
Abstract
Highly purified, subunit, or synthetic viral antigens are known to be weakly immunogenic and potentate only the antibody, rather than cell-mediated immune responses. An alternative approach for inducing protective immunity with small viral peptides would be the direct targeting of viral epitopes to the immunocompetent cells by DNA vaccines encoding antibody fragments specific to activating cell surface co-receptor molecules. Here, we are exploring as a new genetic vaccine, a DNA chimeric molecule encoding a T and B cell epitope-containing influenza A virus hemagglutinin peptide joined to sequences encoding a single-chain variable fragment antibody fragment specific for the costimulatory B cell complement receptors 1 and 2. This recombinant DNA molecule was inserted into eukaryotic expression vector and used as a naked DNA vaccine in WT and CR1/2 KO mice. The intramuscular administration of the DNA construct resulted in the in vivo expression of an immunogenic chimeric protein, which cross-links cell surface receptors on influenza-specific B cells. The DNA vaccination was followed by prime-boosting with the protein-engineered replica of the DNA construct, thus delivering an activation intracellular signal. Immunization with an expression vector containing the described construct and boosting with the protein chimera induced a strong anti-influenza cytotoxic response, modulation of cytokine profile, and a weak antibody response in Balb/c mice. The same immunization scheme did not result in generation of influenza-specific response in mice lacking the target receptor, underlining the molecular adjuvant effect of receptor targeting.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The influenza type A virus is an important pathogen causing an acute respiratory disease in the human population [1, 2]. The virus is responsible for periodic seasonal outbreaks but some strains have shown the potential to raise global pandemics [3–5]. So far vaccination is considered as the most effective way to prevent infection and reduce the complications caused by influenza viruses [4].
The envelope of influenza A viruses contains a surface hemagglutinin (HA), which is a homotrimeric glycoprotein responsible for the initial binding of the influenza virion to the sialic acid receptors on the host cell surface. This binding leads to the entry of the viral genetic material into the cell [2, 6, 7]. The HA molecule is also the primary target of infection- or immunization-induced influenza virus-specific protective antibodies [6, 8, 9].
Both live and subunit (purified glycoprotein) influenza vaccines induce high titers of serum anti-HA antibodies, which directly neutralize viral infectivity by blocking the ability of the virus to attach to the cell surface [4, 7]. Still rapid change of the immunodominant viral antigens HA and NA known as antigenic shift and drift may render any pre-existing immunity useless [6, 7, 10].
A widely used form of human flu vaccine is the traditional trivalent inactivated influenza vaccine (TIV), which incorporates currently circulating viral strains in humans [11]. Anti-viral DNA vaccines are a novel strategy in the vaccine-development field, which basically consists in the administration of expression vectors coding antigen sequences into the host’s cells [12–14]. The gene(s) encoded by the plasmid is then expressed by the host cells transcription/translation machinery that mimics the natural viral infection [13, 15]. When produced under native conditions, the antigens encoded in the DNA vaccines are processed and presented in the context of MHC class I and II molecules and elicit efficient cytotoxic and humoral response [13, 15, 16]. In addition, DNA vaccines are inexpensive and stable at room temperature, which makes them perfectly suitable for rapid production and storage [13, 17, 18]. As DNA vaccine plasmids are unable to replicate or spread into the surrounding cell populations, there is no risk of secondary infection [19].
The intramuscular and intradermal immunizations have proved the most effective approaches for DNA delivery. It has been demonstrated that only a small fraction of the injected plasmid survives after i.m. immunization and is able to transfect the target cells. In spite of the limited quantity of expressed protein and the lack of transfection of professional APC, the i.m. immunization with DNA vectors results in a surprisingly stable generation of virus-specific cytotoxic CD8+ cells, as well as antibodies [18].
Many studies have confirmed that influenza HA-encoding DNA vaccines induce a strong and long-lasting immune response and provide protection against challenge with lethal doses of influenza virus in animals [15, 20–23]. These vaccine preparations induce a particularly efficient cytotoxic T cell response, while eliciting a more limited humoral response [24, 25]. The HA molecule contains many conserved epitopes that might be attractive targets for the design of new and more efficient vaccines, which could induce effective immunity against the virus [26–28].
Complement receptors 1 and 2 (CR1 and CR2) are expressed on B lymphocytes and on follicular dendritic cells in both human and mouse [29, 30]. In the mouse, CR1 and CR2 (CR1/2) are part of the BCR co-receptor complex that also contains CD19 and CD81 [31]. The co-ligation of CR1/2 and the B cell antigen receptor (BCR) reduces the threshold for activation of antigen-specific B cells up to 1,000 times [32, 33].
The generation of a single-chain variable fragment (scFv) permits the construction of completely functional antigen-binding immunoglobulin fragment, comprising the antibody’s heavy and light chains. The engineered scFv antibodies can be genetically fused to particular defined peptide epitopes. The resulting bifunctional chimeric molecule inserted into an expression vector can be used as a naked DNA vaccine to deliver viral epitopes to the antigen-specific cells [34]. Thus, a scFv fragment from an antibody to CR1/2 coupled to the influenza virus-epitope would cross-link the activating mouse CR1/2 to the surface immunoglobulin receptors specific to the virus.
The influenza hemagglutinin peptide HA317-41 (IP) contains an I-Ed restricted T cell epitope and a conformation-dependent B cell epitope [35, 36]. In comparison with the free peptide, the presentation efficiency of the construct may be improved by increasing the local peptide quantity on the cell surface through CR1/CR2 binding.
The effects of a recombinant DNA molecule, encoding an influenza HA peptide and a scFv fragment binding to mouse complement receptor 1/2 (CD21) on antigen-specific B cells have already been tested. The engineered DNA molecule induced a strong, long-lasting CTL and a moderate antibody response against influenza A virus in Balb/c mice [37, 38].
DNA vaccines might be used along with protein ones to combine the advantages of both techniques [28]. Such vaccines can potentially broaden the immune response providing more effective protection against highly variable and elusive pathogens [7, 28]. In the present study, we investigated the response induced after immunization with the DNA vaccine described above, followed by a booster immunization with a protein chimeric molecule, composed of a monoclonal antibody against mouse CR1/2 conjugated to the influenza hemagglutinin peptide HA317-41.
Materials and methods
Genetic work
VL and VH genes of scFv specific for mouse CR1/2 have been described and submitted to GenBank (GenBank ID: AF220555, AF214704). It was shown that the protein fragment generated from these sequences binds the targeted receptors with high specificity [38]. RNA was isolated from the influenza strain A/PR/8/34; then, the HA317-41 fragment was amplified by RT-PCR. An NcoI and an RcaI positions were introduced with sense primer ACCATGGTTACAGGACTAAGGAAC and antisense primer GCTCATGACTCCGCCACCTGAGCCTCCCCCTTCAATAAAACC that allows repeated subcloning into the NcoI position of the pET11c-7G6scFv construct. A dimerization domain from the transcription factor Fos/Jun was fused to the C-terminal end of the scFv by replacing the NotI-BamHI fragment coding the peptide tags with a fragment coding dimerization domain and peptide tags. After the insertion of a linker, containing BamHI, into the bacterial expression vector pET, scFv coding regions were transferred into the eukaryotic expression vector pNut using BamHI and NotI restriction sites. The resulting vectors (pNut-IP-7G6scFv-Fos (Jun)) thus contained the scFv genes fused to a secretion signal peptide at the 5′ ends and a stop codon and bovine polyA region at the 3′ ends, under the control of a metallothionein promoter. The genetic constructs were amplified in a prokaryotic competent culture, isolated and purified by the PureYield™ plasmid Maxiprep system (Promega, Madison, WI).
Enzyme restriction (BamHI/Eco52I, Fermentas) of the vectors was performed for the correct constructs’ size identification. The restriction mixtures were run on 1.2 % agarose gel electrophoresis (BioRad, Richmond, CA).
Cell cultures and antibodies
Rat hybridoma 7G6 [39] (kindly provided by Dr. T. Kinoshita, Osaka University, Osaka, Japan) producing a monoclonal IgG2b antibody specific to murine CR1 and CR2 was cultured on a large scale in the serum-free CHO medium (Gibco, Gaithersburg, MD) at 37 °C/5 % CO2. Antibodies from the supernatant were purified by 50 % ammonium sulfate precipitation and subsequent dialysis.
Rat hybridoma I/9 producing a monoclonal anti-idiotype (anti-B10-anti-(T,G-A-L)) IgG2b antibody (kindly provided by Dr Gloria Laszlo, Immunology Research Group, Hungarian Academy of Sciences, Budapest, Hungary) was cultured under the same conditions, and the purified antibodies (as described above) were used as an isotype control.
The murine B lymphoma cell line IIA1.6 (FcγRII-negative) expressing CR1 and CR2 was cultured in RPMI 1640 medium supplemented with 10 % heat-inactivated FCS, 100 U/ml penicillin, 100 U/ml streptomycin, 2 mM glutamine, and 1 mM sodium pyruvate.
FITC-conjugated 7G6 (Pharmingen BD, San Diego, CA) was used for FACS experiments.
Mice
Female 8 weeks old Balb/c mice were obtained from Harlan Farm, (Blackthorn, UK).
Female C57/B6 CR1/2 KO mice [40] backcrossed to Balb/c background (kindly provided by Prof. Matyas Sandor, Department of Pathology and Laboratory Medicine, University of Wisconsin-Madison, USA) were also used for immunization experiments.
The animals were kept under specific pathogen free (SPF) conditions, and the manipulations were approved by the Animal Care Commission at the Institute of Microbiology in accordance with the national regulations.
Transfection and expression into CHO cells
Chinese hamster ovary (CHO) cells (ATCC Cat. No. CRL-9618) were used to produce the recombinant protein encoded by pNut-IP-7G6scFv-Fos (Jun) DNA constructs. The transfection of DNA into the eukaryotic cells was performed by Lipofectamine™ LTX Reagent (Invitrogen, CA, USA) according the manufacturer’s instructions in a 24-well culture plate. The cells transfected with an empty vector or untransfected cells were used as controls. The cell line was adapted to grow in the serum-free CHO medium at 37 °C, 5 % CO2. pNut-IP-7G6scFv-Fos(Jun) transfectants of the CHO cell line were maintained in the same medium supplemented with methotrexate (10–50 mM; Pharmachemie, Haarlem, The Netherlands) for obtaining stable growth. The expressed recombinant proteins in the supernatants were concentrated and purified by 5 kDa ultrafiltration. After electrophoresis and transfer of samples onto nitrocellulose membrane (0.45 μm, from Sartorius, Germany), the blots were probed with mouse anti-IP-specific monoclonal antibody (IP2-11-1, specific to an influenza A virus hemagglutinin epitope) and goat anti-mouse IgG-HRP (horseradish peroxidase) antibody (Sigma). Films were developed by the enhanced chemiluminescence (ECL) technique.
Construction of protein chimeric molecules
The protein chimeric molecules were constructed as already described [41]. Briefly: the hemagglutinin intersubunit peptide IP (HA317-341 containing T and B cell epitopes) from the influenza virus strain A/PR/8/34 was purchased from Caslo Laboratory (Lyngby, Denmark). The peptide Ac-MVTGLRNIPSIQSRGLFGAIAGFIE-Ahx-K-NH2 was purified (≥98 % purity) by high-performance liquid chromatography.
Copies of IP were coupled to the 7G6 mAb (7G6-IP chimera) and to the irrelevant I/9 mAb (Control chimera). The Ab-IP conjugates were prepared using the classical 1-ethyl-3(3′-dimethylaminopropyl) carbodiimide (EDC)·HCl, (Fluka AG, Buchs, Switzerland) cross-linking technique [42]. During the synthesis of the peptides, an Ahx linker with lysine was added to their C end. Each antibody was mixed with a 20-fold molar excess of the peptide. The chemical reaction was started by the addition of carbodiimide; the mixture was stirred overnight at 4 °C, dialyzed against PBS and concentrated by ultrafiltration.
PAGE and Western blotting
Both constructed protein chimeras were subjected to SDS-PAGE and Western blot analysis. SDS-PAGE was performed using 10 % gels and a MiniProtean II system (BioRad, Richmond, CA) in the presence of 0.1 % SDS. After the electrophoresis, the proteins were transferred to a nitrocellulose membrane (0.45 μm, from Sartorius, Germany) using a MiniTrans Blot device (from BioRad) in a buffer containing 48 mM Tris and 110 mM glycine in the presence of 20 % (v/v) methanol. The membranes were blocked overnight in a TBS buffer containing 0.4 % Tween 20 and were incubated for 1 h at room temperature with mouse anti-IP-specific monoclonal antibody IP2-11-1. After washing, the membranes were incubated in an optimal dilution of an anti-mouse IgG conjugated with alkaline phosphatase (Sigma) for 1 h and the reaction was revealed using the nitro blue tetrazolium/bromo-chloro-indolyl-phosphate substrates (Sigma).
Flow cytometry
The retained ability of the 7G6-IP chimera to interact with CR1/2 was confirmed as it competed successfully with a FITC-conjugated antibody with the same specificity for binding to murine B lymphoma cell line IIA1.6 that expresses CR1/2. The cells were cultured in complete RPMI (Roswell Park Memorial Institute medium) 1640 medium (Gibco, Gaithersburg, MD) containing 10 % FCS (fetal calf serum), 4 mM l-glutamine, 50 μM 2-mercaptoethanol, and antibiotics and were double-washed with ice-cold PBS (containing 2.5 % FCS and 0.05 % sodium azide). Later, the cells were incubated with 7G6-IP chimera (1 μg/106 cells), or with Control chimera, or with PBS alone for 30 min at 4 °C, followed by two washes and a second incubation with a FITC-conjugated 7G6 antibody for 30 min at 4 °C. Ten thousands cells were analyzed from each sample with a BD LSR II flow cytometer using the Diva 6.1.1 software (BD Biosciences, San Jose, CA).
ELISpot assay for counting anti-IP antibody-secreting cells
Female Balb/c mice (group of 10 animals) were immunized i.p. with 20 μg IP peptide emulsified in an equal volume of complete Freund’s adjuvant (CFA, Sigma). The control group of Balb/c mice was immunized with PBS. The re-immunization of animals treated with IP emulsified in CFA was performed with IP peptide emulsified in an incomplete Freund’s adjuvant (IFA). Splenocytes were isolated and cultured (2.106/ml) in complete RPMI 1640 medium (see above) in the presence of increasing concentrations (ranging from 0.04 to 5 μg/ml) of the 7G6-IP chimera or the control chimera or with free IP peptide for 96 h at 37 °C. Cells cultured with 10 μg/ml lipopolysaccharide (LPS) (from E. coli, Sigma, L-2630) or cultured in medium only were used as controls.
Later, 96-well ELISpot plates (Millipore, Bedford, MA, USA) were coated with 10 μg/ml IP peptide for 30 min at room temperature, washed with PBS, and blocked with 1 % gelatin in PBS. The pre-cultured cells were transferred to ELISpot plates with the IP-coated membranes and were further cultured for 4 h in a humidified 5 % CO2 atmosphere at 37 °C. Next, the plates were washed, incubated with an anti-mouse IgG conjugated with alkaline phosphatase (Sigma) for 2 h, and developed by NBT-BCIP substrate (Sigma). The number of spots corresponding to IgG anti-IP antibodies producing cells was counted by C.T.L Immunospot S5 Versa Analyzer (Bonn, Germany).
Antigen presentation assay
Splenocytes from untreated female Balb/c mice were isolated, B cells were purified using a mouse B lymphocyte enrichment set DM (BD IMag™, BD Bioscience) and were used as Antigen Presenting Cells, at 104 cells/well. Splenocytes from IP-CFA immunized Balb/c mice (see the ELISpot assay) were also isolated and T cells were enriched by a mouse CD4+ T cell isolation kit (Miltenyi Biotec, Germany). The enriched T cells were added to the purified B cells at 2 × 104 cells/well. Various concentrations of free IP peptide or 7G6-IP chimera were tested (ranging from 0.04 to 5 μg/ml). The IL-2 production was quantitated in the 24 h culture supernatant by ELISA (enzyme-linked immunosorbent assay) set (BD OptEIA, Bioscience).
Signal transduction
The CR1 and CR2-expressing murine B lymphoma cell line IIA1.6 (1 × 107 cells/ml) was incubated on ice for 20 min in the presence of 30 μg/ml of the protein-engineered 7G6-IP chimera or 30 μg/ml of the control chimera, followed by incubation at 37 °C (for 0, 2, 5 or 10 min.). Cells cultivated in medium only were used as controls. The reaction was stopped with cold PBS, and the cells were centrifuged for 3 min at 400×g. The pellet was treated with lysis buffer (10 mM Tris base pH 7.4, 50 mM NaCl, 1 % Triton X-100, 1 mM sodium orthovanadate, 50 mM NaF, 25 mM sodium pyrophosphate, 5 mM ethylenediaminetetraacetic acid (EDTA), 10 μg/ml Pepstatin-protease inhibitor cocktail tablets) for 45 min at 4 °C. Supernatants were collected after centrifugation for 15 min at 4 °C at 16,000×g. Aliquots of the lysate were pre-cleared on protein G-Sepharose (Amersham Biosciences, UK) for 1 h on a rotating wheel at 4 °C and were subjected to immunoprecipitation with antibodies to CR1/2 bound to protein G-Sepharose beads (from Sigma). Immunoprecipitation was performed overnight on a rotating wheel at 4 °C, and after that the beads were washed three times with lysis buffer. Immunoprecipitates were boiled for 10 min in SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) sample buffer with β-mercaptoethanol, run on two parallel 10 % SDS-PAGE gels and transferred to two nitrocellulose membranes (0.45 μm, Sartorius, Germany). For the detection of phosphorylated tyrosine residues, the first membrane was blocked for 2 h in Tris-buffered saline (TBS), pH 7.4 with 0.05 % Tween 20 and 5 % BSA (bovine serum albumin), incubated further with a mouse anti-phosphotyrosine antibody (from R&D Systems, Minneapolis, MN) and finally with a goat anti-mouse IgG-HRP antibody (Sigma). Tyrosine phosphorylation was evaluated using the ECL technique.
The second membrane was blocked overnight at 4 °C in TBS, pH 7.4, containing 0.05 % Tween 20 and further incubated in an optimal dilution of the 7G6 antibody, followed by goat anti-rat IgG antibody, conjugated to alkaline phosphatase (Sigma) and developed.
Treatment schedule
Two groups (A and B) of Balb/c mice (6–8 mice each) were immunized intramuscularly (i.m.) with the generated DNA constructs pNut-IP-7G6scFv-Fos (Jun) (50 μg per mouse). Immunization was repeated in a 14-days interval. The first group (A) received no further applications. The second group (B) was boosted i.p. with 20 μg/mouse of the protein-engineered 7G6-IP chimera 14 days after the last DNA application, and another protein boost was performed 21 days later.
A third group of Balb/c mice (C) was immunized with the protein chimera 7G6-IP only, and a second immunization was performed 21 days later. Control groups of Balb/c mice were injected (i.m.) with 50 μg of plasmid DNA encoding the influenza HA317-342 (pNut-IP) peptide only, or with 50 μg of plasmid DNA encoding scFv specific for mouse CR1/2 (pNut-7G6scFv), or with 50 μg of the empty plasmid (pNut), or with PBS. All DNA-treated control groups were immunized as described for group A. Two more control groups were treated with free IP peptide (20 μg/mouse), or with free IP peptide emulsified in an equal volume of CFA. Mice were boosted 21 days later with the same doses of IP peptide as described above. The re-immunization of animals treated with IP peptide emulsified in CFA was done with IP peptide emulsified in an incomplete Freund’s adjuvant (IFA).
Similar groups of C57/B6 CR1/2 KO mice were constituted and immunized as described above.
The mice were bled before each immunization and after the last treatment. Collected sera were kept frozen at −70 °C before testing for antibodies and cytokines.
Enzyme-linked immunosorbent assay (ELISA) for anti-IP IgG antibodies
IP peptide diluted to 5 μg/ml in coating buffer (NaHCO3, pH 9.6) was used for coating of microplates (Nunc, Roskilde, Denmark) by incubation overnight at 4 °C. After washing with PBS/0.05 % Tween 20 and blocking with 1 % BSA, serum samples diluted 1:50 for measuring of IgG antibodies were added and incubated for 1 h at room temperature. The plates were then washed and incubated for 1 h at room temperature with alkaline phosphatase-labeled goat anti-mouse IgG (Sigma-Aldrich). After washing, Sigma 104 phosphatase substrate was added and the absorbance was measured at 405 nm. The obtained ELISA results were presented as relative units (RU), corresponding to the titer of anti-IP standard antibodies used for ELISA.
Cytokine detection
IL4, IL10, and IFN-γ levels were measured in mouse sera using commercial ELISA kits (BD Biosciences, USA).
Cytotoxicity assay
The influenza A/Aichi/2/68 (H3N2) strain (The Collection of the Stephan Angeloff Institute of Microbiology, Sofia, Bulgaria) was grown on MDCK cells and after pelleting, the virions were concentrated by ultracentrifugation and suspended in PBS.
Confluent 3T3 (mouse embryo fibroblasts) cell monolayer cultured in DMEM with 5 % FCS was incubated with 102/ml virions for 18 h at 37 °C. Then, cells were washed, trypsinized, and transferred to the wells of a 96-well tissue culture plate (2.105 cells/ml). Freshly isolated spleen cells from immunized mice were used as effector cells in a non-radioactive cytotoxic assay at a ratio between target and effector cells 1:40. After incubation for 4 h at 37 °C, the cells were centrifuged and the lactate dehydrogenase (LDH) concentration in the supernatants was determined by the CytoTox assay (Promega, USA) according to the manufacturer’s instructions. The percentage of specific lysis was calculated as follows: specific lysis (%) = (experimental release − spontaneous release/(maximum release − spontaneous release) × 100. Maximal or spontaneous release was obtained by incubating target cells with 1 % Triton or with medium only.
Statistical analysis
Values in the figures correspond to mean ± SD. All ELISA, cytokine and cytotoxicity samples were triplicated. The unpaired Student t test was used to determine differences between each two groups. The two-tailed Mann–Whitney U test was used when appropriate. A value of p < 0.05 was considered as statistically significant.
Results
Construction of recombinant DNA molecules
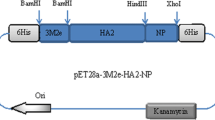
The influenza peptide-7G6scFv-Fos/Jun genetic constructs were engineered and inserted into the eukaryotic expression vector pNut, possessing a viral promotor for foreign protein expression (Fig. 1a). After progressive amplification in competent prokaryotic culture, the DNA chimeric molecules were purified and analyzed by enzyme restriction (BamHI/Eco52I). The correct fragments’ sizes were demonstrated in agarose electrophoresis (Fig. 1b).

Genetic work. a Genetic construction of recombinant DNA molecules: an overview: (for details see the “Materials and methods” section). b Agarose DNA electrophoresis of genetic constructs: lane 1 molecular weight markers; lane 2 pNut-7G6scFv; lane 3 pNut-IP-7G6scFv; lane 4 pNut-IP-7G6scFv-Fos. c Recombinant chimeric molecules retain the IP-B cell epitope after expression in CHO cells. CHO cells transfected with recombinant DNA constructs were cultured with selective marker, and supernatants were concentrated. Purified fusion proteins were subjected to SDS-PAGE and transferred to nitrocellulose membrane. The expressed recombinant proteins were probed with mouse anti-IP-specific monoclonal antibody IP2-11-1, followed by incubation with goat anti-mouse IgG-HRP antibody, and developed by the ECL. Lane 1 empty vector pNut; lane 2 pNut-IP; lane 3 pNut-IP-7G6scFv-Fos; lane 4 untransfected cells. Data are representative of at least 3 independent experiments
Eukaryotic expression of the genetic constructs
To demonstrate the eukaryotic expression of the recombinant genetic constructs, IP-7G6scFv-Fos/Jun CHO cells were transfected with the vector-carrier pNut and the positive clones were selected. Supernatants of cultured transfected CHO cells were tenfold concentrated by ultra-filtration, and the expressed fusion proteins were subjected to Western blot (Fig. 1c).
The protein chimera preparation
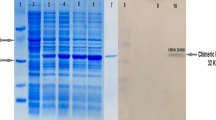
The successful construction of protein chimeras by chemical coupling of influenza peptides to the immunoglobulin molecule was tested by SDS-PAGE and Western blot. It was previously shown by mass-spectral analysis that 14–16 peptides per IgG molecule are bound on a single protein chimera [43]. The recognition of the IP peptide by the IP-specific monoclonal antibody IP2-11-1 in a Western blot confirmed that the peptides coupled to the rat IgG-carrier retained their B cell epitope (Fig. 2a), and it is available for interaction with IP-specific B lymphocytes.

Protein chimeric molecule 7G6-IP retains its functional activity after chemical conjugation. a IP peptides coupled to the anti-CR1/2 antibody are recognized by anti-IP-specific antibodies. After the covalent conjugation of the IP peptides to the CR1/2 antibody, the peptide epitope retains its ability to be recognized by anti-IP antibodies. Samples (20, 10, and 5 μg) from the 7G6-IP chimera (lanes 2–4) or the unconjugated 7G6 antibody (lane 1) were subjected to SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked and incubated with mouse anti-IP-specific monoclonal antibody IP2-11-1, followed by incubation with an anti-mouse IgG conjugated with alkaline phosphatase and developed. Data are representative of at least 3 independent experiments. b The 7G6-IP chimera maintains its ability to bind to CR1/2 on murine B lymphoma cell line IIA1.6 (CR1/2-positive) and competes with FITC-conjugated anti-CR1/2 antibody for the same receptor. Results of flow cytometry analysis are shown. The cells were incubated with the 7G6-IP chimera or with control chimera, washed, and incubated with FITC-conjugated 7G6 antibody. Data are representative of at least 5 independent experiments
The chimera binds to mouse CR1/2
Using flow cytometry, we demonstrated the ability of the 7G6-IP chimera to bind CR1/2 on CR1/2 expressing murine B lymphoma cell line. We assessed the effect of the 7G6-IP chimera in competition with the FITC-conjugated anti-CR1/2 antibody for the same receptor (Fig. 2b).
Anti-IP IgG antibody-secreting plasma cells are affected by 7G6-IP chimera
We found that cultivation of splenocytes from IP-CFA-immunized mice in the presence of the 7G6-IP chimera resulted in an increased number of anti-IP IgG producing plasmocytes. Our data demonstrated that co-crosslinking of BCR and the surface CR1/2 on IP-specific B lymphocytes increases antibody production (Fig. 3a, left panel). The most important increase in the number of plasmocytes secreting IP-specific IgG antibodies was observed after incubation of splenocytes with 5 μg/ml 7G6-IP chimeric molecule. As a control, we performed the same experiment with splenocytes from PBS-treated animals. The presence of IP-specific plasmocytes was not detected (Fig. 3a, right panel).

Engaging of CR1/2 on IP-specific B lymphocytes by the 7G6-IP chimera induces B cell activation. a The co-crosslinking of BCR and CR1/2 on IP-specific B lymphocytes up-regulates antibody production. Splenocytes from IP peptide-immunized (left panel) or untreated Balb/c mice (right panel) were cultured in the presence of the 7G6-IP chimera, or the control chimera, or with free IP peptide. The number of plasma cells secreting anti-IP peptide-specific IgG antibodies was determined using ELISpot assays. Results are expressed as the mean value ± SD of triplicates. Splenocytes from each mouse were tested individually. The numbers of spots in the test wells were compared to control wells containing medium-only-cultured splenocytes (Student t test; *p < 0.05, ***p < 0.001). Data are representative of at least 5 experiments. b CR1/2-mediated presentation of IP by B lymphocytes to IP-specific Th cells. CD4 + T cells isolated from IP-CFA immunized Balb/c mice were incubated with purified B cells from untreated Balb/c mice for 24 h at 37 °C in the presence of various concentrations of 7G6-IP or free IP peptide. The release of IL-2 by the Th cells in the culture supernatants was used as a measure for Ag presentation efficiency. Mean values of one representative experiment of five are shown. c 7G6-IP chimeric molecules trigger signal transduction through CR1/2. Murine CR1/2-positive B lymphoma cell line IIA1.6 was incubated in the presence of the 7G6-IP chimera at 37 °C for 10, 5, 2, or 0 min. Cells cultivated in medium only were used as controls. The cells were lysed, subjected to immunoprecipitation using anti-CR1/2 antibodies, followed by Western blotting with an anti-phosphotyrosine antibody (upper panel) or with the anti-CR1/2 (7G6) antibody (lower panel). Data shown are representative of 3 independent experiments
The IP peptide on the 7G6-IP chimera retained its T cell epitope
Beside a B cell epitope, IP peptide comprises also an I-Ed-restricted T cell epitope (HA317–329). Incubation of APCs (B cells) with the free IP peptide resulted in the activation of peptide-specific T cells. The efficiency of antigen presentation was improved by the targeting of IP peptide to CR1/2 using 7G6-IP chimeric molecule (Fig. 3b).
The 7G6-IP chimera triggers signal through CR1/2
The ability of the constructed protein chimeric molecule to provide signals through the co-stimulatory B cell surface receptors was tested using CR1/2-positive (FcγRIIb-negative) B cell line. The signal transduction was confirmed by the observed tyrosine phosphorylation of the receptor in a CR1/2-expressing B cell line after incubation with 7G6-IP chimeric molecule (Fig. 3c).
Anti-IP antibody production
Blood samples were collected by retro-orbital puncture, and mouse sera were tested for IgG anti-IP antibodies by ELISA. Sera obtained at the end of the experiment (day 90) were also tested, and significant differences were registered between the groups.
The 7G6-IP treated Balb/c group produced high levels of anti-IP antibodies after the second immunization and further reached the level registered in Balb/c group injected with IP + CFA (Fig. 4, left panel). The IP-immunized Balb/c group produced moderate levels of IgG antibodies after the first and the second immunization, while in the group vaccinated with pNut-IP-7G6scFv-Fos/Jun IgG antibody levels raised 5 weeks after the second DNA immunization and were not detected in the end point of observation. Induction of IgG anti-IP antibodies was not observed in the Balb/c group immunized with pNut-IP-7G6scFv-Fos/Jun + 7G6-IP.

Immunization with protein chimera 7G6-IP indices humoral immune response in Balb/c mice. Anti-IP IgG antibody titers in mice injected with PBS or with 50 μg of pNut-IP-7G6scFv (i.m.), or with 20 μg IP peptide, or with 20 μg IP peptide in CFA, or with 20 μg 7G6-IP (i.p.). The results are presented as relative units (RU). They were calculated using standard IgG antibodies against IP. The titers obtained from the standard’s dilutions were used to create a curve from 0 to 400.0 RU. Results are expressed as RU obtained from individual sera collected before each immunization and 3 weeks after the last one. Each point at the x axis indicated the injections as described in “Materials and methods” section. The data are represented as mean ± SEM; p values are calculated using the Mann–Whitney U test (*p < 0.05; **p < 0.01), in comparison with PBS-treated controls
No anti-IP IgG antibodies were detected in the sera of C57/B6 CR1/2 KO mice injected either with DNA chimeric molecules or with protein-engineered construct (Fig. 4, right panel).
Cytokine profile
IL4, IL10, and IFN-γ levels were measured in mice sera after immunization with different constructs using ELISA kits. The Th1 cytokine IFN-γ is important for the efficient cellular immune response after vaccination (including DNA vaccination), and high levels in mouse serum correlate with the generation of antigen-specific IFN-γ producing CD8+ cells. As expected, the groups of Balb/c mice treated with pNut-IP-7G6scFv-Fos/Jun produced high serum levels of IFN-γ as compared to the PBS-injected mice only after double DNA immunization, while 7G6-IP treated mice produced high IFN-γ levels starting from the first immunization.
The boost with 7G6-IP in group B significantly decreased IFN-γ production as compared to group A.
We did not observe significant differences in IL-4 and IL-10 production between the groups of animals immunized with DNA and/or protein chimeric molecules and PBS-treated mice (Fig. 5).

DNA immunization with pNut-IP-7G6scFv induces IFN-γ production in treated mice. Serum levels of IFN-γ in Balb/c mice treated as described in Fig. 4 were measured by sandwich ELISA. The data are represented as mean ± SEM of 6–8 mice per group; p values were calculated in comparison with PBS-treated controls using the Mann–Whitney U test (*p < 0.05; **p < 0.01; ***p < 0,002)
Generation of cytotoxic T cells
We examined the CTL activity of freshly isolated spleen cells from all animals against influenza virus-infected 3T3 cells 2 months after the last immunization. Immunization with 7G6-IP, IP alone, or IP + CFA induced a low cytotoxic activity, while pNut-IP gave a moderate result (Fig. 6). The cytotoxic activity in the pNut-IP-7G6scFv-Fos/Jun and pNut-IP-7G6scFv-Fos/Jun + 7G6-IP-immunized groups was significantly higher as compared to the other treated groups.

CTL activity of splenocytes isolated at day 30 after the last immunization. 3T3 cells pulsed with the influenza virus were cultured with effector spleen cells at a ratio 1:40 for 4 h at 37 °C. LDH concentration in the supernatants was determined in triplicates by a commercial CytoTox assay. Results are expressed as the mean ± SEM comparing immunized and PBS-only-treated mice using the Mann–Whitney U test (*p < 0.05; **p < 0.01; ***p < 0.001). Data are representative of at least 3 independent experiments
Discussion
Prevention or reduction of pathogenic diseases by vaccination is one of the most important achievements of medicine. Influenza epidemics have emerged as a world health problem with serious social impact over last decades. The development of effective influenza vaccine is still an urgent challenge for biomedical industry [2, 4]. Protein-engineered or gene-engineered chimeric molecules targeting antigens to non-antigen-binding surface receptors are a tool for directed immune response modulation. In both cases, antibodies (or their fragments) are used, which transport epitopes to the cells of interest when targeted to their natural ligands and could modulate the immune response to the desired course.
The role of co-stimulatory surface receptors in the amplification of the immune response is well recognized [29, 44]. Complement receptors 1 and 2 (CD35 and CD21) are expressed on B lymphocytes and regulate B cell activation. The co-crosslinking of BCR and complement receptors reduces the threshold for B cells activation, leading to enhanced antigen presentation by B cells [33, 40].
DNA-based vaccines provide protective immunity by triggering the expression of the vector-coded antigen and inducing an efficient immune response. Similar to protein subunit vaccines, DNA-based vaccines present only the required antigen for immunization, and in addition benefit of the in vivo antigen synthesis. DNA vaccines contain a powerful promoter for eukaryotic expression and unmethylated CpG motifs acting as adjuvants. These CpG sequences activate target cells by TLR9 recognition and enhance the expression of the vaccine-coded proteins. Thus, after the DNA enters host cells, the encoded antigen is expressed and presented to the immune system as if during a natural infection [18].
The most important advantage of genetic vaccines is the induction of both humoral and cellular immune responses without the risks associated with live attenuated vaccines. In general, DNA vaccines predominantly evoke CTL generation, while protein subunit vaccines trigger a humoral response [16, 18].
A number of DNA vaccines encoding the main influenza antigens (HA and NA) induce protective immunity against virus infection [11, 15, 17, 21–23, 25, 28]. In the present study, however, we used a small 25-amino acid peptide as a part of viral HA that can modify the immune response compared to the whole HA.
The generation of genetic or protein chimeric molecules using antibody carrier is an alternative way for antigen delivery to the cells of interest [37, 41, 43, 44]. A scFv with Ag includes and preserves the properties of native antibodies, but could be also used as naked DNA vaccine. Tchorbanov et al. [37] have already constructed a chimeric DNA molecule, containing influenza virus hemagglutinin peptide and a scFv antibody fragment binding to complement receptors 1 and 2. It was shown that single or double immunization with a scFv-IP DNA vaccine leads to high CTL anti-influenza activity, but did not result in significant antibody production against IP peptide. In the present study, we constructed the protein replica of the genetic construct by the covalent binding of IP peptides to a native anti-CR1/2 antibody. The chimeric molecule was used for prime-boosting after double DNA vaccination or for immunization alone.
Post-expression activity of DNA chimeric molecules is questionable, which requires proving of the activity of protein chimeric molecules in vitro. The ability of the 7G6-IP chimera to bind to CR1/2 on mouse B cells was demonstrated by flow cytometric analysis. It was also proven that the IP peptide retains its B and T cell epitopes after conjugation to the antibody carrier and could be recognized by IP-specific B and T cells. The assembly of IP peptide with targeting antibody was able to trigger signal transduction through CR1/2 on a CR1/2-positive B cell line. We expected to enhance the humoral response against the IP peptide in vivo, because the 7G6-IP chimera constructed by us would bind preferentially the targeted available IP-specific B cells. Indeed, administration and boosting with the protein chimeric molecule 7G6-IP of Balb/c mice increased the level of anti-IP IgG antibodies as compared to immunization with free IP peptide. IP alone provoked high antibody titers when administered in CFA, while the naked pNut-IP-7G6scFv-Fos/Jun DNA vaccine produced moderate titers long after the second DNA administration. Interestingly, the group of animals double-immunized with pNut-IP-7G6scFv-Fos/Jun and prime-boosted with the protein chimera 7G6-IP exhibited a very low level of anti-IP IgG as compared to the animals immunized with the free IP peptide. This was not unexpected, having in mind that the pNut-IP-7G6scFv chimera does not encode the whole HA molecule, but only a small viral peptide [11, 17, 22, 25]. Even prime-boosting with the protein chimera 7G6-IP could not turn the immune response to the humoral course, while the independent protein chimera 7G6-IP administration generated high levels of specific antibodies.
There are several possible mechanisms by which 7G6-dependant targeting of IP peptide to CR1/2 could lead to an enhanced Ab response. The binding of the 7G6-IP chimeric molecules to CR1/2 may increase the local Ag concentration at the surface of IP-specific B cells, thus, providing the interaction between IP peptide and its specific BCR. Another possibility is the cross-linking of CR1/2 and BCR on IP-specific B cells, leading to intracellular signal transduction. Even the interaction between the 7G6-IP and CR1/2 on IP-non-specific B cells could proceed to Ag presentation, supporting anti-IP antibody production [44]. The immunization of CR1/2 KO mice with 7G6-IP chimeric molecule did not lead to anti-IP antibody production, confirming the essential role of CR1/2 for a normal humoral immune response.
The cytokine production supports the obtained results. We did not observe significant differences in the IL-4 and IL-10 production between the immunized groups of animals, but IFN-γ levels indicated the prevalence of the Th1 immune response in the groups vaccinated with protein chimera 7G6-IP, and with the pNut-IP-7G6scFv DNA vaccine with or without protein boosting. As expected, high levels of IFN-γ were measured in the IP-CFA group, but it was not associated with significant anti-IP-specific CTL response.
The most promising property of DNA vaccines is the effective induction of long-lasting CTL memory responses. In previous studies, we reported the presence of CD8+ cytotoxic T lymphocytes persisting for at least 6 months after a single or repeated immunization with pNut-IP-7G6scFv. In the present, study we obtained even higher CTL activity after prime-boosting with the protein construct 7G6-IP. Moderate CTL anti-influenza activity was also detected in the animal groups immunized with DNA encoding pNut-IP and free IP peptide in CFA. These results are in accordance with data that the conventional vaccine combined with CFA can prime CD8+ cytotoxic T cells and induce cytotoxic immunity [37].
Our results point out that co-immunization with both the pNut-IP-7G6scFv DNA chimeric molecule and the protein version of the vaccine further increases the intensity of the CTL response against the influenza virus. The protein chimera alone produced high titers of anti-influenza IgG antibodies. The joining of sequence encoding a scFv fragment of an antibody to a stimulatory co-receptor and the influenza peptide epitope potentiated CTL response, while prime-boosting with the protein chimera could not switch the immune response to the humoral course.
References
Nicholson KG, Webster RG, Malden MA, Hay AJ, editors. Textbook of influenza. Oxford: Black-well Science; 1998.
Bouvier NM, Palese P. The biology of influenza viruses. Vaccine. 2008;26:D49–53.
Mishra AC, Chadha MS, Choudhary ML, Potdar VA, et al. Pandemic influenza (H1N1) 2009 is associated with severe disease in India. PLoS ONE. 2010;5:e10540.
Ellebedy AH, Webby RJ. Influenza vaccines. Vaccine. 2009;27:D65–8.
Sims LD, Domenech J, Benigno C, Kahn S, Kamata A, Lubroth J, et al. Origin and evolution of highly pathogenic H5N1 avian influenza in Asia. Vet Rec. 2005;157:159–64.
Ellebedy AH, Ahmed R. Re-engaging cross-reactive memory B cells: the influenza puzzle. Front Immunol. 2012;3:1–7.
Ze C, Kurata T, Tamura S. Identification of effective constituents of influenza vaccine by immunization with plasmid DNAs encoding viral proteins. Jpn J Infect Dis. 2000;53:219–28.
Wilson IA, Cox NJ. Structural basis of immune recognition of influenza virus hemagglutinin. Annu Rev Immunol. 1990;8:737–71.
Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–69.
Carrat F, Flahault A. Influenza vaccine: the challenge of antigenic drift. Vaccine. 2007;25:6852–62.
Wang S, Taaffe J, Parker C, Solorzano A, Cao H, Garcia-Sastre A, et al. Hemagglutinin (HA) proteins from H1 and H3 serotypes of influenza A viruses require different antigen designs for the induction of optimal protective antibody responses as studied by codon-optimized HA DNA vaccines. J Virol. 2006;80:11628–37.
Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH, Felgner PL, Dwarki VJ, et al. Heterologous protection against influenza by injection of DNA encoding a viral protein. Science. 1993;259:1745–9.
Fynan EF, Webster RG, Fuller DH, Haynes JR, Santoro JC, Robinson HL, et al. DNA vaccines: a novel approach to immunization. Int J Immunopharmacol. 1995;17:79–83.
Cox GJ, Zamb TJ, Babiuk LA. Bovine herpesvirus 1: immune responses in mice and cattle injected with plasmid DNA. J Virol. 1993;67:5664–7.
Operschall E, Pavlovic J, Nawrath M, Molling K. Mechanism of protection against influenza A virus by DNA vaccine encoding the hemagglutinin gene. Intervirology. 2000;43:322–30.
Donnelly JJ, Wahren B, Liu MA. DNA vaccines: progress and challenges. J Immunol. 2005;175:633–9.
Kim JH, Jacob J. DNA vaccines against influenza viruses. Curr Top Microbiol Immunol. 2009;333:197–210.
Liu MA. DNA vaccines: a review. J Intern Med. 2003;253:402–10.
Schalk JA, Mooi FR, Berbers GA, van Aerts LA, Ovelgonne H, Kimman TG. Preclinical and clinical safety studies on DNA vaccines. Hum Vaccine. 2006;2:45–53.
Chen Z, Sahashi Y, Matsuo K, Asanuma H, Takahashi H, Iwasaki T, et al. Comparison of the ability of viral protein-expressing plasmid DNAs to protect against influenza. Vaccine. 1998;16:1544–9.
Tan L, Lu H, Zhang D, Wang K, Tian M, Liu C, et al. Efficacy of seasonal pandemic influenza hemagglutinin DNA vaccines delivered by electroporation against a seasonal H1N1 virus challenge in mice. Sci China Life Sci. 2011;54:293–9.
Kadowaki S, Chen Z, Asanuma H, Aizawa C, Kurata T, Tamura S. Protection against influenza virus infection in mice immunized by administration of hemagglutinin-expressing DNAs with electroporation. Vaccine. 2000;18:2779–88.
Fang J, Chen Z, Liu X, Li H, Wang J, Shen X, et al. Immunization with a low dose of hemagglutinin-encoding plasmid protects against 2009 H1N1 pandemic influenza virus in mice. J Virol Methods. 2011;173:314–9.
Fynan EF, Webster RG, Fuller DH, Haynes JR, Santoro JC, Robinson HL. DNA vaccines: protective immunizations by parenteral, mucosal, and gene-gun inoculations. Proc Nat Acad Sci U S A. 1993;90:11478–82.
Johnson PA, Conway MA, Daly J, Nicolson C, Robertson J, Mills KH. Plasmid DNA encoding influenza virus haemagglutinin induces Th1 cells and protection against respiratory infection despite its limited ability to generate antibody responses. J Gen Virol. 2000;81:1737–45.
Wilson JC, von Itzstein M. Recent strategies in the search for new anti-influenza therapies. Curr Drug Targets. 2003;4:389–408.
Munoz ET, Deem MW. Epitope analysis for influenza vaccine design. Vaccine. 2005;23:1144–8.
Tan L, Lu H, Zhang D, Tian M, Hu B, Wang Z, et al. Protection against H1N1 influenza challenge by a DNA vaccine expressing H3/H1 subtype hemagglutinin combined with MHC class II-restricted epitopes. Virol J. 2010;7:363.
Carroll MC. The complement system in B cell regulation. Mol Immunol. 2004;41:141–6.
Molina H, Wong W, Kinoshita T, Brenner C, Foley S, Holers VM. Distinct receptor and regulatory properties of recombinant mouse complement receptor 1 (CR1) and Crry, the two genetic homologues of human CR1. J Exp Med. 1992;175:121–9.
Matsumoto AK, Martin DR, Carter RH, Klickstein LB, Ahearn JM, Fearon DT. Functional dissection of the CD21/CD19/TAPA-1/Leu-13 complex of B lymphocytes. J Exp Med. 1993;178:1407–17.
Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271:348–50.
Carter RH, Fearon DT. CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science. 1992;256:105–7.
Ahmad Z, Yeap S, Ali A, Ho W-Y, Noorjahan B, Hamid M. scFv antibody: principles and clinical application. Clin Dev Immunol. 2012;2012:1–15.
Nagy Z, Rajnavolgyi E, Hollosi M, Toth GK, Varadi G, Penke B, et al. The intersubunit region of the influenza virus haemagglutinin is recognized by antibodies during infection. Scand J Immunol. 1994;40:281–91.
Rajnavolgyi E, Horvath A, Gogolak P, Toth GK, Fazekas G, Fridkin M, et al. Characterizing immunodominant and protective influenza hemagglutinin epitopes by functional activity and relative binding to major histocompatibility complex class II sites. Eur J Immunol. 1997;27:3105–14.
Ivanovska N, Tchorbanov A, Prechl J, Maximova V, Voynova E, Vassilev TL. Immunization with a DNA chimeric molecule encoding a hemagglutinin peptide and a scFv CD21-specific antibody fragment induces long-lasting IgM and CTL responses to influenza virus. Vaccine. 2006;24:1830–7.
Prechl J, Tchorbanov A, Horvath A, Baiu DC, Hazenbos W, Rajnavolgyi E, et al. Targeting of influenza epitopes to murine CR1/CR2 using single-chain antibodies. Immunopharmacology. 1999;42:159–65.
Kinoshita T, Takeda J, Hong K, Kozono H, Sakai H, Inoue K. Monoclonal antibodies to mouse complement receptor type 1 (CR1): their use in a distribution study showing that mouse erythrocytes and platelets are CR1-negative. J Immunol. 1988;140:3066.
Molina H, Holers VM, Li B, Fang Y-F, Mariathasan S, Goellner J, et al. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc Natl Acad Sci USA. 1996;93:3357.
Tchorbanov A, Voynova E, Mihaylova N, Todorov T, Nikolova M, Yomtova VM, et al. Selective silencing of DNA-specific B lymphocytes delays lupus activity in MRL/lpr mice. Eur J Immunol. 2007;37:3587–96.
Bauminger S, Wilchek M. The use of carbodiimides in the preparation of immunizing conjugates. Methods Enzymol. 1980;70:151–9.
Mihaylova N, Voynova E, Tchorbanov A, Dolashka-Angelova P, Bayry J, Devreese B, et al. Simultaneous engagement of FcgammaIIb and CD22 inhibitory receptors silences targeted B cells and suppresses autoimmune disease activity. Mol Immunol. 2009;47:123–30.
Baiu D, Prechl J, Tchorbanov A, Molina H, Erdey A, Capel P, et al. Modulation of antibody responses by antibody-mediated antigen targeting to complement and Fc receptors. J Immunol. 1999;162:3125–30.
Acknowledgments
The work was supported by Grant DTK 02/19 from the National Science Fund, Bulgaria to A. Tchorbanov.
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kerekov, N.S., Ivanova, I.I., Mihaylova, N.M. et al. Built-in adjuvanticity of genetically and protein-engineered chimeric molecules for targeting of influenza A peptide epitopes. Immunol Res 60, 23–34 (2014). https://doi.org/10.1007/s12026-014-8489-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-014-8489-0