on Experiments on Animals (CPCSEA, India).
Histopathological Analysis
Mice were perfused transcardially with PBS followed by PBS containing 4% paraformaldehyde (PFA) at day 5 p.i. Brain tissue was collected, post-fixed in 4% PFA overnight, and embedded in paraffin [28]. Brains were sectioned at 5 μm and stained with hematoxylin and eosin (H&E) to evaluate inflammation or meningitis. The paraffin-embedded slides were first deparaffinized on a hot plate followed by xylene treatment for 10 mins. Sections were then rehydrated gradually through different alcohol grades from 100 to 50%. Sections were then treated with hematoxylin for 1 min and excess stain was washed under running tap water. Subsequently, sections were dehydrated by passing through 50 to 95% ethanol. Sections were then stained with eosin stain for 30 s and washed with 95% ethanol twice. Further dehydration was carried out with 100% ethanol and xylene. Sections were mounted with Refrax mounting medium (Anatech Ltd., MI, USA) and observed under the upright light microscope (Nikon Eclipse 50i) and analyzed with Nikon imaging software (NIS, Nikon Corp. Tokyo, Japan).
Immunofluorescence and Confocal Microscopy of Brain Cryosections
Mock and MHV-infected mice at day 5 p.i. were transcardially perfused and fixed with 4% PFA. Brain tissues were further post-fixed in 4% PFA overnight and isotonically equilibrated in 30% PBS sucrose. Tissues were embedded in OCT cryomatrix (Tissue Tek, Hatfield, PA) and sagittal sections (10 μm) were done on cryostat (Thermo Scientific). Tissue sections were mounted on charged slides and processed as described earlier [23]. Briefly, sections were incubated with 1 M glycine in PBS for 1 h at room temperature (RT) to reduce non-specific cross-linking and subsequently treated with 1 mg/ml NaBH4 in PBS for 10 min at RT to reduce autofluorescence. Sections were washed in PBS and incubated with blocking serum containing PBS with 0.5% Triton X-100 and 2% normal goat serum. Sections were incubated overnight at 4 °C with anti-Cx43 in combination with either anti-vimentin or anti-viral nucleocapsid (N) antisera prepared in blocking serum at dilutions listed in Table 1. The following day, the sections were washed and incubated in appropriate FITC or Texas Red conjugated fluorescent secondary antibody (1:200; Jackson ImmunoResearch Inc., PA, USA) for 1 h at RT. All incubations were done in humid chambers. Slides were washed and mounted in Vectashield® mounting media containing DAPI (Vector Laboratories, CA, USA). Sections were visualized and imaged using a Zeiss® confocal microscope (LSM710; Carl Zeiss AG, Germany).
Primary Meningeal Culture
Primary meningeal cultures from day 0–1 C57BL/6 mouse pup brains were prepared as described earlier, with minor modifications [29]. Briefly, the meninges were carefully removed, homogenized in Hank’s Balanced Salt solution (HBSS), and passed through a 70-μm cell strainer. The resulting flow-through were collected and centrifuged at 1000 rpm for 10 mins to pellet the meningeal cells. The cell pellet was washed twice with HBSS and re-suspended in growth medium containing Dulbecco’s minimal essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% l-glutamine, and 1% penicillin–streptomycin. The cells were plated and allowed to grow at 37 °C in a humidified CO2 incubator. After 72 h, all non-adherent cells were removed and fresh medium was added. Adherent cells were maintained as meningeal fibroblast culture in growth medium till confluency with a media change every 2–3 days.
Mixed Glia Cultures
Primary cultures of mixed glia from newborn (day 0–1) C57BL/6 mouse pup brains were prepared as described earlier [23]. Briefly, the meninges were removed and brain tissue was minced and incubated in a shaking water bath at 37 °C for 30 min in HBSS containing 300 mg/ml DNase I (Sigma®) and 0.25% trypsin (Sigma®). Enzyme dissociated cells were triturated with 0.25% of FBS, washed and pelleted at 300×g for 10 min. The pellet was resuspended in HBSS and passed through a 70-μm strainer and centrifuged at 300×g for 10 min. The pellet was resuspended in astrocyte-specific medium (DMEM containing 1% l-glutamine, 10% FBS, and 1% penicillin–streptomycin), plated in T25 flasks, and allowed to grow at 37 °C in a humidified CO2 incubator. After 24 h, all non-adherent cells were removed and fresh astrocyte-specific medium was added. Adherent cells were allowed to grow to confluence with a media change every 3 to 4 days.
Flow Cytometry
Confluent cultures were trypsinized and washed twice in flow buffer (PBS containing 2% FBS). Following centrifugation (300×g, 5 min), the cell pellet was resuspended in flow buffer and approximately 1 × 106 cells were distributed per polystyrene round-bottom 5-ml flow tubes. Cells were fixed in 100 μl Cyto-Fix buffer (BD Biosciences, San Jose, CA) for 15 min at RT. Following a wash in flow buffer, cells were incubated with anti-vimentin and anti-glial fibrillary acidic protein (GFAP) antibody diluted (1:40) in 1× BD Perm/Wash Buffer for 30 mins at RT. Cells were washed, centrifuged, and labeled with goat anti-mouse TRITC and goat anti-rabbit FITC diluted (1:40) in BD Perm/Wash buffer and incubated for 30 mins at RT. After a final wash, all tubes were resuspended in 500 mL of flow buffer and subjected to flow cytometry in a BD FACS Verse™ flow cytometer and analyzed using BD FACSuite™ software [30].
Infection of Primary Meningeal Culture
Primary meningeal fibroblasts were infected with MHV-A59 for investigating the effect of virus infection on meningeal gap junction protein Cx43, following previously published protocols [23]. In brief, meningeal cultures were infected with MHV-A59 diluted in inoculation medium (DMEM containing 2% FBS and 1% penicillin–streptomycin) at a multiplicity of infection of 2 and incubated for 1 h at 37 °C in a humidified CO2 chamber. After initial viral adsorption for 1 h, medium was changed and infected cells were maintained in normal growth medium for 24 h. For mock infection, parallel cultures were initially incubated in the inoculation medium for 1 h followed by normal growth medium. All cells were collected 24 h p.i. and used for immunolabeling and biochemical assays.
Immunofluorescence and Confocal Microscopy
Immunofluorescence staining of primary meningeal fibroblasts was performed as described previously [31], with minor modifications. In brief, cells fixed with 4% PFA for 15 mins at RT were permeabilized in PBS containing 0.5%Triton X-100 and incubated in blocking serum (PBS containing 0.2%TritonX-100 and 2.5% goat serum) for 1 h. Cells were incubated for 1 h at RT with a combination of anti-Cx43, anti-vimentin, anti-N anti-GFAP, and anti-calnexin antisera at dilutions listed in Table 1. Subsequently, the cells were washed and incubated for 1 h at RT with appropriate combination of FITC or Texas Red conjugated secondary antibody (1:200) diluted in blocking serum. Cells were washed and mounted in Vectashield mounting media containing DAPI. Cells were visualized in Zeiss® Confocal Microscope (LSM710) or Axio observer microscope with apoptome module (Carl Zeiss, Germany) and images were acquired and processed with Zen 2012 (Carl Zeiss AG, Germany) software.
Western Blotting
Mock and virus-infected cells (24 h p.i.) were washed with PBS and homogenized in ice-cold protein extraction buffer (25 mM Tris (pH 7.6), 1 mM MgCl2, 1% Triton X-100, 0.5% SDS with 1X EDTA-free complete Protease Inhibitor (Roche, Mannheim, Germany) and phosphatase inhibitors (1 mM NaVO4 and 10 mM NaF)) by mild vortexing at regular time interval for 1 h. Lysed cell suspension was centrifuged at 4 °C for 10 min at 13,200 rpm using an Eppendorf® 5415 R centrifuge. The resulting supernatant was quantified for total protein concentration using a Pierce® BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL, USA). Equal amount (5 μg) of total protein was resolved by 12% SDS-PAGE [32]. Proteins were transferred to PVDF membrane in transfer buffer (25 mM Tris, 192 mM glycine, and 20% methanol). Membrane was blocked with 5% skimmed milk in Tris-buffered saline containing 0.1% v/v Tween-20 (TBST) for 1 h at RT and incubated overnight at 4 °C with anti-Cx43 and anti-vimentin antibodies at dilutions listed in Table 1. Membranes were then washed with TBST and incubated with appropriate horseradish peroxidase-conjugated secondary antibody (1:2000; Jackson ImmunoResearch Inc., PA, USA) for 2 h at RT. Membranes were developed using Super Signal West Pico chemiluminescent substrate (Thermo Fisher Scientific, Rockford, IL, USA) and images were captured using Syngene G: Box™ ChemiDoc system and GENEsys software. Membranes were subsequently re-probed with γ-actin antibody and densitometric analysis of immunoreactive bands was carried out using ImageJ software.
Lucifer Yellow Scrape-Loading and Dye Transfer Assay
To evaluate gap junction coupling, meningeal fibroblasts were grown on 35-mm culture dishes to 100% confluency. Permeability mediated by gap junctions under control and MHV-A59-infected conditions (24 h p.i.) was determined by gap junction permeable Lucifer yellow (LY) dye transfer experiment, using a protocol described previously [33], with minor modifications. LY dye (Sigma, Saint Louis, MO, USA) dissolved in PBS (4 mg/ml) was scrape loaded onto confluent monolayer of meningeal fibroblasts. After 1 min of incubation, LY was thoroughly washed with PBS, fresh 10% DMEM was added, and dye spreading from scrape line was imaged using an Olympus IX-81 microscope system appended with a Hamamatsu Orca-1 CCD camera. Images were processed and analyzed using ImagePro (Media Cybernetics) and ImageJ software and the distance of dye spread from the scrape-loading line was measured.
In Situ TritonX-100 Solubilization
Cells plated on coverslips were washed with PBS and treated with 1%Triton X-100 at 4 °C for 30 mins to solubilize the Cx43 molecules not involved in gap junction plaques while the Cx43 molecules involved in cell surface gap junction plaques remain insoluble. Upon thorough washing and extraction of solubilized Cx43 molecules, cells were fixed and immunofluorosence labelling was performed as described previously [31]. Cells were visualized in Axio observer microscope with apoptome module (Carl Zeiss, Germany) and images acquired and processed with Zen 2012 (Carl Zeiss AG, Germany) software.
Statistical Analysis
Data are presented as mean ± SEM. Significance of the difference between two experiment groups for the LY dye transfer assay and western blotting was determined by unpaired two-tailed Student’s t test. The data from cell enrichment experiment were analyzed by one-way analysis of variance, and pairwise comparisons were made using the post hoc Tukey method for multiple testing. Statistical significance was set at p < 0.05. All statistical analysis were carried out using GraphPad prism (ver. 7) software (GraphPad Software, Inc).
Results
Altered Cx43 Distribution and Inflammation of the Meninges in MHV-A59-Infected Mouse Brains
Four-week old C57BL/6 mice were intracranially inoculated with MHV-A59 or mock-infected with PBS-BSA as controls. Mice were sacrificed on day 5 p.i., the time point previously reported to result in peak CNS inflammation [25]. Paraffin-embedded brain sections were stained with hematoxylin and eosin to visualize inflammation in different brain regions (Fig. 1a–f). The mock-infected mouse brain sections showed no signs of inflammation and exhibited intact meningeal layer lining the cortex and choroid plexus in the ventricular spaces (Fig. 1a–e). In contrast, acute meningitis and inflammatory tissue damage was evident in the corresponding brain regions from MHV-A59-infected mice (Fig. 1b–f).

MHV-A59 infection causes meningitis, choroid plexitis, and ependymitis in the brain of C57BL/6 mice (a–f). Representative images of the cortical meningeal lining (a, b) and choroid plexus in the fourth ventricle (c, d) and lateral-ventricular region (e, f) from mock (a, c, e) and MHV-A59-infected (b, d, f) mice at day 5 p.i. stained with hematoxylin and eosin. Control mice show no sign of inflammation. Inflammation was noted in the cortical meningeal covering (b), choroid plexus (d), and ependyma (f) in infected mouse brains compared to control. Original images have been taken at magnifications of × 40 (a–d) and × 10 (e, f). Arrows in b, d, and f indicate inflamed leptomeninges, choroid plexus, and ependyma in MHV-A59-infected brain sections
Previous studies suggested that the meningeal cells express high levels of Cx43 and are tightly coupled by gap junctions both in developing and adult brain [13]. Moreover, Cx-mediated gap junction communications are known to be affected in several CNS-associated injuries and pathologies. Therefore, to investigate the effect of viral infection on Cx43 expression and distribution in the infected meningeal cells of MHV-A59-infected mouse brains, we performed double-immunolabeling with a combination of anti-Cx43 and anti-viral nucleocapsid (N; Fig. 2a–l) or anti-vimentin antibody (Fig. 3a–l). Our results clearly showed that MHV-A59 infection altered the expression pattern of Cx43 in infected meningeal cells. Interestingly, we observed a marked downregulation in the overall Cx43 staining in infected and vimentin-positive cells of the meninges (Fig. 2a–f, Fig. 3a–f), choroid plexus (Fig. 2g–l), and perivascular region (Fig. 3g–l) in infected brains compared to controls. Additionally, while the control brain sections showed a characteristic punctate distribution of Cx43 (Fig. 2b, h), the infected and vimentin-positive meningeal fibroblasts in the infected mouse brains showed loss of its characteristic punctate staining (Fig. 2e, k).

Reduced Cx43 punctate staining in MHV-A59-infected brain cryosections compared to mock-infected control mice brain section (a–f). Confocal images of 10-μm brain cryosections from the cortical meningeal region of mock-infected control (a–c) and MHV-A59-infected (d–f) brain sections double-immunolabeled with anti-viral nucleocapsid (N, green) and anti-Cx43 (red) showing reduced Cx43 punctate staining. Control sections (b, c) displaying a characteristic punctate Cx43 staining (b, c: arrow and inset), while the infected brains (e, f) exhibiting diffused Cx43. Merged image (f: arrow) showing an infected cell with internalized Cx43 staining in perinuclear region; inset showing an enlarged view of the infected cell. (g–l) Confocal images of 10-μm thin brain cryosections from the choroid plexus region of mock-infected control (g–i) and MHV-A59-infected (j–l) brain sections double-immunolabeled with anti-viral nucleocapsid (N, green) and anti-Cx43 (red) showing reduced Cx43 punctate staining. Control sections showing a characteristic punctate Cx43 honeycomb like pattern (h, i: arrow and inset), which appear as diffused staining in infected brains (k, l). Merged image (l: arrow) showing infected cells with reduced Cx43 punctate staining; inset showing an enlarged view of the infected cell. No anti-viral N staining was noted in the control brain sections (a–c, g–i). All sections were counterstained with DAPI (blue) to visualize nuclear localisation

Confocal images showing reduced Cx43 punctate staining in vimentin-positive fibroblasts in the meninges and perivascular region (a–l). Confocal images of 10-μm thin brain cryosections double-immunolabeled with anti-vimentin (red) and anti-Cx43 (green) showing reduced Cx43 staining in the inflamed cortical meningeal layer and perivascular region of MHV-A59-infected (d–f, j–l) mouse brains compared to mock-infected controls (a–c, g–i). Sections were counterstained with DAPI (blue) to visualize nuclear localisation. Meningeal fibroblasts in the cortical and perivascular region of MHV-infected brains (d–f, j–l: arrow) depict reduction in Cx43 immunostaining compared to respective mock-infected brain regions (a–c, g–i: arrow). Insets in f and l demonstrate reduced Cx43 immunoreactivity in a magnified view of fibroblast. Note the higher intensity of Cx43 staining is noted in vimentin-positive fibroblasts in representative brain images from the mock-infected cortical meningeal layer and perivascular region (a, c and g, i)
Retention of Cx43 in Perinuclear Compartments of Primary Meningeal Cultures Infected with MHV-A59
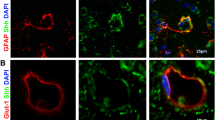
To determine the mechanisms by which viral infection can modulate Cx43-mediated gap junction communication particularly in the meningeal cells, we established a complimentary in vitro model of primary meningeal cultures infected with MHV-A59. Purity of the primary cultures was first determined by flow cytometry, using vimentin as fibroblast marker [1] and GFAP as astrocyte marker (Fig. 4a). Viable cell population (P1) was normalized for each marker expression with isotype controls. As expected, majority of the cells (92.4%) in our established cultures were vimentin-positive meningeal fibroblasts. In addition, a small population (0.04%) of the cells were found to be GFAP-positive, and a few (1.83%) were positive for both GFAP and vimentin and probably represents stem/progenitor cells with a potential to differentiate to either astrocytes or fibroblasts at a later stage in culture. Supporting this, our double-immunostaining of primary meningeal cultures with anti-GFAP and anti-vimentin antibodies also showed that most of the cells in cultures expressed vimentin only (Fig. 4b, arrows). However, a few GFAP-positive and GFAP and vimentin dual positive cells were also observed. Visual counting of the cells from three independent cultures (with 30 different fields from each set) demonstrated that about 74.83% cells were positive for vimentin only, while 4.6% cells expressed GFAP only and 11.7% stained for both markers (Fig. 4c). Additionally, we also quantified the vimentin expression in our primary meningeal cultures. Our immunoblotting experiments clearly showed that the primary meningeal cultures expressed a significantly higher level of the fibroblast marker vimentin compared to mixed glial cultures demonstrating the enrichment for meningeal fibroblasts in the primary cultures (Fig. 4d).

Characterization of primary meningeal fibroblasts by flow cytometry and immunofluorescence. a Flow cytometric analysis of primary meningeal cultures double-immunolabeled with anti-vimentin and anti-GFAP antisera. Gating (P1) of cell population is carried out from side scattering (SSC) vs forward scattering (FSC) plot of all recorder events. Majority of the gated cells (92.4%) in cultures were vimentin-positive and GFAP-negative, as seen in the lower right (LR) quadrant of the gated plot (P1). Histograms (lower panels) depict higher mean intensity of vimentin expression compared to that of GFAP. b Photomicrographs of primary meningeal culture cells double-immunolabeled for anti-vimentin (red) and anti-GFAP (green) antibodies. Cells were counterstained with DAPI (blue) to visualize nuclear staining. Arrows indicate vimentin-positive meningeal fibroblasts. c Histogram represents quantitation of the double-immunolabeled cells showing around 74.83% cells are positive for vimentin only, whereas 11.7% cells are positive for both markers and about 4.6% are GFAP only. The values are mean ± SEM from three independent experiments (having 30 different fields each) for the mentioned types of cells (one-way ANOVA; ****p < 0.0001). d Representative immunoblot showing higher level of vimentin expression in primary meningeal culture compared to mixed glial culture. All blots were re-probed with γ-actin to monitor equal protein loading
To determine if viral infection can alter cellular expression/distribution of the Cx43 in primary meningeal fibroblasts, we infected the primary meningeal cultures with MHV-A59, a neurotropic demyelinating strain of mouse hepatitis virus [27]. At 24 h p.i., cells were double-labeled with a combination of anti-Cx43 and anti-N or anti-vimentin antibodies (Fig. 5a–l). In the mock-infected meningeal fibroblast cultures, vimentin positive cells expressed profuse Cx43 at the cell surface, demonstrated by its characteristic punctate staining (Fig. 5a–c, g–i; arrow and inset). Interestingly, in MHV-A59-infected cultures, vimentin-positive fibroblasts displayed Cx43 staining predominantly localized in the intracellular compartments with a typical perinuclear distribution (Fig. 5d–f, j–l; arrow and inset). Interestingly, most of these intracellular Cx43 puncta in MHV-infected cultures was found to co-localize with the endoplasmic reticulum marker, calnexin (Fig. 6a–f).

MHV-A59 infection reduced Cx43 expression at cell surface and retained Cx43 in the perinuclear region of infected primary meningeal fibroblasts (a–l) Double-immunolabeling for Cx43 (green) and vimentin or viral nucleocapsid (red) showing punctate staining of Cx43 mostly at the cell surface of mock-infected vimentin-positive primary meningeal fibroblasts (a–c, g–i). Virus-infected and vimentive-positive fibroblasts show a predominant intracellular localization of Cx43 (d–f, j–l; arrows). Note the colocalization of Cx43 with viral nucleocapsid in infected cells (f: arrow) surrounding the nucleus. Perinuclear distribution of Cx43 in MHV-A59-infected cultures is also evident in vimentin-positive meningeal fibroblasts (l: arrow). Insets depict magnified view showing Cx43 perinuclear staining in infected (f, l) cells and characteristic punctate cell surface localization in mock-infected controls (c, i). Cells were counterstained with DAPI (blue) to visualize nuclear localization in merged images

Intracellular retention of Cx43 in endoplasmic reticulum of MHV-A59-infected primary meningeal fibroblast cultures (a–f). Meningeal fibroblasts were mock-infected (a–c) or infected with MHV-A59 (d–f), and double-immunostained for calnexin (green: b, e) and Cx43 (red: a, d). In infected cultures (d–f), Cx43 puncta are mostly seen in the perinuclear region which colocalize with ER marker calnexin (f; arrows). In control cells (a–c), most of the Cx43 staining is observed towards the periphery (c; arrowheads) with only a few Cx43 puncta seen to colocalize with calnexin (c; arrow). Cells were counterstained with DAPI (blue) to visualize nuclear localization in merged images
Reduced Cx43 Expression and Functional Gap Junction Communication in Infected Primary Meningeal Cultures
Earlier studies have shown that MHV-A59 infection can reduce Cx43 protein levels in the CNS and mouse primary astrocytes [23]. Moreover, our immunofluorescence studies also suggest a possible reduction in total Cx43 levels in vimentin-labeled cells of virus-infected mouse brains and altered cellular distribution in primary meningeal fibroblasts. To quantify the observed change, we measured total Cx43 levels in primary meningeal cultures infected with MHV-A59. Cx43 levels were found to be significantly downregulated in the infected cultures compared to control (− 50.50 ± 10.77%; p < 0.01) (Fig. 7a; lower panel). To determine if reduced Cx43 expression together with its increased intracellular retention affected functional gap junction communication in infected meningeal cells, we performed LY dye transfer in a scrape-loading assay. Cx43 channels are permeable to LY, a small molecular dye that moves from cells to the neighboring cells via gap junctions [34]. Primary meningeal fibroblasts were either mock-infected or infected with MHV-A59 and gap junction activity was assayed by scrape-loading dye transfer 24 h p.i. MHV-A59 infection prevented cultured meningeal fibroblasts from transferring the dye to neighboring cells, as the distance of dye spread beyond the injured cells at the scratch boundary was significantly less compared to the control cultures (Fig.7b). Control meningeal fibroblasts exhibited active gap junction communication, while infected fibroblasts displayed less gap junction communication, as evident from shorter dye travel distance (control: 287.5 ± 24.35 μm vs. infected: 139.8 ± 5.19 μm; p < 0.0001) (Fig. 7b; lower panel).

MHV-A59 infection reduced Cx43 expression at total protein level and decreased functional gap junctional communication. a Immunoblot showing a reduction in total Cx43 expression in primary meningeal fibroblast cultures infected with MHV-A59 at MOI of 2 and 24 h p.i. compared to mock-infected controls. Blots were re-probed with γ-actin to monitor equal protein loading. Histogram (lower panel) depicts percentage values (mean ± SEM) of Cx43 protein expression (Student’s t test; **p < 0.01). b Representative photomicrograph and histogram depicting a significant reduction in functional gap junction communication in primary meningeal fibroblast cultures infected with MHV-A59 compared to control as detected by LY dye transfer in a scrape-loading assay. Arrow indicates the site of scrape loading with LY and distance of dye spread. Histogram (lower panel) represents quantitation of data from three independent experiments. Values are mean ± SEM (Student’s t test; ****p < 0.0001)
Reduced Gap Junction Plaques in Infected Primary Meningeal Cultures
Musil and Goodenough demonstrated that Cx43 assembled into the gap junction plaques are resistant to solubilization by 1% Triton X-100 at 4 °C, while Cx43 monomers are mostly solubilized under such conditions [35]. In order to understand the assembly of Cx43 into plaques, mock- and MHV-A59-infected cells were treated in situ with 1% Triton X-100 at 4 °C, 24 h p.i., and double immunofluorescence labelling was performed for anti-Cx43 and anti-vimentin (Fig. 8a–i) or anti-N (Fig. 8j–l). Interestingly, Triton X-100 solubilization resulted in a marked decrease in Cx43 staining in mock-infected cultures compared to non-Triton X-100-treated experimental controls (Fig. 8a–f). This might be because intracellular soluble monomers of Cx43 were extracted by Triton X-100 treatment leaving behind the Cx43 puncta that were assembled in gap junction plaques and resistant to Triton X-100 solubilization. In contrast, MHV-infected meningeal fibroblasts showed mostly intracellular accumulation of Cx43 in the perinuclear region (Fig. 8g–l; arrows in h, i, k, and l). This might be due to aggregation of misfolded protein or increased amount of prematurely oligomerized and Triton X-100-resistant Cx43 that failed to traffic to the cell surface to form gap junction plaques.

MHV-A59 infection increased retention of Triton X-100 insoluble intracellular aggregates of Cx43 (a–l). Representative confocal images of MHV-A59-infected (g–l) and mock-infected primary meningeal fibroblasts (a–f) double-immunolabeled with anti-Cx43 (red: b, e, h, k) and anti-vimentin (green: a, d, g) or anti-viral nucleocapsid (N; green: j). Control cells not subjected to Triton X-100 solubilization (−Triton X-100; a–c), exhibited characteristic punctate Cx43 immunostaining (b, c; arrow). Upon Triton X-100 solubilization (+TritonX-100; d–f), mock-infected control cells displayed a few Cx43 puncta representative of Triton X-100-insoluble gap junction plaques at the cell surface (e, f; arrow). In contrast, vimentin-positive primary fibroblasts in MHV-A59-infected cultures (g–i) displayed increased immunostaining for Cx43 mostly in the perinuclear regions following Triton X-100 solubilization (+Triton X-100) suggesting intracellular cytosolic aggregates of Cx43 (h, i; arrow). Anti-N staining illustrating that most of these intracellular cytosolic aggregates of Cx43 noted in +Triton X-100 condition are in MHV-infected cells (j–l). Cells were counterstained with DAPI (blue) to visualize nuclear localization
Discussion
Using mouse brain slices and cultured primary meningeal fibroblasts, we show that MHV-A59 can directly infect meningeal cells in vivo in mice as well as in culture conditions. Our results reveal that (i) MHV infection reduces total Cx43 protein levels in primary meningeal fibroblasts, (ii) majority of Cx43 in infected, viral antigen-positive fibroblasts show intracellular perinuclear staining, both in vivo and in vitro, (iii) MHV infection causes retention of Triton X-100 insoluble intracellular aggregates of Cx43 which may not be able to assemble into gap junctions, and (iv) the observed alterations in Cx43 expression and cellular distribution is associated with loss of functional gap junction communication between fibroblasts in primary culture as demonstrated by LY scrape-loading and dye transfer assay. Taken together, our results suggest that MHV-A59 infection can directly affect expression and intracellular distribution of Cx43 in meningeal fibroblasts which might play an important role in altered CNS homeostasis during acute infection.
Earlier studies suggest that MHV hijacks host translational machinery to produce its own proteins during viral multiplication thereby arresting the synthesis of large number of host cellular proteins [36]. Virus-induced host translational shutdown and mRNA decay can affect molecules particularly with shorter half-life such as Cx43 [37, 38] resulting in reduced Cx43 levels. In addition, it has been shown that ER stress-mediated unfolded protein response can result in downregulation of Cx43 expression at both mRNA and protein levels [39]. Thus, it is also possible that MHV-induced ER stress can lead to accumulation of misfolded Cx43 in the perinuclear compartments, subsequently leading to downregulation of Cx43 expression and reduced trafficking to cell surface. This is further supported by our Triton X-100 solubilization experiment showing that infected cells have increased intracellular accumulation of Cx43 in the perinuclear region that was not extracted by Triton X-100 treatment. In contrast, the control cells showed Cx43 puncta only at the cell surface present in gap junction plaques and no intracellular monomers that were supposedly removed by the Triton X-100.
Neurovirulent strains of MHV are preferentially transported by microtubules in neurons [40]. Interestingly, Cx43 is also known to directly bind to microtubules [41] and transported to the cell surface along microtubules as conduit [42]. Thus, virus transport using the cellular microtubule network might possibly hinder microtubule-dependent Cx43 trafficking to the cell surface. In this regard, our previous studies have shown that depolymerization of the microtubule network by colchicine in primary astrocytes can lead to similar retention of Cx43 in the perinuclear compartments as in virus-infected cells [23]. Thus, it is possible that virus-specific utilization of the microtubules in the infected meningeal fibroblasts resulted in reduced trafficking and assembly of Cx subunits into GJ plaques causing loss of gap junctional communication. Nevertheless, further studies are needed to understand the underlying regulatory mechanisms of Cx43 association with the microtubule network in infected meningeal cells and its oligomerization into gap junction plaques.
Impaired gap junction communication in meningeal fibroblasts can contribute to disruption of functional link to the neural tissue. It is reported that meningeal cells can communicate with astrocytes via Ca2+ signaling [43]. Interestingly, ablation of astrocytic Cx30 has been shown to cause nearly complete depletion of Cx26 immunoreactivity in leptomeninges [11]. Moreover, the meninges are important in maintaining the functional coupling among the glia limitans astrocytes, which are more coupled than the astrocytes in the molecular layer [44]. During injury-induced parenchymal reaction, reactive astrocytes and the meningeal cells form a glia–fibroblast interface that forms new basal lamina which along with astrocytic endfeet reforms the glial limitans [45, 46]. This process is crucial for restoring the BBB function and re-establishing CNS homeostasis. Thus, Cx channels play an important role in maintaining BBB function, and loss of these junctional communications might affect BBB permeability. The effect of Cx43 on BBB may be mediated via tight junction proteins such as claudin, occludin, and zona occludens (ZO-1). There are reports of Cx43 interacting with tight junction protein ZO-1 through its carboxyl terminus [47]. The PDZ domain of ZO-1 serves to recruit several signaling molecules and also provides anchorage to several cytoskeletal elements. Moreover, the dynamic association of ZO-1 with Cx43 also modulates the stability of gap junction channel and regulates its internalization and endosomal turnover [48]. ZO-1 and other tight junction proteins are reported to be downregulated in BBB leakage during different pathological conditions [49]. Hence, the virus-induced retention of Cx43 might alter the stability of membrane resident tight junction proteins and disrupt the BBB integrity.
Increased permeability of the BBB is a pathological hallmark in several neurological disorders such as multiple sclerosis, bacterial meningitis, and neurotropic virus infections including human immunodeficiency virus, measles virus, and Japanese encephalitis virus [50,51,52]. Experimental intracranial inoculation with MHV might increase BBB permeability, leading to rapid viral dissemination and enhanced transmigration of activated immune cells into the brain. Although the homing mechanism of immune cells and molecular mechanisms of BBB disruption in MHV infection are not well understood, viral infection-associated loss of Cx-mediated intercellular communication might play an important role in altered BBB permeability and altered CNS homeostasis during neuroinflammation.
In summary, we have shown that MHV infection in brain can reduce Cx43 expression in meningeal fibroblasts and induce intracellular retention of Cx43, which might contribute to reduced gap junction communication. Such loss of junctional communication might have a potential role in modulating coupling between fibroblasts of the meningeal layer and at fibroblast–astrocyte interface of glial limitans superficialis subsequently leading to altered BBB function.
References
Decimo I, Fumagalli G, Berton V, Krampera M, Bifari F (2012) Meninges: from protective membrane to stem cell niche. Am J Stem Cells 1(2):92–105
Weller RO (2005) Microscopic morphology and histology of the human meninges. Morphologie : bulletin de l'Association des anatomistes 89(284):22–34. https://doi.org/10.1016/S1286-0115(05)83235-7
Shearer MC, Niclou SP, Brown D, Asher RA, Holtmaat AJ, Levine JM, Verhaagen J, Fawcett JW (2003) The astrocyte/meningeal cell interface is a barrier to neurite outgrowth which can be overcome by manipulation of inhibitory molecules or axonal signalling pathways. Mol Cell Neurosci 24(4):913–925. https://doi.org/10.1016/j.mcn.2003.09.004
Wanner IB, Deik A, Torres M, Rosendahl A, Neary JT, Lemmon VP, Bixby JL (2008) A new in vitro model of the glial scar inhibits axon growth. Glia 56(15):1691–1709. https://doi.org/10.1002/glia.20721
Tabernero A, Giaume C, Medina JM (1996) Endothelin-1 regulates glucose utilization in cultured astrocytes by controlling intercellular communication through gap junctions. Glia 16(3):187–195
Charles AC, Naus CC, Zhu D, Kidder GM, Dirksen ER, Sanderson MJ (1992) Intercellular calcium signaling via gap junctions in glioma cells. J Cell Biol 118(1):195–201. https://doi.org/10.1083/jcb.118.1.195
Finkbeiner S (1992) Calcium waves in astrocytes-filling in the gaps. Neuron 8(6):1101–1108. https://doi.org/10.1016/0896-6273(92)90131-V
Saez JC, Connor JA, Spray DC, Bennett MV (1989) Hepatocyte gap junctions are permeable to the second messenger, inositol 1,4,5-trisphosphate, and to calcium ions. Proc Natl Acad Sci U S A 86(8):2708–2712. https://doi.org/10.1073/pnas.86.8.2708
Arishima H, Sato K, Kubota T (2002) Immunohistochemical and ultrastructural study of gap junction proteins connexin26 and 43 in human arachnoid villi and meningeal tumors. J Neuropathol Exp Neurol 61(12):1048–1055. https://doi.org/10.1093/jnen/61.12.1048
Mercier F, Hatton GI (2001) Connexin 26 and basic fibroblast growth factor are expressed primarily in the subpial and subependymal layers in adult brain parenchyma: roles in stem cell proliferation and morphological plasticity? J Comp Neurol 431(1):88–104
Lynn BD, Tress O, May D, Willecke K, Nagy JI (2011) Ablation of connexin30 in transgenic mice alters expression patterns of connexin26 and connexin32 in glial cells and leptomeninges. Eur J Neurosci 34(11):1783–1793. https://doi.org/10.1111/j.1460-9568.2011.07900.x
Nagy JI, Li X, Rempel J, Stelmack G, Patel D, Staines WA, Yasumura T, Rash JE (2001) Connexin26 in adult rodent central nervous system: demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J Comp Neurol 441(4):302–323. https://doi.org/10.1002/cne.1414
Spray DC, Moreno AP, Kessler JA, Dermietzel R (1991) Characterization of gap junctions between cultured leptomeningeal cells. Brain Res 568(1–2):1–14. https://doi.org/10.1016/0006-8993(91)91373-9
Lerner DL, Beardslee MA, Saffitz JE (2001) The role of altered intercellular coupling in arrhythmias induced by acute myocardial ischemia. Cardiovasc Res 50(2):263–269. https://doi.org/10.1016/S0008-6363(00)00301-1
Masaki K, Suzuki SO, Matsushita T, Matsuoka T, Imamura S, Yamasaki R, Suzuki M, Suenaga T et al (2013) Connexin 43 astrocytopathy linked to rapidly progressive multiple sclerosis and neuromyelitis optica. PLoS One 8(8):e72919. https://doi.org/10.1371/journal.pone.0072919
Nagy JI, Li W, Hertzberg EL, Marotta CA (1996) Elevated connexin43 immunoreactivity at sites of amyloid plaques in Alzheimer’s disease. Brain Res 717(1–2):173–178. https://doi.org/10.1016/0006-8993(95)01526-4
Rufer M, Wirth SB, Hofer A, Dermietzel R, Pastor A, Kettenmann H, Unsicker K (1996) Regulation of connexin-43, GFAP, and FGF-2 is not accompanied by changes in astroglial coupling in MPTP-lesioned, FGF-2-treated parkinsonian mice. J Neurosci Res 46 (5):606–617. doi:https://doi.org/10.1002/(SICI)1097-4547(19961201)46:5<606::AID-JNR9>3.0.CO;2-N
Crow DS, Beyer EC, Paul DL, Kobe SS, Lau AF (1990) Phosphorylation of connexin43 gap junction protein in uninfected and Rous sarcoma virus-transformed mammalian fibroblasts. Mol Cell Biol 10(4):1754–1763. https://doi.org/10.1128/MCB.10.4.1754
Faccini AM, Cairney M, Ashrafi GH, Finbow ME, Campo MS, Pitts JD (1996) The bovine papillomavirus type 4 E8 protein binds to ductin and causes loss of gap junctional intercellular communication in primary fibroblasts. J Virol 70(12):9041–9045
Fatemi SH, Folsom TD, Reutiman TJ, Sidwell RW (2008) Viral regulation of aquaporin 4, connexin 43, microcephalin and nucleolin. Schizophr Res 98(1–3):163–177. https://doi.org/10.1016/j.schres.2007.09.031
Koster-Patzlaff C, Hosseini SM, Reuss B (2007) Persistent Borna disease virus infection changes expression and function of astroglial gap junctions in vivo and in vitro. Brain Res 1184:316–332. https://doi.org/10.1016/j.brainres.2007.09.062
Orellana JA, Saez JC, Bennett MV, Berman JW, Morgello S, Eugenin EA (2014) HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J Neurochem 128(5):752–763. https://doi.org/10.1111/jnc.12492
Basu R, Banerjee K, Bose A, Das Sarma J (2015) Mouse hepatitis virus infection remodels connexin43-mediated gap junction intercellular communication in vitro and in vivo. J Virol 90(5):2586–2599. https://doi.org/10.1128/JVI.02420-15
Lavi E, Gilden DH, Wroblewska Z, Rorke LB, Weiss SR (1984) Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34(5):597–603. https://doi.org/10.1212/WNL.34.5.597
Das Sarma J (2010) A mechanism of virus-induced demyelination. Interdiscip Perspect Infect Dis 2010:109239–109228. https://doi.org/10.1155/2010/109239
De Bock M, Vandenbroucke RE, Decrock E, Culot M, Cecchelli R, Leybaert L (2014) A new angle on blood-CNS interfaces: a role for connexins? FEBS Lett 588(8):1259–1270. https://doi.org/10.1016/j.febslet.2014.02.060
Das Sarma J, Fu L, Tsai JC, Weiss SR, Lavi E (2000) Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J Virol 74(19):9206–9213. https://doi.org/10.1128/JVI.74.19.9206-9213.2000
Chatterjee D, Biswas K, Nag S, Ramachandra SG, Das Sarma J (2013) Microglia play a major role in direct viral-induced demyelination. Clin Dev Immunol 2013:510396–510312. https://doi.org/10.1155/2013/510396
Struckhoff G (1995) Cocultures of meningeal and astrocytic cells—a model for the formation of the glial-limiting membrane. Int J Dev Neurosci: Off J Int Soc Dev Neurosci 13(6):595–606. https://doi.org/10.1016/0736-5748(95)00040-N
Marek R, Caruso M, Rostami A, Grinspan JB, Das Sarma J (2008) Magnetic cell sorting: a fast and effective method of concurrent isolation of high purity viable astrocytes and microglia from neonatal mouse brain tissue. J Neurosci Methods 175(1):108–118. https://doi.org/10.1016/j.jneumeth.2008.08.016
Das Sarma J, Meyer RA, Wang F, Abraham V, Lo CW, Koval M (2001) Multimeric connexin interactions prior to the trans-Golgi network. J Cell Sci 114(Pt 22):4013–4024
Lampe PD, Kurata WE, Warn-Cramer BJ, Lau AF (1998) Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J Cell Sci 111(Pt 6):833–841
Li W, Hertzberg EL, Spray DC (2005) Regulation of connexin43-protein binding in astrocytes in response to chemical ischemia/hypoxia. J Biol Chem 280(9):7941–7948. https://doi.org/10.1074/jbc.M410548200
Hanani M (2012) Lucifer yellow—an angel rather than the devil. J Cell Mol Med 16(1):22–31. https://doi.org/10.1111/j.1582-4934.2011.01378.x
Musil LS, Goodenough DA (1991) Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol 115(5):1357–1374. https://doi.org/10.1083/jcb.115.5.1357
Raaben M, Groot Koerkamp MJ, Rottier PJ, de Haan CA (2007) Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell Microbiol 9(9):2218–2229. https://doi.org/10.1111/j.1462-5822.2007.00951.x
Lin PC, Shen CC, Liao CK, Jow GM, Chiu CT, Chung TH, JC W (2013) HYS-32, a novel analogue of combretastatin A-4, enhances connexin43 expression and gap junction intercellular communication in rat astrocytes. Neurochem Int 62(6):881–892. https://doi.org/10.1016/j.neuint.2013.02.027
Fong JT, Kells RM, Falk MM (2013) Two tyrosine-based sorting signals in the Cx43 C-terminus cooperate to mediate gap junction endocytosis. Mol Biol Cell 24(18):2834–2848. https://doi.org/10.1091/mbc.E13-02-0111
Huang T, Wan Y, Zhu Y, Fang X, Hiramatsu N, Hayakawa K, Paton AW, Paton JC et al (2009) Downregulation of gap junction expression and function by endoplasmic reticulum stress. J Cell Biochem 107(5):973–983. https://doi.org/10.1002/jcb.22202
Biswas K, Das Sarma J (2014) Effect of microtubule disruption on neuronal spread and replication of demyelinating and nondemyelinating strains of mouse hepatitis virus in vitro. J Virol 88(5):3043–3047. https://doi.org/10.1128/JVI.02545-13
Giepmans BN, Verlaan I, Hengeveld T, Janssen H, Calafat J, Falk MM, Moolenaar WH (2001) Gap junction protein connexin-43 interacts directly with microtubules. Current biology : CB 11(17):1364–1368. https://doi.org/10.1016/S0960-9822(01)00424-9
Francis R, Xu X, Park H, Wei CJ, Chang S, Chatterjee B, Lo C (2011) Connexin43 modulates cell polarity and directional cell migration by regulating microtubule dynamics. PLoS One 6(10):e26379. https://doi.org/10.1371/journal.pone.0026379
Grafstein B, Liu S, Cotrina ML, Goldman SA, Nedergaard M (2000) Meningeal cells can communicate with astrocytes by calcium signaling. Ann Neurol 47(1):18–25
Anders JJ, Salopek M (1989) Meningeal cells increase in vitro astrocytic gap junctional communication as measured by fluorescence recovery after laser photobleaching. J Neurocytol 18(2):257–264. https://doi.org/10.1007/BF01206666
Abnet K, Fawcett JW, Dunnett SB (1991) Interactions between meningeal cells and astrocytes in vivo and in vitro. Brain research Dev Brain Res 59 (2):187–196, DOI: https://doi.org/10.1016/0165-3806(91)90099-5
Bundesen LQ, Scheel TA, Bregman BS, Kromer LF (2003) Ephrin-B2 and EphB2 regulation of astrocyte-meningeal fibroblast interactions in response to spinal cord lesions in adult rats. J Neurosci: Off J Soc Neurosci 23(21):7789–7800
Giepmans BN, Moolenaar WH (1998) The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Current biology : CB 8(16):931–934. https://doi.org/10.1016/S0960-9822(07)00375-2
Gilleron J, Carette D, Fiorini C, Benkdane M, Segretain D, Pointis G (2009) Connexin 43 gap junction plaque endocytosis implies molecular remodelling of ZO-1 and c-Src partners. Commun Integr Biol 2(2):104–106. https://doi.org/10.4161/cib.7626
Chai Q, He WQ, Zhou M, Lu H, Fu ZF (2014) Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J Virol 88(9):4698–4710. https://doi.org/10.1128/JVI.03149-13
Minagar A, Alexander JS (2003) Blood-brain barrier disruption in multiple sclerosis. Mult Scler 9(6):540–549. https://doi.org/10.1191/1352458503ms965oa
Spindler KR, Hsu TH (2012) Viral disruption of the blood-brain barrier. Trends Microbiol 20(6):282–290. https://doi.org/10.1016/j.tim.2012.03.009
van Sorge NM, Doran KS (2012) Defense at the border: the blood-brain barrier versus bacterial foreigners. Future Microbiol 7(3):383–394. https://doi.org/10.2217/fmb.12.1
Acknowledgements
Authors thank IISER-K confocal facility and Mr. Ritabrata Ghosh for his technical assistance with microscopy.
Funding
This work is supported by research grants (BT/PR14260/MED/30/437/2010 and BT/PR4530/MED/30/715/2012 from Department of Biotechnology (DBT), India; RG3774A2/1 from National Multiple Sclerosis Society (NMSS), USA) and start up research fund from Indian Institute of Science Education and Research Kolkata (IISER-K), India, to JDS. AB is supported by IISER-K Integrated PhD program. RB is a recipient of senior research fellowship award from Council for Scientific and Industrial research (CSIR), India. MM is a recipient of Young Scientist award from Science and Engineering research Board (SERB), India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animal experiments were performed following the approved guidelines of the Institutional Animal Ethical Committeee and Committee for the Purpose of Control and Supervision on Experiments on Animals (CPCSEA, India).
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Bose, A., Basu, R., Maulik, M. et al. Loss of Cx43-Mediated Functional Gap Junction Communication in Meningeal Fibroblasts Following Mouse Hepatitis Virus Infection. Mol Neurobiol 55, 6558–6571 (2018). https://doi.org/10.1007/s12035-017-0861-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0861-3