
Abstract
Purpose of Review
Over the last years, the focus of clinical and animal research in subarachnoid hemorrhage (SAH) shifted towards the early phase after the bleeding based on the association of the early injury pattern (first 72 h) with secondary complications and poor outcome. This phase is commonly referenced as early brain injury (EBI). In this clinical review, we intended to overview commonly used definitions of EBI, underlying mechanisms, and potential treatment implications.
Recent Findings
We found a large heterogeneity in the definition used for EBI comprising clinical symptoms, neuroimaging parameters, and advanced neuromonitoring techniques. Although specific treatments are currently not available, therapeutic interventions are aimed at ameliorating EBI by improving the energy/supply mismatch in the early phase after SAH.
Summary
Future research integrating brain-derived biomarkers is warranted to improve our pathophysiologic understanding of EBI in order to ameliorate early injury patterns and improve patients’ outcomes.
Similar content being viewed by others
Introduction to the Concept of EBI
Aneurysmal subarachnoid hemorrhage (SAH) accounts for 5–7% of all stroke types [1] and primarily affects young patients at their most productive years. Despite improved neurocritical care management with a decrease in case fatality over the last decades, SAH is still a devastating disease with high long-term morbidity [2]. Only two-thirds of survivors regain functional independence at 1 year after the bleeding [3]. Mechanisms of secondary brain injury after SAH are multifactorial. Although the incidence of vasospasm was successfully decreased in clinical trials, the translation into improved functional outcome failed [4]. In the last years, the focus of experimental and clinical research shifted towards pathophysiologic mechanisms in the first 72 h after the bleeding, commonly referred to as “early brain injury” (EBI). EBI was first described in 2004 by Kusaka et al. [5] and is more and more recognized as an important denominator related to delayed cerebral ischemia (DCI) and long-term morbidity and mortality after SAH [6••, 7].
In this review, we aimed (1) to summarize the current understanding of underlying pathophysiologic mechanisms of EBI, (2) to comment on various definitions of EBI commonly used in literature, and (3) to discuss current and potential future treatment implications. We decided to focus on available studies in humans after SAH to make this review suitable for clinicians in the care of SAH patients.
Mechanisms of EBI
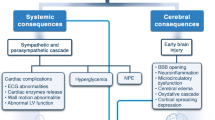
There is increasing evidence that pathophysiologic mechanisms of brain injury start immediately after the bleeding. A sudden increase in intracranial pressure (ICP) caused by the extravasation of blood in the subarachnoid space provokes a decrease in cerebral perfusion (cerebral perfusion pressure, CPP), impairment of autoregulation [8•], and in severe cases, transient or persistent ischemia [9]. Neuronal cell death and endothelial damage result in cytotoxic edema and blood-brain barrier (BBB) breakdown [10,11,12], which aggravates the development of vasogenic edema [13, 14]. Cell death is furthermore believed to be a consequence of microcirculatory failure, microthrombosis, altered ionic homeostasis, excitotoxicity, oxidative stress, and neuronal swelling [7, 15]. Besides ischemia, “nonischemic” mechanisms such as energy dysfunction secondary to cortical spreading depolarizations (SDs) [16] or mitochondrial dysfunction [17] are also considered to be an important mitigator of EBI [18•]. Finally, the blood in the subarachnoid space and intracerebral hemorrhage itself may aggravate brain injury by microglial activation and initiation of a proinflammatory response [19] (Fig. 1).

The complex pathophysiologic mechanisms contributing to early brain injury (EBI) after subarachnoid hemorrhage (SAH). Definitions of EBI used in the clinical setting are heterogenous and include clinical symptoms, neuroimaging parameters, and advanced neuromonitoring techniques. ICP = intracranial pressure, CPP = cerebral perfusion pressure, BBB = blood-brain barrier
Definitions
So far, no consensus exists on a uniform definition of EBI which has led to an uncritical use of the term “EBI” in clinical practice and research. In the following section, we aimed to describe different approaches used for the definition of EBI based on clinical parameters, neuroimaging, and advanced neuromonitoring techniques (Fig. 1).
EBI Based on Clinical Signs and Symptoms
Clinical evaluation of disease severity soon after the bleeding is a mainstay in the definition of EBI. Commonly used grading scales include the Hunt&Hess grade [20] or WFNS scale [21]. Poor-grade patients (commonly defined as H&H or WFNS grade 4–5) with prolonged loss of consciousness [22••] more likely suffer from EBI. It remains questionable whether patients with early obstructive hydrocephalus who mimic a severe disease and rapidly improve after the insertion of an external ventricular drain also qualify for this definition [23]. Together with a poor clinical grade on admission, ictal loss of consciousness (LOC) may best reflect EBI as a clinical equivalent of global cerebral ischemia or progressive intracranial hypertension [22••, 24]. In this context, it is important to mention that early-onset seizures with prolonged time to regain consciousness may again mimic EBI leading to a misclassification of the clinical grade soon after the bleeding. This is supported by the finding that SAH patients with early-onset seizures more often achieve good outcomes as compared with poor-grade patients without early seizures [25]. Still, early-onset seizures may also aggravate EBI, especially if seizures are accompanied by hemodynamic instability and increased energy demand.
Another clinical manifestation suggestive of EBI is early neuroworsening. This may be associated with a high blood burden in the subarachnoid, intraventricular or intraparenchymal space [26], and other factors such as rebleeding [27] early obstructive hydrocephalus, and early-onset seizures.
EBI Based on Neuroimaging
One important neuroimaging biomarker of EBI is the amount of the intracranial blood volume early after the bleeding. Semiquantitative grading scales evaluating the blood burden in the subarachnoid space and the presence of intraventricular or intraparenchymal bleeding are commonly integrated in the prediction of DCI and outcome after SAH [28,29,30]. The association of a higher intracranial blood volume with poor clinical grades and poor outcome [26]suggests its significant contribution to the pathophysiologic concept of EBI. A more sophisticated approach to quantify the amount of blood in the subarachnoid space using a semiautomated process was not better in the prediction of hospital complications and poor outcome [31].
Another commonly used neuroimaging biomarker to EBI is the presence of brain edema early after SAH. While admission global cerebral edema (GCE) correlates with early disease severity and links EBI with secondary brain ischemia and poor outcome, this construct lacks sensitivity in the identification of unilateral or focal brain edema [32, 33]. More recently, a simple semiquantitative score grading both focal and global brain edema (SEBES, Subarachnoid Hemorrhage Early Brain Edema Score) was introduced [6••]. The SEBES is a CT-based evaluation of the absence of visible sulci caused by effacement of sulci or disruption of the gray-white matter junction at 2 predefined brain tissue levels (basal ganglia and centrum semiovale) in each hemisphere. The score ranges from 0 (no edema) to 4 (GCE) and may therefore better describe the transition from focal edema to global brain swelling. Admission high-grade SEBES (3–4 points) was furthermore associated not only with clinical disease severity but also with DCI and poor functional outcome [6••]. Moreover, GCE was quantified by selective sulcal volumes (SSV) using a semiautomatic approach. Smaller SSV suggestive of GCE were also associated with worse outcomes and may be a candidate marker of EBI [34].
Another way to quantify parenchymal pathologies and therefore EBI with advanced neuroimaging techniques comprises the early use of brain MRI by identifying the ischemic lesion load as well as vasogenic and cytotoxic edema [14, 35, 36].
In summary, neuroimaging techniques are useful to quantify EBI in a standardized way and may help to discriminate patients with a higher injury load early after the bleeding. Modern MRI techniques and the use of serial imaging may further elucidate the amount of axonal damage and the result of microvascular ischemic injury after SAH.
EBI Based on Invasive Neuromonitoring
Invasive multimodal neuromonitoring techniques provide a unique opportunity to monitor pathophysiologic mechanisms of EBI at the cellular level in unconscious patients with SAH [37••]. It is important to mention that the selection of poor-grade patients for invasive monitoring limits the generalizability of these results to intermediate- or good-grade patients. The interpretation of study results using invasive neuromonitoring techniques is further limited by on the lack of high spatial resolution because neither unilateral hemispheric damage nor the global pathology may be identified. Integration of the probe location is therefore of paramount importance in the interpretation of brain oxygenation, CBF (cerebral blood flow), and metabolic changes [38]. Analysis of trend statistics is also useful to overcome this limitation and to early identify tissue at risk before a permanent damage occurs [39]. Another noteworthy limitation of data interpretation in the early phase of monitoring is the risk of insertion injury during probe placement. Therefore, the initial hours of monitoring should be interpreted with caution.
So far, only single-center observational trials investigated the early phase after SAH, mostly when the aneurysm has already been secured [18•, 37••, 40, 41]. These data suggest that brain oxygenation is diminished in the early phase after the bleeding.
Reasons for this can be multifactorial, including a compromised oxygen delivery secondary to an increase in ICP, stunned myocardium with decrease in cardiac output, and an increased brain oxygen consumption. The metabolic correlate can be quantified by an increase in the lactate-to-pyruvate ratio (CMD-LPR), a decrease in brain tissue glucose, and a profound excitatory response [37••]. Cell damage may be assessed by increased CMD-glycerol levels [41]. In a study including 18 poor-grade SAH patients, brain metabolic changes in the first 72 h revealed elevated levels of CMD-LPR, CMD-glutamate, and CMD-glycerol in the presence of a normal or even hyperemic state, evaluated by brain computed tomography (CT) perfusion scans [18•]. This finding is important and implicates that impaired cerebral metabolism explaining EBI is not only a consequence of brain tissue hypoxia but also reflects nonischemic metabolic distress including mitochondrial dysfunction [42].
Cerebral microdialysis furthermore allows the analysis of brain extracellular biomarkers, e.g., of axonal injury or neuroinflammation. Recently, higher levels of brain extracellular CMD-total-TAU protein indicating severe axonal damage were linked to poor neurologic outcome at 1 year [43]. These results highlight the potential of this method in further understanding mechanisms of EBI.
EBI Based on Neurohemoinflammation and Systemic Inflammation
There is increasing evidence that neurohemoinflammation occurs early after SAH and is one potential mechanism of EBI [37••]. In a multimodal neuromonitoring study including 26 poor-grade SAH patients, the proinflammatory cytokine CMD-IL6 was highest in the initial phase after SAH and higher in patients with aneurysm rebleeding, GCE, and episodes of CPP < 70 mmHg [37••]. The proposed pathophysiologic mechanism includes early brain edema secondary to the disruption of the BBB and neuronal apoptosis by proinflammatory cytokines [10, 44]. Concordantly, CMD-MMP-9 levels were upregulated in the first 12 h after monitoring start, especially in patients with LOC, poor clinical grade, and initial brain tissue hypoxia [37••]. MMP-9 is known to be involved in endothelial basal membrane damage, neuroinflammation, and apoptosis and may therefore play an important role in the pathogenesis of EBI [45]. Neither CMD-IL6 nor CMD-MMP-9 was associated with systemic inflammatory markers underlining the idea of compartmentalization of the central nervous system. Still, there is evidence that also increased peripheral levels of cytokines are associated with EBI [46••]. In specific, IL-6, IL-10, and MIP1ß have recently been identified as an indicator of EBI using correlation network analysis in humans [46••]. Moreover, early platelet activation, systemic inflammation, and SIRS (systemic inflammatory response syndrome) are common in the early phase after SAH and associated with disease severity and poor functional outcome [47, 48•].
EBI Based on Cortical Spreading Depolarizations
Recent evidence suggests that electrical brain failure may contribute to brain tissue injury after SAH [49••, 50••]. Cortical spreading depolarizations (SDs) are self-propagating waves of neuronal and glial electrical depolarization [51], which can be recorded using subdural strip electrodes in humans [52]. Although SDs may be associated with vasodilation in healthy subjects, their occurrence after SAH is commonly associated with inverse neurovascular coupling leading to hypoperfusion, brain tissue hypoxia, and metabolic derangement. SDs have been identified as a determinant of EBI in patients after SAH although the proof of causality needs further confirmation. In a recent study including 23 poor-grade SAH patients, the presence of ischemic and/or hemorrhagic lesions in the frontal cortex was associated with a higher incidence of SDs [49••]. The association between SDs and early focal brain injury is further supported by a larger study conducted by Eriksen et al. [50••]. While 33/37 (89%) patients with early focal brain injury exhibited SDs in the first 4 days, only 7/17 (41%) patients without early focal brain injury had SDs. Moreover, SDs correlated with the volume of early focal brain injury during the acute phase after SAH in this study.
EBI Based on EEG Findings
Immediate or early-onset seizures commonly described as convulsive seizures occur at ictus and within the first 12 to 24 h after SAH [53]. Convulsive seizures in the very early phase were reported in 4.8% of SAH patients in a systematic review including 14 studies [53]. Although not entirely understood, they may be triggered by transient biochemical changes following the bleeding. Associated factors with ictal and early seizures include raised ICP, direct toxicity of the blood, neuroinflammation, vasospasm, and SDs. From a clinical point of view it can be difficult to differentiate between true seizures and nonepileptic movements related to raised ICP or herniation in the initial phase after SAH. As already mentioned, clinical reevaluation after prehospital seizures is important since seizures may lead to misclassification of clinical grades. The value of continuous EEG (cEEG) monitoring in the early phase after SAH needs further confirmation based on prospective multicenter studies. The primary goal of cEEG monitoring in SAH patients lies in the detection of subclinical seizures and nonconvulsive status epilepticus (NCSE) [54]. Around 3 to 19% of SAH patients develop NC seizures and 11% have NCSE [55,56,57].
Current Implications for Clinical Practice
It is important to separate primary brain damage from mechanisms leading to secondary brain injury, which are potentially amenable to specific treatment strategies. Currently, there is no therapy available specifically targeting EBI. Management strategies to ameliorate EBI in the early phase after SAH primarily aim to provide sufficient energy supply to the brain and normalizing pathological parameters which are known to interfere with aggravation of EBI.
ICP Control
Early intracranial hypertension is strongly interrelated with the pathogenesis of EBI and can result from multiple factors including GCE, acute hydrocephalus, intraparenchymal hematoma, and intraventricular hemorrhage. A stepwise approach to decrease ICP depending on the need of neurosurgical interventions and other common treatment strategies should be followed to decrease the risk of brain injury. Recent studies suggest that the amount of raised ICP above a certain threshold (ICP-burden) is more important than absolute ICP levels [58].
Approximately 50% of patients are admitted with obstructive hydrocephalus [59] and urgently need sufficient cerebrospinal fluid (CSF) drainage [60, 61]. Although the early development of hydrocephalus may not primarily be regarded as a marker of EBI, inadequate or delayed treatment can easily aggravate EBI and result in secondary brain damage. Other interventions to decrease ICP include optimal positioning of the patient, ventilation strategies targeting normocapnia or short-term hyperventilation, adequate sedation, and analgesia and surgical hematoma evacuation in case of a mass lesion [7, 62]. Osmotherapy is commonly used to decrease ICP although the effect on outcome is less clear. Only case series addressing the impact of hypertonic saline on ICP treatment could be identified with solely one study showing outcome improvement in poor-grade SAH patients [63]. In a multimodal neuromonitoring study, a potential benefit of normothermia due to lowering of ICP and ameliorating metabolic distress was suggested [64]. As a last tier treatment option of refractory ICP, hypothermia [65], barbiturate coma, or decompressive craniectomy [66, 67] may be considered.
CPP Optimization
The optimal range of CPP levels in different phases after SAH is less clear. While a systolic blood pressure below 140 mmHg should be targeted before aneurysm obliteration, permissive hypertension is recommended when DCI is diagnosed [60, 61]. It is important to mention that a CPP targeted management with pressure calibration at the level of foramen of Monro is recommended when ICP is measured continuously. Recent data derived from observational studies using invasive multimodal monitoring techniques suggest that a higher CPP (> 70 mmHg) may ameliorate brain tissue hypoxia and metabolic distress in poor-grade SAH patients [37••, 68]. Still, a large interindividual variability may exist depending on the status of cerebral autoregulation (CA). CA is commonly impaired in the early phase after SAH, although recent data argue against the use of a predefined cutoff level for diagnosis [8•]. Systemic application of erythropoietin within 72 h of bleeding shortened the time with impaired autoregulation and reduced the risk of developing DCI after SAH in a prospective randomized trial (EPO trial) [69]. Because evidence of individualized CPP targets based on the CA status in acutely brain-injured patients is lacking, a phase II trial is currently investigating the safety and feasibility of such an approach in traumatic brain injury (TBI) patients (COGITATE trial: NCT02982122).
Further studies are needed to support the idea of individualized CPP goals as part of personalized medicine integrating the information of multimodal neuromonitoring techniques in the management of poor-grade SAH patients at high risk of EBI.
Management of GCE
There is limited literature how to best treat patients with admission GCE or high-grade SEBES. Admission GCE was associated with hypermetabolism and metabolic distress arguing for interventions targeting at sufficient energy supply and amelioration of metabolic demand [33, 70]. In this line, CPP levels above 90 mmHg were associated with improved brain metabolism in an observational trial in patients with GCE [33]. Further studies are needed to support such an invasive treatment approach. Another potential intervention to target GCE is hypothermia [71], which is endorsed by animal data and needs confirmation in clinical trials. For now, targeted temperature management (TTM) in the sense of normothermia is commonly applied in many centers taking care of SAH patients (see next paragraph) [72].
Targeted Temperature Management
Up to 60% of critically ill brain-injured patients experience fever during the first 24 h after admission [73]. Based on the known deleterious effect of fever on outcome [73, 74], normothermia is currently recommended [75, 76]. So far, there is no high-grade evidence that normothermia improves outcome in SAH patients. A prospective, randomized, multicentre study currently investigates the impact of normothermia on functional outcome (INTREPID: NCT02996266) after acute brain injury including SAH. As mentioned previously, hypothermia was only tested in the setting of refractory ICP and GCE in small single-center trials in combination with barbiturate coma [65, 71].
Management of Intracerebral Blood Burden
Since the intracranial blood burden is associated with poor outcome, trials aimed at aggressive neurosurgical clearance of blood with conflicting results which is therefore not recommended by guidelines [60, 61]. There is some evidence that a reduction of the postoperative clot volume may be associated with a reduction in secondary complications, including DCI; however, this approach has to be tested prospectively [77].
Management of Cortical Spreading Depolarizations
Addressing the cessation of SDs remains challenging and raises questions about the optimal pharmacological agent. Case reports and a retrospective review [78] suggest that ketamine has a dose-dependent suppressive effect on SDs. A first prospective small pilot study in ten TBI and SAH patients confirmed an effective inhibition of SDs by ketamine over wide ranges of dosage [79]. The effect on outcome improvement needs further investigations. Other potential treatment candidates include hypothermia and nimodipine. For now, targeting normothermia and normotension and providing sufficient energy supply to the brain are recommended by experts since fever, hypotension, and hypoglycemia may trigger SDs in acutely brain-injured patients [80].
Management of Seizures
Antiepileptic therapy should be initiated in patients with seizures; however, a prophylactic use is not recommended [60, 61].
Treatment of Aneurysms
Optimal patient management with early aneurysm securing aims at the prevention of rebleeding [60, 61]. Most of rebleeding events occur in the very early phase after SAH and result in poor functional outcome or death [27]. Although early obliteration of aneurysms might be beneficial, conflicting data exist for best timing of aneurysm treatment [81].
Management of Neuroinflammation
Several agents including ASS, NSAIDs, thromboxane synthase inhibitors, steroids, nitric oxide donors, and immunosuppressant therapies have been tested to treat neuroinflammation after SAH; however, none of these drugs can be recommended as routine treatment so far [82••]. In a multimodal neuromonitoring study including 24 poor-grade SAH patients, brain interstitial CMD-IL 6 levels decreased following the administration of diclofenac [83]. These findings suggest a positive effect of parenteral diclofenac on the extracellular proinflammatory response in these patients.
Conclusion
In summary, pathophysiologic mechanisms in the first 72 h after SAH gained clinical and research interest in the past decade. Based on the association of a more severe injury pattern early after the bleeding with secondary complications and poor outcome, future research should focus on EBI by integrating brain-derived biomarkers in order to improve the critical care management after SAH. An agreement on the use of a common definition is strongly needed.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2003;2(1):43–53.
Nieuwkamp DJ, Setz LE, Algra A, Linn FH, de Rooij NK, Rinkel GJ. Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta-analysis. Lancet Neurol. 2009;8(7):635–42. https://doi.org/10.1016/S1474-4422(09)70126-7.
Rinkel GJ, Algra A. Long-term outcomes of patients with aneurysmal subarachnoid haemorrhage. Lancet Neurol. 2011;10(4):349–56. https://doi.org/10.1016/S1474-4422(11)70017-5.
Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 2011;10(7):618–25. https://doi.org/10.1016/S1474-4422(11)70108-9.
Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2004;24(8):916–25. https://doi.org/10.1097/01.WCB.0000125886.48838.7E.
•• Ahn SH, Savarraj JP, Pervez M, Jones W, Park J, Jeon SB, et al. The subarachnoid hemorrhage early brain edema score predicts delayed cerebral ischemia and clinical outcomes. Neurosurgery. 2018;83(1):137–45. https://doi.org/10.1093/neuros/nyx364 The authors developed a new radiographic score grading cerebral edema in SAH patients and found an association with DCI and poor functional outcome.
de Oliveira Manoel AL, Goffi A, Marotta TR, Schweizer TA, Abrahamson S, Macdonald RL. The critical care management of poor-grade subarachnoid haemorrhage. Crit Care. 2016;20:21. https://doi.org/10.1186/s13054-016-1193-9.
• Gaasch M, Schiefecker AJ, Kofler M, Beer R, Rass V, Pfausler B, et al. Cerebral autoregulation in the prediction of delayed cerebral ischemia and clinical outcome in poor-grade aneurysmal subarachnoid hemorrhage patients. Crit Care Med. 2018;46(5):774–80. https://doi.org/10.1097/CCM.0000000000003016 Impairement of cerebral autoregulation was evident in the intial phase after SAH reflecting early brain injury. Patients with DCI had higher pressure reactivity index values in the first 72 hours.
Grote E, Hassler W. The critical first minutes after subarachnoid hemorrhage. Neurosurgery. 1988;22(4):654–61.
Ostrowski RP, Colohan AR, Zhang JH. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28(4):399–414. https://doi.org/10.1179/016164106X115008.
Thal SC, Sporer S, Klopotowski M, Thal SE, Woitzik J, Schmid-Elsaesser R, et al. Brain edema formation and neurological impairment after subarachnoid hemorrhage in rats. J Neurosurg. 2009;111(5):988–94. https://doi.org/10.3171/2009.3.JNS08412.
Hayman EG, Wessell A, Gerzanich V, Sheth KN, Simard JM. Mechanisms of global cerebral edema formation in aneurysmal subarachnoid hemorrhage. Neurocrit Care. 2017;26(2):301–10. https://doi.org/10.1007/s12028-016-0354-7.
Keep RF, Andjelkovic AV, Stamatovic SM, Shakui P, Ennis SR. Ischemia-induced endothelial cell dysfunction. Acta Neurochir Suppl. 2005;95:399–402.
Weimer JM, Jones SE, Frontera JA. Acute cytotoxic and vasogenic edema after subarachnoid hemorrhage: a quantitative MRI study. AJNR Am J Neuroradiol. 2017;38(5):928–34. https://doi.org/10.3174/ajnr.A5181.
Chou SH, Feske SK, Simmons SL, Konigsberg RG, Orzell SC, Marckmann A, et al. Elevated peripheral neutrophils and matrix metalloproteinase 9 as biomarkers of functional outcome following subarachnoid hemorrhage. Transl Stroke Res. 2011;2(4):600–7. https://doi.org/10.1007/s12975-011-0117-x.
Sakowitz OW, Santos E, Nagel A, Krajewski KL, Hertle DN, Vajkoczy P, et al. Clusters of spreading depolarizations are associated with disturbed cerebral metabolism in patients with aneurysmal subarachnoid hemorrhage. Stroke. 2013;44(1):220–3. https://doi.org/10.1161/STROKEAHA.112.672352.
Jacobsen A, Nielsen TH, Nilsson O, Schalen W, Nordstrom CH. Bedside diagnosis of mitochondrial dysfunction in aneurysmal subarachnoid hemorrhage. Acta Neurol Scand. 2014;130(3):156–63. https://doi.org/10.1111/ane.12258.
• Carteron L, Patet C, Solari D, Messerer M, Daniel RT, Eckert P, et al. Non-ischemic cerebral energy dysfunction at the early brain injury phase following aneurysmal subarachnoid hemorrhage. Front Neurol. 2017;8:325. https://doi.org/10.3389/fneur.2017.00325 This multimodal neuromonitoring study indicates that cerebral energy dysfunction (CMD-LPR >40 or CMD-lactate >4 mmol/L) was associated with normal or hyperemic CBF in the early phase after SAH.
Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl Stroke Res. 2013;4(4):432–46. https://doi.org/10.1007/s12975-013-0257-2.
Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair of intracranial aneurysms. J Neurosurg. 1968;28(1):14–20. https://doi.org/10.3171/jns.1968.28.1.0014.
Teasdale GM, Drake CG, Hunt W, Kassell N, Sano K, Pertuiset B, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry. 1988;51(11):1457.
•• Suwatcharangkoon S, Meyers E, Falo C, Schmidt JM, Agarwal S, Claassen J, et al. Loss of consciousness at onset of subarachnoid hemorrhage as an important marker of early brain injury. JAMA Neurol. 2016;73(1):28–35. https://doi.org/10.1001/jamaneurol.2015.3188 The results of this study show that loss of consciousness at ictus was associated with markers of EBI including a poor clinical grade, intracerebral blood burden, and global cerebral edema. LOC was associated with poor functional outcome.
Ransom ER, Mocco J, Komotar RJ, Sahni D, Chang J, Hahn DK, et al. External ventricular drainage response in poor grade aneurysmal subarachnoid hemorrhage: effect on preoperative grading and prognosis. Neurocrit Care. 2007;6(3):174–80. https://doi.org/10.1007/s12028-007-0019-7.
Hayashi T, Suzuki A, Hatazawa J, Kanno I, Shirane R, Yoshimoto T, et al. Cerebral circulation and metabolism in the acute stage of subarachnoid hemorrhage. J Neurosurg. 2000;93(6):1014–8. https://doi.org/10.3171/jns.2000.93.6.1014.
Fung C, Balmer M, Murek M, Z’Graggen WJ, Abu-Isa J, Ozdoba C, et al. Impact of early-onset seizures on grading and outcome in patients with subarachnoid hemorrhage. J Neurosurg. 2015;122(2):408–13. https://doi.org/10.3171/2014.10.JNS14163.
Helbok R, Kurtz P, Vibbert M, Schmidt MJ, Fernandez L, Lantigua H, et al. Early neurological deterioration after subarachnoid haemorrhage: risk factors and impact on outcome. J Neurol Neurosurg Psychiatry. 2013;84(3):266–70. https://doi.org/10.1136/jnnp-2012-302804.
Zhao B, Fan Y, Xiong Y, Yin R, Zheng K, Li Z, et al. Aneurysm rebleeding after poor-grade aneurysmal subarachnoid hemorrhage: predictors and impact on clinical outcomes. J Neurol Sci. 2016;371:62–6. https://doi.org/10.1016/j.jns.2016.10.020.
Frontera JA, Claassen J, Schmidt JM, Wartenberg KE, Temes R, Connolly ES Jr, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhage: the modified fisher scale. Neurosurgery. 2006;59(1):21–7; discussion −7. https://doi.org/10.1227/01.NEU.0000218821.34014.1B.
Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery. 1980;6(1):1–9.
Hijdra A, van Gijn J, Nagelkerke NJ, Vermeulen M, van Crevel H. Prediction of delayed cerebral ischemia, rebleeding, and outcome after aneurysmal subarachnoid hemorrhage. Stroke. 1988;19(10):1250–6.
Ko SB, Choi HA, Carpenter AM, Helbok R, Schmidt JM, Badjatia N, et al. Quantitative analysis of hemorrhage volume for predicting delayed cerebral ischemia after subarachnoid hemorrhage. Stroke. 2011;42(3):669–74. https://doi.org/10.1161/STROKEAHA.110.600775.
Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002;33(5):1225–32.
Helbok R, Ko SB, Schmidt JM, Kurtz P, Fernandez L, Choi HA, et al. Global cerebral edema and brain metabolism after subarachnoid hemorrhage. Stroke. 2011;42(6):1534–9. https://doi.org/10.1161/STROKEAHA.110.604488.
Choi HA, Bajgur SS, Jones WH, Savarraj JP, Ko SB, Edwards NJ, et al. Quantification of cerebral edema after subarachnoid hemorrhage. Neurocrit Care. 2016;25(1):64–70. https://doi.org/10.1007/s12028-015-0229-3.
De Marchis GM, Filippi CG, Guo X, Pugin D, Gaffney CD, Dangayach NS, et al. Brain injury visible on early MRI after subarachnoid hemorrhage might predict neurological impairment and functional outcome. Neurocrit Care. 2015;22(1):74–81. https://doi.org/10.1007/s12028-014-0008-6.
Frontera JA, Ahmed W, Zach V, Jovine M, Tanenbaum L, Sehba F, et al. Acute ischaemia after subarachnoid haemorrhage, relationship with early brain injury and impact on outcome: a prospective quantitative MRI study. J Neurol Neurosurg Psychiatry. 2015;86(1):71–8. https://doi.org/10.1136/jnnp-2013-307313.
•• Helbok R, Schiefecker AJ, Beer R, Dietmann A, Antunes AP, Sohm F, et al. Early brain injury after aneurysmal subarachnoid hemorrhage: a multimodal neuromonitoring study. Crit Care. 2015;19:75. https://doi.org/10.1186/s13054-015-0809-9 In this multimodal neuromonitoring study, the authors describe the brain metabolic profile early after SAH. They found that metabolic distress, brain tissue hypoxia, and a higher inflammatory response were common and were associated with poor functional outcome.
Kofler M, Gaasch M, Rass V, Schiefecker AJ, Ianosi B, Lindner A, et al. The importance of probe location for the interpretation of cerebral microdialysis data in subarachnoid hemorrhage patients. Neurocrit Care. 2019. https://doi.org/10.1007/s12028-019-00713-8.
Le Roux P, Menon DK, Citerio G, Vespa P, Bader MK, Brophy GM, et al. Consensus summary statement of the International Multidisciplinary Consensus Conference on Multimodality Monitoring in Neurocritical Care: a statement for healthcare professionals from the Neurocritical Care Society and the European Society of Intensive Care Medicine. Neurocrit Care. 2014;21(Suppl 2):S1–26. https://doi.org/10.1007/s12028-014-0041-5.
Helbok R, Kofler M, Schiefecker AJ, Gaasch M, Rass V, Pfausler B, et al. Clinical use of cerebral microdialysis in patients with aneurysmal subarachnoid hemorrhage-state of the art. Front Neurol. 2017;8:565. https://doi.org/10.3389/fneur.2017.00565.
Sarrafzadeh A, Haux D, Kuchler I, Lanksch WR, Unterberg AW. Poor-grade aneurysmal subarachnoid hemorrhage: relationship of cerebral metabolism to outcome. J Neurosurg. 2004;100(3):400–6. https://doi.org/10.3171/jns.2004.100.3.0400.
Hutchinson PJ, Jalloh I, Helmy A, Carpenter KL, Rostami E, Bellander BM, et al. Consensus statement from the 2014 International Microdialysis Forum. Intensive Care Med. 2015;41(9):1517–28. https://doi.org/10.1007/s00134-015-3930-y.
Helbok R, Schiefecker A, Delazer M, Beer R, Bodner T, Pfausler B, et al. Cerebral tau is elevated after aneurysmal subarachnoid haemorrhage and associated with brain metabolic distress and poor functional and cognitive long-term outcome. J Neurol Neurosurg Psychiatry. 2015;86(1):79–86. https://doi.org/10.1136/jnnp-2013-307326.
Sozen T, Tsuchiyama R, Hasegawa Y, Suzuki H, Jadhav V, Nishizawa S, et al. Role of interleukin-1beta in early brain injury after subarachnoid hemorrhage in mice. Stroke. 2009;40(7):2519–25. https://doi.org/10.1161/STROKEAHA.109.549592.
Guo Z, Sun X, He Z, Jiang Y, Zhang X. Role of matrix metalloproteinase-9 in apoptosis of hippocampal neurons in rats during early brain injury after subarachnoid hemorrhage. Neurol Sci : official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology. 2010;31(2):143–9. https://doi.org/10.1007/s10072-009-0192-x.
•• Savarraj J, Parsha K, Hergenroeder G, Ahn S, Chang TR, Kim DH, et al. Early brain injury associated with systemic inflammation after subarachnoid hemorrhage. Neurocrit Care. 2018;28(2):203–11. https://doi.org/10.1007/s12028-017-0471-y Systemic inflammatory markers including IL-6 and MIP1β were significantly associated with global cerebral edema and poor clinical grade reflective of EBI in this study.
Rass V, Gaasch M, Kofler M, Schiefecker AJ, Ianosi BA, Rhomberg P, et al. Systemic inflammatory response syndrome as predictor of poor outcome in nontraumatic subarachnoid hemorrhage patients. Crit Care Med. 2018;46(12):e1152–e9. https://doi.org/10.1097/CCM.0000000000003429.
• Frontera JA, Provencio JJ, Sehba FA, McIntyre TM, Nowacki AS, Gordon E, et al. The role of platelet activation and inflammation in early brain injury following subarachnoid hemorrhage. Neurocrit Care. 2017;26(1):48–57. https://doi.org/10.1007/s12028-016-0292-4 In this study, including 127 SAH patients, early platelet activation and increased CRP were associated with poor clinical grades, delayed cerebral edema, and poor functional outcome.
•• Hartings JA, York J, Carroll CP, Hinzman JM, Mahoney E, Krueger B, et al. Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction. Brain : a journal of neurology. 2017;140(10):2673–90. https://doi.org/10.1093/brain/awx214 In this study, including 23 poor-grade SAH patients, the authors could show that the presence of ischemic and/or hemorrhagic lesions in the frontal cortex was associated with a higher incidence of SDs.
•• Eriksen N, Rostrup E, Fabricius M, Scheel M, Major S, Winkler MKL, et al. Early focal brain injury after subarachnoid hemorrhage correlates with spreading depolarizations. Neurology. 2019;92(4):e326–e41. https://doi.org/10.1212/WNL.0000000000006814 This study links early focal brain injury with SDs. 89% patients with early focal brain injury exhibited SDs and only 41% patients without early focal brain injury had SDs.
Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med. 2011;17(4):439–47. https://doi.org/10.1038/nm.2333.
Dreier JP, Fabricius M, Ayata C, Sakowitz OW, William Shuttleworth C, Dohmen C, et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: review and recommendations of the COSBID research group. J Cereb Blood Flow Metab : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2017;37(5):1595–625. https://doi.org/10.1177/0271678X16654496.
Raper DM, Starke RM, Komotar RJ, Allan R, Connolly ES Jr. Seizures after aneurysmal subarachnoid hemorrhage: a systematic review of outcomes. World Neurosurg. 2013;79(5–6):682–90. https://doi.org/10.1016/j.wneu.2012.08.006.
Claassen J, Hirsch LJ, Frontera JA, Fernandez A, Schmidt M, Kapinos G, et al. Prognostic significance of continuous EEG monitoring in patients with poor-grade subarachnoid hemorrhage. Neurocrit Care. 2006;4(2):103–12. https://doi.org/10.1385/NCC:4:2:103.
Claassen J, Mayer SA, Hirsch LJ. Continuous EEG monitoring in patients with subarachnoid hemorrhage. J Clin Neurophysiol : official publication of the American Electroencephalographic Society. 2005;22(2):92–8.
Claassen J, Albers D, Schmidt JM, De Marchis GM, Pugin D, Falo CM, et al. Nonconvulsive seizures in subarachnoid hemorrhage link inflammation and outcome. Ann Neurol. 2014;75(5):771–81. https://doi.org/10.1002/ana.24166.
Claassen J, Taccone FS, Horn P, Holtkamp M, Stocchetti N, Oddo M, et al. Recommendations on the use of EEG monitoring in critically ill patients: consensus statement from the neurointensive care section of the ESICM. Intensive Care Med. 2013;39(8):1337–51. https://doi.org/10.1007/s00134-013-2938-4.
Helbok R, Meyfroidt G, Beer R. Intracranial pressure thresholds in severe traumatic brain injury: con : the injured brain is not aware of ICP thresholds! Intensive Care Med. 2018. https://doi.org/10.1007/s00134-018-5249-y.
Hellingman CA, van den Bergh WM, Beijer IS, van Dijk GW, Algra A, van Gijn J, et al. Risk of rebleeding after treatment of acute hydrocephalus in patients with aneurysmal subarachnoid hemorrhage. Stroke. 2007;38(1):96–9. https://doi.org/10.1161/01.STR.0000251841.51332.1d.
Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G, et al. European Stroke Organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis. 2013;35(2):93–112. https://doi.org/10.1159/000346087.
Connolly ES Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2012;43(6):1711–37. https://doi.org/10.1161/STR.0b013e3182587839.
Smith ER, Carter BS, Ogilvy CS. Proposed use of prophylactic decompressive craniectomy in poor-grade aneurysmal subarachnoid hemorrhage patients presenting with associated large sylvian hematomas. Neurosurgery. 2002;51(1):117–24 discussion 24.
Tseng MY, Al-Rawi PG, Czosnyka M, Hutchinson PJ, Richards H, Pickard JD, et al. Enhancement of cerebral blood flow using systemic hypertonic saline therapy improves outcome in patients with poor-grade spontaneous subarachnoid hemorrhage. J Neurosurg. 2007;107(2):274–82. https://doi.org/10.3171/JNS-07/08/0274.
Oddo M, Frangos S, Milby A, Chen I, Maloney-Wilensky E, Murtrie EM, et al. Induced normothermia attenuates cerebral metabolic distress in patients with aneurysmal subarachnoid hemorrhage and refractory fever. Stroke. 2009;40(5):1913–6. https://doi.org/10.1161/STROKEAHA.108.534115.
Seule MA, Muroi C, Mink S, Yonekawa Y, Keller E. Therapeutic hypothermia in patients with aneurysmal subarachnoid hemorrhage, refractory intracranial hypertension, or cerebral vasospasm. Neurosurgery. 2009;64(1):86–92; discussion −3. https://doi.org/10.1227/01.NEU.0000336312.32773.A0.
Stuart RM, Claassen J, Schmidt M, Helbok R, Kurtz P, Fernandez L, et al. Multimodality neuromonitoring and decompressive hemicraniectomy after subarachnoid hemorrhage. Neurocrit Care. 2011;15(1):146–50. https://doi.org/10.1007/s12028-009-9264-2.
Schirmer CM, Hoit DA, Malek AM. Decompressive hemicraniectomy for the treatment of intractable intracranial hypertension after aneurysmal subarachnoid hemorrhage. Stroke. 2007;38(3):987–92. https://doi.org/10.1161/01.STR.0000257962.58269.e2.
Schmidt JM, Ko SB, Helbok R, Kurtz P, Stuart RM, Presciutti M, et al. Cerebral perfusion pressure thresholds for brain tissue hypoxia and metabolic crisis after poor-grade subarachnoid hemorrhage. Stroke. 2011;42(5):1351–6. https://doi.org/10.1161/STROKEAHA.110.596874.
Tseng MY, Hutchinson PJ, Richards HK, Czosnyka M, Pickard JD, Erber WN, et al. Acute systemic erythropoietin therapy to reduce delayed ischemic deficits following aneurysmal subarachnoid hemorrhage: a phase II randomized, double-blind, placebo-controlled trial. J Neurosurg. 2009;111(1):171–80. https://doi.org/10.3171/2009.3.JNS081332.
Zetterling M, Hallberg L, Ronne-Engstrom E. Early global brain oedema in relation to clinical admission parameters and outcome in patients with aneurysmal subarachnoid haemorrhage. Acta Neurochir. 2010;152(9):1527–33; discussion 33. https://doi.org/10.1007/s00701-010-0684-8.
Gasser S, Khan N, Yonekawa Y, Imhof HG, Keller E. Long-term hypothermia in patients with severe brain edema after poor-grade subarachnoid hemorrhage: feasibility and intensive care complications. J Neurosurg Anesthesiol. 2003;15(3):240–8.
Picetti E, Oddo M, Prisco L, Helbok R, Taccone FS. A survey on fever monitoring and management in patients with acute brain injury: the SUMMA study. J Neurosurg Anesthesiol. 2018. https://doi.org/10.1097/ANA.0000000000000536.
Rincon F, Patel U, Schorr C, Lee E, Ross S, Dellinger RP, et al. Brain injury as a risk factor for fever upon admission to the intensive care unit and association with in-hospital case fatality: a matched cohort study. J Intensive Care Med. 2015;30(2):107–14. https://doi.org/10.1177/0885066613508266.
Wartenberg KE, Schmidt JM, Claassen J, Temes RE, Frontera JA, Ostapkovich N, et al. Impact of medical complications on outcome after subarachnoid hemorrhage. Crit Care Med. 2006;34(3):617–23 quiz 24.
Diringer MN, Bleck TP, Claude Hemphill J 3rd, Menon D, Shutter L, Vespa P, et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society’s Multidisciplinary Consensus Conference. Neurocrit Care. 2011;15(2):211–40. https://doi.org/10.1007/s12028-011-9605-9.
Cariou A, Payen JF, Asehnoune K, Audibert G, Botte A, Brissaud O, et al. Targeted temperature management in the ICU: guidelines from a French expert panel. Ann Intensive Care. 2017;7(1):70. https://doi.org/10.1186/s13613-017-0294-1.
Ota N, Matsukawa H, Kamiyama H, Tsuboi T, Noda K, Hashimoto A, et al. Preventing cerebral vasospasm after aneurysmal subarachnoid hemorrhage with aggressive cisternal clot removal and nicardipine. World Neurosurg. 2017;107:630–40. https://doi.org/10.1016/j.wneu.2017.08.088.
Hertle DN, Dreier JP, Woitzik J, Hartings JA, Bullock R, Okonkwo DO, et al. Effect of analgesics and sedatives on the occurrence of spreading depolarizations accompanying acute brain injury. Brain. 2012;135(Pt 8):2390–8. https://doi.org/10.1093/brain/aws152.
Carlson AP, Abbas M, Alunday RL, Qeadan F, Shuttleworth CW. Spreading depolarization in acute brain injury inhibited by ketamine: a prospective, randomized, multiple crossover trial. J Neurosurg. 2018;130:1–7. https://doi.org/10.3171/2017.12.JNS171665.
Helbok R, Hartings JA, Schiefecker A, Balanca B, Jewel S, Foreman B, et al. What should a clinician do when spreading depolarizations are observed in a patient? Neurocrit Care. 2019. https://doi.org/10.1007/s12028-019-00777-6.
Rawal S, Alcaide-Leon P, Macdonald RL, Rinkel GJ, Victor JC, Krings T, et al. Meta-analysis of timing of endovascular aneurysm treatment in subarachnoid haemorrhage: inconsistent results of early treatment within 1 day. J Neurol Neurosurg Psychiatry. 2017;88(3):241–8. https://doi.org/10.1136/jnnp-2016-314596.
•• de Oliveira Manoel AL, Macdonald RL. Neuroinflammation as a target for intervention in subarachnoid hemorrhage. Front Neurol. 2018;9:292. https://doi.org/10.3389/fneur.2018.00292 In this review, anti-inflammatory drugs tested after SAH are discussed.
Schiefecker AJ, Rass V, Gaasch M, Kofler M, Thome C, Humpel C, et al. Brain extracellular interleukin-6 levels decrease following antipyretic therapy with diclofenac in patients with spontaneous subarachnoid hemorrhage. Ther Hypothermia Temp Manag. 2018;9:48–55. https://doi.org/10.1089/ther.2018.0001.
Funding
Open access funding provided by University of Innsbruck and Medical University of Innsbruck.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Raimund Helbok received speaker’s honoraria of BARD Medical and ZOLL Medical and serves in the advisory board of the Intrepid trial (Bard Medical). Verena Rass declares no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Critical Care
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rass, V., Helbok, R. Early Brain Injury After Poor-Grade Subarachnoid Hemorrhage. Curr Neurol Neurosci Rep 19, 78 (2019). https://doi.org/10.1007/s11910-019-0990-3
Published:
DOI: https://doi.org/10.1007/s11910-019-0990-3