
Abstract
Purpose of Review
Advancements in our understanding of the hepatitis B viral (HBV) life cycle have paved the way for novel approaches to treat HBV infection. This review summarizes the various strategies being pursued to achieve a functional cure, defined as loss of hepatitis B surface antigen (HBsAg) and absence of viral replication 6 months off-therapy.
Recent Findings
Direct acting antiviral, host targeting antiviral, and immunological approaches are in various stages of development as treatment for chronic HBV infection.
Summary
Novel treatments are being developed in pursuit of a cure for HBV. Current evidence suggests a single therapeutic agent alone may be insufficient, necessitating the need for combination therapy targeting HBV and the host immune response. Ongoing research focused on identifying the best therapeutic combination holds promise in achieving functional cure for HBV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite the availability of a safe and effective vaccine for over four decades, the global health burden of hepatitis B virus (HBV) infection remains high. Recent estimates suggest there are ~ 300 million people with chronic infection resulting in 820,000 deaths annually, primarily due to complications of cirrhosis and hepatocellular carcinoma (HCC) [1]. Additionally, there are approximately 1.5 million incident infections annually. Acknowledging this substantial health burden, the World Health Organization (WHO) has set an ambitious goal of reducing HBV incidence by 90% and HBV-related mortality by 65% by 2030 compared to 2015 levels [2].
Of the two approved classes of therapy for chronic hepatitis B (CHB), peginterferon alfa (Peg-IFN) and nucleos(t)ide analogs (NAs), NAs are the mainstay of treatment. NAs effectively suppress viral replication, improve hepatic inflammation, and reduce progression to cirrhosis and HCC [3]. However, only a small percentage of patients (≤ 1%) achieve the desired endpoint of functional cure, defined as the loss of hepatitis B surface antigen (HBsAg) and HBV DNA below the level of quantitation, that permits safe discontinuation of therapy [4, 5•]. Consequently, most patients require long-term therapy. This is because NAs do not directly target the two sources of HBsAg in serum-covalently closed circular DNA (cccDNA), a non-integrated form of the viral DNA located within the hepatocyte nucleus that serves as the template for viral transcription, and integrated HBV DNA [6, 7]. Although NAs have an excellent safety profile [8], long-term use is associated with increased healthcare costs, medication noncompliance, and risk of renal and bone toxicity. Therefore, there is a need for a novel, finite duration therapy that can achieve high rates of functional cure [9•].
This review focuses on novel therapies in development that hold promise for achieving functional cure. Advancements in therapeutics in combination with more widespread use of HBV vaccination will be crucial for reducing the global burden of HBV infection and achieving the WHO’s goal of HBV eradication.
The HBV Viral Life Cycle
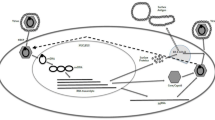
HBV is a small hepatotropic DNA virus with a partially double-stranded circular genome of approximately 3.2 kilobases. The infectious virion or Dane particle consists of an outer lipid envelope made up of large, middle, and small HBsAg and an inner icosahedral viral nucleocapsid, consisting of core protein monomers with the viral DNA and HBV polymerase [10]. The viral life cycle and druggable targets are shown in Fig. 1.

HBV life cycle and sites of drug targets. The viral life cycle begins with virion attachment to the hepatocyte surface via the sodium taurocholate co-transporting polypeptide (NTCP), which facilitates viral entry via endocytosis. After entry, there is uncoating and release of the partially double-stranded rcDNA, which is transported to the hepatocyte nucleus, where host cellular enzymes repair the rcDNA to form the cccDNA. cccDNA serves as the transcriptional template for all mRNAs, including the pregenomic RNA (pgRNA). In the cytoplasm, core proteins self-assemble and, through an encapsidation reaction, the pregenomic RNA and viral polymerase are packaged to form the nucleocapsid. Viral replication occurs within the nucleocapsid through reverse transcription to first form the negative strand then positive strand synthesis. The mature viral capsids containing rcDNA are then enveloped with the surface proteins in the endoplasmic reticulum (ER) and secreted from the infected cell as intact virions (Dane particle) or transported back to the nucleus to replenish the cccDNA pool. Drug targets (1) target viral entry, (2) cccDNA inhibition, (3) targeting viral translation, (4) capsid assembly modulators, (5) targeting the HBV polymerase (HBV Pol), and (6) targeting HBsAg secretion
Treatment Goals and Endpoints
The ideal endpoint of treatment for HBV is a sterilizing cure. This is defined as the elimination of all traces of the virus including cccDNA and integrated HBV DNA [11]. However, achieving a sterilizing cure for HBV is currently not attainable. Therefore, the current focus of HBV treatment is aimed at obtaining a functional cure defined as sustained HBsAg loss for 24 weeks off-treatment, with or without seroconversion to anti-HBs, and HBV DNA below the limit of quantitation [12, 13]. A functional cure is associated with the reduction in clinical outcomes (cirrhosis, decompensated liver disease, and HCC), is durable off-treatment, and is easy to assess [14•, 15]. However, recent studies revealing that HBsAg can originate from both cccDNA and integrated HBV DNA highlight the complexity of achieving a functional cure [16, 17, 18••].
Given the challenges of achieving a functional cure, some have advocated for using a more achievable endpoint, partial cure, defined as HBsAg < 100 IU/mL, negative HBeAg status, and persistent HBV DNA levels below the lower limit of detection (LLOD) after 24 weeks off treatment [12]. Although a partial cure is also associated with the improvement in clinical outcomes, it is not a durable off-treatment endpoint. Additionally, it does not result in an HBsAg loss which is important for removing stigma for many patients. Thus, a partial cure is considered an intermediate endpoint, and a functional cure continues to be the preferred endpoint of novel therapies in the development for CHB [12].
Novel Therapies in Development
As shown in Table 1, novel therapies in development for CHB can be categorized into two main groups: those that target key steps in the viral lifecycle, referred to as direct acting antiviral agents, and those that target the immune response, referred to as immunomodulators.
Direct Antiviral Therapies
-
1.
Inhibitors of viral entry
Targeting the viral entry can block the infection of uninfected hepatocytes and thereby prevent the replenishment of the cccDNA pool. Bulevirtide, a small synthetic myristoylated lipopeptide mimicking the pre-S1 sequence of the large S protein, is an entry inhibitor which blocks HBV entry into hepatocytes by binding to the sodium taurocholate co-transporting polypeptide (NTCP) receptor [37]. Although there is evidence on the safety and efficacy of bulevirtide among subjects with chronic hepatitis D virus infection, there is only a limited data among patients with HBV mono-infection [19]. Bulevirtide has minimal effect on HBsAg levels, suggesting that its effectiveness in achieving a functional cure over a short dosing period may be limited. Hepalatide is another NTCP receptor blocker currently under evaluation [22].
Monoclonal and polyclonal antibodies that specifically bind to the N-terminal region of pre-S1 and block HBV attachment to hepatocytes are under development [38]. VIR-3434 is a monoclonal antibody designed to block the HBV entry into hepatocytes and reduce the level of virions and subviral particles. It may also function as a T cell vaccine against HBV. In a proof-of-principle study, VIR-3434 demonstrated a rapid HBsAg decline of ≥ 1 log IU/mL in virally suppressed subjects [21]. Studies in combination with pegIFN with or without a small interfering RNA (siRNA) and NA are ongoing [39]. Additionally, GC1102, a recombinant hepatitis B immunoglobulin, showed HBsAg loss in 2 (22%) out of 9 participants with low pretreatment HBsAg levels [22].
-
2.
Inhibitors of viral translation
Targeting HBV mRNA using siRNA or antisense oligonucleotides (ASO) are promising approaches to lower viral antigen production and indirectly inhibit HBV replication. siRNAs are short, double-stranded RNAs that lead to gene silencing in a sequence-specific manner by utilizing the endogenous RNA interference pathway. A single siRNA can target multiple HBV mRNAs due to shared terminal 3′ sequences in all HBV mRNAs. siRNAs require a carrier vehicle for their uptake as they do not readily cross the vascular endothelium. ASOs are single-stranded DNA or RNA sequences that bind to complementary sequences of mRNAs and degrade the mRNAs via a ribonuclease H–dependent mechanism. ASOs have the advantage of targeting pre-mRNA, enhancing their specificity and reducing off-target effects without the need for carrier vehicles [40].
siRNAs
First-generation siRNAs, such as ARC-520, demonstrated the proof-of-principle reduction in HBsAg levels in chimpanzee and human studies. Notably, greater declines in HBsAg levels were observed in HBeAg-positive compared to HBeAg-negative chimpanzees. This was later shown to be as a result of changes in the target sequence of the mRNA encoded by integrated but not cccDNA-derived mRNA. In clinical trials, first-generation siRNAs (ARC-520 and ARC-521) showed modest reduction in HBsAg (~ 0.5 log IU/mL) in both HBeAg-positive and HBeAg-negative virally suppressed subjects [41]. However, the development of ARC-520/521 was halted due to hepatotoxicity attributed to the delivery vehicle.
Second-generation, GalNAc-conjugated siRNAs (JNJ-3989, VIR-2218, RG-6346, and AB-729) were developed to target mRNAs transcribed from cccDNA and integrated DNA and improve delivery. These agents have shown promising results with no significant safety concerns [20, 42, 43]. A 2–2.5 log10 IU/mL decline in HBsAg levels can be achieved after one to four doses, which persisted for months post-treatment. Almost all patients achieve at least a 1 log10 IU/mL reduction in HBsAg levels, which may be sustained up to 1 year after the last dose. However, the decline in the HBsAg level was observed to plateau around 20 weeks of treatment, raising concerns about the long-term efficacy of siRNA-based approaches. In a trial involving AB-729, the initial decline in HBsAg plateaued after a few weeks, and no participant achieved HBsAg loss. Mild ALT elevation and increased HBV-specific T cell activation markers were associated with HBsAg decline in some participants [23].
Given the limitations and absence of a functional cure with siRNA monotherapy, combination therapy is a logical next step. Combining AB-729 with NAs in HBeAg-negative participants demonstrated a marked and sustained decline in HBsAg and HBV DNA levels without ALT flares in participants who achieved HBsAg < 100 IU/mL [44]. In another study combining VIR-2218 with the monoclonal antibody VIR-3434, most participants achieved HBsAg level of < 10 IU/mL at the end of treatment, but none achieved a functional cure [45]. Another trial combining VIR-2218 with PegIFNα resulted in a greater decrease in HBsAg levels, compared to VIR-2218 monotherapy, 2.55 vs. 1.89 log10 IU/mL, with 15.6% of subjects achieving HBsAg loss by the end of treatment [24].
Many questions remain to be clarified with the use of siRNA in chronic HBV infection. The optimal duration is unknown, which agent to combine it with and in what sequence. Not all combinations will result in synergy as was observed in a trial combining a siRNA and capsid assembly modulator [46]. The results of several ongoing combination trials with various agents are eagerly awaited.
ASOs
In preclinical studies, ASOs have shown promise in reducing serum HBsAg and HBV DNA levels [47]. In clinical trials, GalNAc-conjugated ASOs, RG6004 and GSK3389404, only resulted in moderate dose-dependent decline in HBsAg levels. These reductions were transient, and HBsAg levels rebounded to baseline within a few weeks after therapy cessation. Consequently, these ASOs are no longer in development [48, 49].
In a phase 2 trial of bepirovirsen, an unconjugated ASO, at a dose of 300 mg per week for 24 weeks, sustained HBsAg, and HBV DNA loss was observed in 9 to 10% of participants [25•]. ALT flares were noted in 40% of patients receiving bepirovirsen monotherapy. This was an important study demonstrating that a functional cure could be achieved with short-duration finite monotherapy. Similar to siRNAs, ASOs are safe and well tolerated.
Whether the sustained reduction in HBsAg levels achieved with siRNAs and ASOs will result in HBsAg clearance over time remains to be determined. Nevertheless, these are promising agents that likely will be part of a functional cure regimen.
-
3.
Inhibitors of capsid assembly
Capsid assembly modulators (CAMs), also known as core protein allosteric modulators (CpAMs), target the HBV core protein. By modifying nucleocapsid assembly, the site of genome replication, CAMs disrupt the viral life cycle and inhibit HBV replication. These compounds bind to a specific hydrophobic pocket between core protein dimers, inducing the formation of aberrant nucleocapsids or empty nucleocapsids, depending on the type of CAM [50]. Two types of CAMs have been described based on their mechanism of action: Type 1 CAMs (CAM-As) induce the formation of aberrantly assembled nucleocapsids, while type 2 CAMs (CAM-Es) result in the formation of morphologically normal but empty nucleocapsids [51].
CAMs offer several therapeutic advantages over other novel agents in development, including an oral route of administration and potent inhibition of viral replication across different genotypes. CAM-As, such as RO7049389, have shown significant reductions in HBV DNA and RNA levels in HBeAg-positive subjects [52]. CAM-Es, including vebicorvir, JNJ-6379, and JNJ-0440, have demonstrated similar declines in HBV DNA and RNA without significant effects on HBsAg levels [27, 53, 54]. CAMs alone do not have a measurable impact on HBV antigens and carry an increased risk of resistance, necessitating combination therapy with other antiviral agents. Similar to NAs, discontinuation of CAM treatment can lead to viral relapse and hepatitis flares.
Current CAMs must invariably be used as combination therapy due to concerns for resistance. When combined with NAs, CAMs demonstrate faster and greater inhibition of viral replication and transcription compared to NA alone. They can also achieve a deeper suppression of HBV DNA and HBV RNA in subjects already receiving NA treatment [55]. However, minimal changes were noted on HBeAg and HBsAg levels with vebicorvir alone or in combination with entecavir [55]. Unfortunately, vebicorvir is no longer in development because of recognized hepatotoxicity [56]. However, several other more potent second-generation CAMs including ABI-H2158, ABI-H3733, AB-836, ALG-000184 [57], and VNRX-9945 are currently under development and hold promise for greater efficacy and improved safety [58, 59].
CAMs have also been combined with siRNAs and NAs. The REEF1 trial evaluated a triple combination regimen of a siRNA (JNJ-3989), with a CAM (bersacapavir, formerly JNJ-6379), and NA. Bersacapavir alone demonstrated only minimal declines in HBsAg levels. Surprisingly, the triple combination regimen resulted in a lower reduction in HBsAg levels compared to the dual combination of siRNA and NA, after 48 weeks of treatment [60]. In the REEF-2 trial, non-cirrhotic HBeAg-negative HBV subjects on NAs received add-on treatment with bersacapavir and JNJ-3989 or placebo for 48 weeks. At the end of the treatment, the active treatment group showed a significant reduction in HBsAg levels, while the placebo group had minimal reduction. Although 71% of participants in the active treatment group achieved HBsAg levels below 100 IU/mL, none achieved HBsAg clearance. One subject experienced a severe hepatitis flare following withdrawal of NA therapy and required liver transplantation [61]. A promising third-generation CAM, AB-836 with potent inhibition of HBV replication (declines of 3.04 to 3.55 log10 IU/mL at day 28) depending on AB-836 dose, without significant HBsAg decline has been discontinued due to hepatotoxicity [59].
The lack of reduction of HBeAg and HBsAg with CAMs is a major limitation of this class of drugs. Thus, the exact role of CAMs in a curative regimen remains to be determined.
-
4.
Inhibitors of cccDNA
Permanent silencing or elimination of cccDNA is the sine qua non for achieving a cure for HBV infection and preventing the risk of reactivation [62]. Targeting cccDNA is a challenge due to its nuclear location. Nevertheless, there are several promising approaches in development. One is to inhibit the multiple steps in its formation from rcDNA, another is to prevent the nuclear import of rcDNA by capsid-targeting drugs. An alternate approach is to silence its transcriptional activity by inducing the host cell’s epigenetic machinery, or by blocking the de-silencing activity of HBV X protein (HBx). Finally, existing cccDNA may be degraded directly using genome editing with designer nucleases or indirectly through immune-mediated mechanisms, such as APOBEC enzymes [63].
Several candidate molecules have been shown in vitro to disrupt the conversion of rcDNA to cccDNA. CCC-0975, a disubstituted sulfonamide inhibitor, may inhibit cccDNA formation [64]. However, it primarily affects new cccDNA formation and does not impact the established pool of cccDNA. Another potential candidate, CCC_R08, is an oral cccDNA inhibitor that has shown sustained reductions in HBsAg and cccDNA in mouse models [65]. Further research is needed to explore the clinical effectiveness of such a strategy in the clinic.
Interferon-alpha and lymphotoxin-β-receptor activation have been shown to degrade cccDNA via APOBEC 3A and 3B-mediated deamination of the negative genomic strand. The Farnesoid X receptor (FXR-α) was shown to be a proviral host factor for HBV. Paradoxically, FXR-α agonists were shown to decrease cccDNA levels [66]. In early clinical studies, it was shown that the combination of vonafexor (EYP001), an FXR agonist, and Peg-IFN α may enhance the action of Peg-IFN α, based on the magnitude in reduction of HBsAg levels. This remains to be proven as there was no Peg-IFN α control arm [67].
cccDNA is organized into a chromatin-like structure, making it amenable to epigenetic manipulation. Gene editing methods, including zinc finger nucleases, transcription activator–like effector nucleases, and the CRISPR/Cas9 system, have been shown to degrade cccDNA and are promising strategies under investigation [68, 69]. However, challenges include the delivery of the editing tools to all infected hepatocytes, cleaving integrated HBV DNA, and the risk of off-target effects.
Targeting HBX could offer a durable approach to silencing cccDNA. Nitazoxanide, an antiparasitic agent, and pevonedistat, a neddylation inhibitor, have shown potential in inhibiting cccDNA formation by targeting HBX, a protein essential for viral transcription, and its interaction with DDB1 [70].
-
5.
Inhibitors of HBV polymerase
The HBV polymerase, the sole viral protein with enzymatic action, plays a critical role in viral replication. NAs widely used in HBV treatment primarily target the reverse transcriptase activity of the HBV polymerase [5•]. However, despite their effectiveness in inhibiting viral replication, they do not affect cccDNA and have a limited impact on HBsAg. Therefore, replication rapidly returns in a majority of patients once the NA is discontinued necessitating long-term treatment. Current efforts are focused on improving potency and reducing metabolite toxicity of newer NAs such as pradefovir [30, 31]. Targeting the other enzymatic function of the HBV polymerase, its RNAseH function, is being explored. RNAseH degrades the pgRNA after it serves as the template for negative strand synthesis. Although many RNAseH inhibitors have been identified, none is currently in clinical development.
-
6.
Inhibitors of HBsAg release
Clearance of HBsAg is the current goal of HBV therapies in development. Consequently, there is interest in developing agents that can reduce HBsAg levels. Nucleic acid polymers (NAPs) [33] are one potential approach being explored [71].
NAPs are synthetic oligonucleotides that selectively target the assembly and secretion of HBV subviral particles. They have little to no effect on mature virion secretion. By reducing circulating HBsAg levels, NAPs are believed to restore immunomodulation and promote host-mediated clearance [72]. In a small study, the addition of NAPs (REP 2139 or REP 2165) to a regimen of tenofovir and PEG-IFN resulted in sustained suppression of HBsAg to undetectable levels in 44% of participants. These participants also developed anti-HBs antibodies, suggesting either a potential restoration of the immune response or a shifting of the balance between levels of HBsAg and anti-HBs in circulation [33]. However, it is important to note that a majority of NAP-treated subjects experienced grade 3–4 ALT flares. Larger controlled studies are needed to further evaluate the efficacy and safety of NAPs.
A similar compound, S-antigen transport–inhibiting oligonucleotide polymers (STOPS), which is a locked nucleic acid-modified version of a NAP, is no longer in development due to the minimal effect on HBsAg levels [73].
Immunomodulators
In chronic HBV infection, patients often have impaired innate and adaptive immune responses. High levels of viral antigens, such as HBsAg, can lead to the exhaustion of HBV-specific T cells, further compromising the immune response [74]. To achieve long-term control of HBV infection, it is crucial to restore immune function.
-
1.
Inducing innate immunity
In chronic HBV infection, the innate immune system has difficulty sensing and effectively responding to the HBV. Additionally, HBV may actively evade the innate immune response leading some to consider it a stealth virus [74]. A number of therapeutic approaches are being developed to restore the innate immune response. These include Toll-like receptor (TLRs) agonists, retinoid acid–inducible gene-I (RIG-I), and stimulator of interferon genes (STING), which aim to induce the production of interferons and proinflammatory cytokines that can help clear HBV [75].
TLRs play a critical role in the early innate immune response by sensing highly conserved bacterial or viral products such as LPS, bacterial DNA, and viral double-stranded RNA. The stimulation of TLRs results in a variety of cellular responses that lead to the production of IFNs, pro-inflammatory cytokines, and effector cytokines that eliminate pathogens. The TLR-7 agonists vesatolimod (GS-9620), RO7020531 and JNJ-4964, and TLR-8 agonist selgantolimod (GS-9688) [34, 76] have shown promise in preclinical trials by activating intrahepatic dendritic cells and triggering the production of interferons, as well as activate natural killer (NK) cells and T cells [77, 78]. However, clinical trials with vesatolimod and selgantolimod did not show substantial benefit in reducing HBsAg levels [34, 76]. In a phase 2 study, selgantolimod led to HBsAg loss in 5% of virally suppressed subjects and a modest reduction in HBsAg levels [34].
Inarigivir (SB9200), an oral agonist of RIG-I and NOD2 that induces interferon-mediated antiviral immune responses in virus-infected cells, is no longer being developed due to hepatotoxicity and death of a research subject [79], 80.
Further research is needed to define the optimal innate immune targets, and to establish the role of agonists of innate immunity in combination with antiviral therapy. Moreover, understanding the timing and duration of immune modulation in relation to viral inhibition will be important for achieving a functional cure.
-
2.
Inducing adaptive immunity
Restoring an adequate adaptive immune response will likely be necessary for controlling and potentially curing HBV infection.
Checkpoint inhibition, using antibodies targeting the programmed death receptor (PD-1) or programmed death ligand (PD-L1), is one non-specific approach to restore T cell function. PD-1 and its ligands downregulate T cell activation. PD-1 has been shown to be overexpressed by HBV-specific T cells. Thus, agents that block PD-1 and its ligand checkpoint inhibitors represent a therapeutic strategy to restore function of HBV-specific T cells and enhancing antibody production [81]. Proof-of-concept has been demonstrated in cancer treatment. There is, however, a concern for autoimmunity, and an uncontrolled immune response may lead to flares of hepatitis. In a pilot study, the use of PD-1 inhibitor nivolumab with or without GS-4774, a therapeutic vaccine in virally suppressed subjects, resulted in only modest declines in HBsAg levels. HBsAg seroclearance was achieved in one patient in the nivolumab-only group [82]. In another phase 2 study using the PD-L1 antibody envafolimab (ASC22), 3 out of 7 subjects (43%) with baseline HBsAg levels below 100 IU/mL achieved HBsAg loss [35]. Oral, liver-specific checkpoint inhibitors are in development. The optimal duration and sequence of checkpoint inhibition in a therapeutic regimen remain to be determined.
Strategies to restore T cell responsiveness include creation of genetically engineered T cells. T cell receptors cloned from HBV-reactive cells can be introduced into T cells. Alternatively, T cells can be modified to express chimeric antigen receptors that specifically target HBV antigens on the hepatocyte surface [83]. These genetically modified T cells have been shown to effectively eliminate HBV-infected cells in vitro and have shown safety in animal models [84, 85]. The safety and efficacy of using genetically engineered T cells were shown in a small study in which adoptive transfer of autologous T cells expressing HBV-specific T cell receptors was achieved in participants with HBV-related HCC [86]. This may open the possibility of using this approach in patients with CHB.
-
3.
Therapeutic vaccines
Therapeutic vaccination aims to induce both B and T cell immune responses, as the resolution of acute HBV infection is associated with lifelong immunity mediated by these components of the immune system. Previous attempts to develop a therapeutic vaccine using a variety of antigens, protein-based, protein-antibody complex–based, and DNA-based vaccines, have been unsuccessful [87]. Novel vaccination strategies are exploring use of viral recombinant vectors, dendritic cell vaccines, and mRNA vaccines. Viral vector-based vaccines, in particular, offer the advantage of intracellular antigen expression, triggering a robust cytotoxic T lymphocyte response [88].
TG1050, an adenovirus 5–based vaccine expressing HBV antigens, and lentiviral vectors encoding HBV antigens, have demonstrated the induction of specific T cell responses but only modest declines in HBsAg levels [89]. Studies involving GS-4774, a yeast-based vaccine expressing HBsAg, HBcAg, and HBX, have shown minimal reduction in serum HBsAg levels, despite robust cytokine production by HBV-specific CD8 T cells [90].
Innovative strategies are being developed to enhance the immunogenicity of peptide-based vaccines. These include the utilization of novel nasal formulations such as NASVAC and derivatives of Sci-B-Vac, like BRII-179 [36, 91]. Clinical trials evaluating the safety and efficacy of these approaches have yielded promising results. The mucoadhesive carboxy-vinyl polymer (CVP-NASVAC) nasal therapeutic vaccine, containing HBsAg and HBcAg, has demonstrated significant reductions in HBsAg levels in participants receiving both NA therapy and HBV carriers. Notably, 9.5% of subjects (6 out of 71) achieved a functional cure [91]. Another study investigating BRII-179, which encompasses all three HBV surface envelope proteins, has shown moderate antibody responses; however, it did not lead to a noticeable reduction in HBsAg levels in virally suppressed subjects under NA therapy [36].
Prime-boost vaccination, involving sequential administration of vaccines with different antigens and delivery systems, is one of the more promising approaches to overcome HBV-specific tolerance and have been shown to generate high levels of memory T cells in preclinical studies. Additionally, combining viral antigenemia knockdown using siRNAs with therapeutic vaccination has resulted in the development of polyfunctional HBV-specific CD8 + T cells and elimination of HBV [92].
An advantage of vaccination is its theoretical safety, as it specifically stimulates HBV-specific immunity without non-specific activation of innate responses or autoimmunity. However, important considerations include determining the optimal vaccine composition, use of T or B cell adjuvants, the vaccine regimen, and the mode of administration need to be addressed in future studies.
Combination Therapy
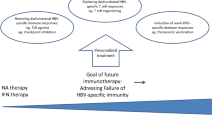
Success with combination therapy for other infectious diseases such as those caused by human immunodeficiency virus and hepatitis C virus implies that a similar approach will be needed to achieve a functional cure of chronic HBV infection. Although over 40 compounds with multiple mechanisms of action are currently in development, Table 1, combination approaches are largely empirical due to the lack of knowledge of the mechanisms required to achieve viral control and HBsAg loss. The most promising approaches focus on agents that inhibit viral replication, reduce antigenic burden, and partially enhance/restore immune control against HBV, Fig. 2 [24, 25•, 33, 35, 45]. However, determining the optimal combinations, timing and sequence of use, and treatment duration remains a challenge. Personalized treatment approaches may be necessary based on factors such as HBeAg status, viral load, HBsAg levels, and presence of cirrhosis.

Possible therapeutic combinations for achieving a functional cure. Ideal combination is yet to be determined
Conclusion
Combination therapy with currently approved agents peg-IFN and NAs can achieve functional cure in ~ 10% of patients. However, many patients decline or are ineligible to receive peg-IFN due to its poor tolerability. Functional cure rates with NAs alone range from 1 to 3%. This highlights the need for newer therapy that can achieve higher rates of a functional cure to reduce the morbidity and mortality associated with chronic HBV infection. Given that the bulk of patients with chronic HBV infection reside in low- and middle-income countries, the ideal regimen would be one that is of finite duration, orally administered, easy to manufacture, and scalable. To realize this goal, continued research, investment, and commitment from both the industry and the scientific community will be crucial.
Abbreviations
- AASLD:
-
American Association for the Study of Liver Disease
- ALT:
-
Alanine aminotransferase
- ASO:
-
Antisense oligonucleotides
- AST:
-
Aspartate aminotransferase
- CAMs:
-
Capsid assembly modulators
- CHB:
-
Chronic hepatitis B
- cccDNA:
-
Covalently closed circular DNA
- DNA:
-
Deoxyribonucleic acid
- dslDNA:
-
Double-stranded linear DNA
- EASL:
-
European Association for the Study of the Liver
- FXR-α:
-
Farnesoid X receptor
- HBcrAg:
-
Hepatitis B core antigen
- HBeAg:
-
Hepatitis B e antigen
- HBsAg:
-
Hepatitis B surface antigen
- HBV:
-
Hepatitis B virus
- HBx:
-
Hepatitis B virus X protein
- HCC:
-
Hepatocellular carcinoma
- HSPG:
-
Heparan sulfate proteoglycans
- IU:
-
International units
- IVDU:
-
Intravenous drug users
- kb:
-
Kilobase pairs
- LLOD:
-
Lower limit of detection
- mL:
-
Milliliters
- NAs:
-
Nucleos(t)ide analogs
- NTCP:
-
Sodium taurocholate co-transporting polypeptide
- NAPs:
-
Nucleic acid polymers
- pgRNA:
-
Pre-genomic RNA
- PEG-IFN α:
-
Pegylated interferon alfa
- PCR:
-
Polymerase chain reaction
- RIG-I:
-
Retinoid acid inducible gene-I
- RNA:
-
Ribonucleic acid
- rcDNA:
-
Relaxed circular DNA
- STING:
-
Stimulator of interferon genes
- STOPS:
-
S-Antigen transport-inhibiting oligonucleotide polymers
- siRNA:
-
Small interfering RNA
- TLR:
-
Toll-like receptor
- WHO:
-
World Health Organization
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
World Health Organization. Hepatitis B fact sheet WHO press, Geneva, Switzerland. Available from: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b. Accessed 26 Oct 2023.
Cox AL, El-Sayed MH, Kao JH, Lazarus JV, Lemoine M, Lok AS, et al. Progress towards elimination goals for viral hepatitis. Nat Rev Gastroenterol Hepatol. 2020;17(9):533–42.
Terrault NA, Lok ASF, McMahon BJ, Chang KM, Hwang JP, Jonas MM, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67(4):1560–99.
Ghany MG. Current treatment guidelines of chronic hepatitis B: the role of nucleos(t)ide analogues and peginterferon. Best Pract Res Clin Gastroenterol. 2017;31(3):299–309.
• Marcellin P, Wong DK, Sievert W, Buggisch P, Petersen J, Flisiak R, et al. Ten-year efficacy and safety of tenofovir disoproxil fumarate treatment for chronic hepatitis B virus infection. Liver Int. 2019;39(10):1868–75. Seminal paper which showed long term safety and efficacy of Tenofovir in treatment of hepatitis B.
Chen CH, Hu TH, Wang JH, Lai HC, Hung CH, Lu SN, et al. Comparison of HBsAg changes between HBeAg-negative patients who discontinued or maintained entecavir therapy. Hepatol Int. 2020;14(3):317–25.
Hirode G, Choi HSJ, Chen CH, et al. Off-Therapy Response After Nucleos(t)ide Analogue Withdrawal in Patients With Chronic Hepatitis B: An International, Multicenter, Multiethnic Cohort (RETRACT-B Study). Gastroenterology. 2022;162(3):757-771.e4.
Hou JL, Zhao W, Lee C, Hann HW, Peng CY, Tanwandee T, et al. Outcomes of long-term treatment of chronic HBV infection with entecavir or other agents from a randomized trial in 24 countries. Clin Gastroenterol Hepatol. 2020;18(2):457-67.e21.
• Yardeni D, Chang KM, Ghany MG. Current best practice in hepatitis B management and understanding long-term prospects for cure. Gastroenterology. 2023;164(1):42-60.e6. Comprehensive review on chronic hepatitis B management.
Tsukuda S, Watashi K. Hepatitis B virus biology and life cycle. Antiviral Res. 2020;182: 104925.
Lok AS, Zoulim F, Dusheiko G, Ghany MG. Hepatitis B cure: from discovery to regulatory approval. J Hepatol. 2017;67(4):847–61.
Ghany MG, Buti M, Lampertico P, Lee HM; 2022 AASLD-EASL HBV-HDV Treatment Endpoints Conference Faculty. Guidance on treatment endpoints and study design for clinical trials aiming to achieve cure in chronic hepatitis B and D: Report from the 2022 AASLD-EASL HBV-HDV Treatment Endpoints Conference. Hepatology. 2023;78(5):1654-1673.
Cornberg M, Lok AS, Terrault NA, Zoulim F; 2019 EASL-AASLD HBV Treatment Endpoints Conference Faculty. Guidance for design and endpoints of clinical trials in chronic hepatitis B - Report from the 2019 EASL-AASLD HBV Treatment Endpoints Conference. Hepatology. Published online November 12, 2019.
• Yip TC, Wong GL, Chan HL, Tse YK, Lam KL, Lui GC, et al. HBsAg seroclearance further reduces hepatocellular carcinoma risk after complete viral suppression with nucleos(t)ide analogues. J Hepatol. 2019;70(3):361–70. Important study which showed HBsAg seroclearance was associated with decreased HCC risk.
Alawad AS, Auh S, Suarez D, Ghany MG. Durability of spontaneous and treatment-related loss of hepatitis B s antigen. Clin Gastroenterol Hepatol. 2020;18(3):700-9 e3.
Martinez MG, Boyd A, Combe E, Testoni B, Zoulim F. Covalently closed circular DNA: the ultimate therapeutic target for curing HBV infections. J Hepatol. 2021;75(3):706–17.
Wong DK, Seto WK, Fung J, Ip P, Huang FY, Lai CL, et al. Reduction of hepatitis B surface antigen and covalently closed circular DNA by nucleos(t)ide analogues of different potency. Clin Gastroenterol Hepatol. 2013;11(8):1004-10 e1.
•• Wooddell CI, Yuen MF, Chan HL, Gish RG, Locarnini SA, Chavez D, Ferrari C, Given BD, Hamilton J, Kanner SB, Lai CL, Lau JYN, Schluep T, Xu Z, Lanford RE, Lewis DL. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci Transl Med. 2017 Sep 27;9(409):eaan0241. Important paper which showed that source of HBsAg is not only from cccDNA but also from integrated HBV DNA.
Wedemeyer H, Aleman S, Brunetto MR, Blank A, Andreone P, Bogomolov P, et al. A phase 3, randomized trial of bulevirtide in chronic hepatitis D. N Engl J Med. 2023.
Gane E, Locarnini S, Lim TH, Strasser S, Sievert W, Cheng W, et al. Short interfering RNA JNJ-3989 combination therapy in chronic hepatitis B shows potent reduction of all viral markers but no correlate was identified for HBsAg reduction and baseline factors. J Hepatol. 2021;75:S289–90.
Agarwal K, Yuen MF, Wedemeyer H, Arizpe A, Shen L, Cloutier D, et al. Rapid HBsAg reduction in chronic hepatitis B virus infection: preliminary results from a PHASE 1 study evaluating a single dose of VIR-3434, a novel neutralizing vaccinal monoclonal antibody. Hepatology. 2021;74:514a-a.
Clinical trials. Available from: https://clinicaltrials.gov/. Accessed 26 Oct 2023.
Yuen MF, Berliba E, Sukeepaisarnjaroen W, Strasser S, Tangkijvanich P, Holmes J, et al. Repeat dosing of the GalNAc-siRNA AB-729 in subjects with chronic hepatitis B results in robust and sustained HBsAg suppression. J Hepatol. 2021;75:S203–4.
Yuen MF, Lim YS, Cloutier D, Thanawala V, Shen L, Elie SE, et al. Preliminary 48-week safety and efficacy data of Vir-2218 alone and in combination with pegylated interferon alfa in participants with chronic HBV infection. Hepatology. 2022;76:S19–20.
• Yuen MF, Lim SG, Plesniak R, Tsuji K, Janssen HLA, Pojoga C, Gadano A, Popescu CP, Stepanova T, Asselah T, Diaconescu G, Yim HJ, Heo J, Janczewska E, Wong A, Idriz N, Imamura M, Rizzardini G, Takaguchi K, Andreone P, Arbune M, Hou J, Park SJ, Vata A, Cremer J, Elston R, Lukić T, Quinn G, Maynard L, Kendrick S, Plein H, Campbell F, Paff M, Theodore D; B-Clear Study Group. Efficacy and Safety of Bepirovirsen in Chronic Hepatitis B Infection. N Engl J Med. 2022 Nov 24;387(21):1957-1968. Phase 2 trial showing effectiveness of bepirovirsen in management of hepatitis B.
Zhang H, Wang F, Zhu X, Chen Y, Chen H, Li X, et al. Antiviral activity and pharmacokinetics of the hepatitis B virus (HBV) capsid assembly modulator GLS4 in patients with chronic HBV infection. Clin Infect Dis. 2021;73(2):175–82.
Zoulim F, Lenz O, Vandenbossche JJ, Talloen W, Verbinnen T, Moscalu I, et al. JNJ-56136379, an HBV capsid assembly modulator, is well-tolerated and has antiviral activity in a phase 1 study of patients with chronic infection. Gastroenterology. 2020;159(2):521-+.
Zhang W, Guo L, Liu H, Wu G, Shi H, Zhou M, et al. Discovery of Linvencorvir (RG7907), a hepatitis B virus core protein allosteric modulator, for the treatment of chronic HBV infection. J Med Chem. 2023;66(6):4253–70.
Jia H, Mai J, Wu M, Chen H, Li X, Li C, et al. Safety, tolerability, pharmacokinetics, and antiviral activity of the novel core protein allosteric modulator ZM-H1505R (Canocapavir) in chronic hepatitis B patients: a randomized multiple-dose escalation trial. BMC Med. 2023;21(1):98.
Gao Y, Kong F, Song X, Shang J, Yao L, Xia J, et al. Pradefovir treatment in patients with chronic hepatitis B: week 24 results from a multicenter, double-blind, randomized, noninferiority, phase 2 trial. Clin Infect Dis. 2021.
Zhang H, Hu Y, Wu M, Liu J, Zhu X, Li X, et al. Randomised clinical trial: safety, efficacy and pharmacokinetics of HS-10234 versus tenofovir for the treatment of chronic hepatitis B infection. Aliment Pharmacol Ther. 2021;53(2):243–52.
Squires KE, Ogilvie L, Jucov A, Anastasiy I, Ghicavii N, Huguet J, et al. A randomized phase 1b trial of the active site polymerase inhibitor nucleotide ATI-2173 in patients with chronic hepatitis B virus infection. J Viral Hepat. 2023;30(1):19–28.
Bazinet M, Pântea V, Placinta G, Moscalu I, Cebotarescu V, Cojuhari L, et al. Safety and efficacy of 48 weeks REP 2139 or REP 2165, tenofovir disoproxil, and pegylated interferon alfa-2a in patients with chronic HBV infection naïve to nucleos(t)ide therapy. Gastroenterology. 2020;158(8):2180–94.
Gane E, Dunbar PR, Brooks A, Zhao Y, Tan S, Lau A, et al. Efficacy and safety of 24 weeks treatment with oral TLR8 agonist, selgantolimod, in virally-suppressed adult patients with chronic hepatitis B: a phase 2 study. J Hepatol. 2020;73:S52-S.
Wang GQ, Qian JD, Cui YM, Yan YM, He HD, Wu JZ. A phase IIa trial of subcutaneously administered PD-L1 antibody ASC22 (ENVAFOLIMAB) in patients with chronic hepatitis B. Hepatology. 2021;74:62a-a.
Ma H, Lim TH, Leerapun A, Weltman M, Jia J, Lim YS, et al. Therapeutic vaccine BRII-179 restores HBV-specific immune responses in patients with chronic HBV in a phase Ib/IIa study. JHEP Rep. 2021;3(6):100361.
Urban S, Bartenschlager R, Kubitz R, Zoulim F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology. 2014;147(1):48–64.
Hehle V, Beretta M, Bourgine M, Ait-Goughoulte M, Planchais C, Morisse S, et al. Potent human broadly neutralizing antibodies to hepatitis B virus from natural controllers. J Exp Med. 2020;217(10).
Vir Biotechnology announces first patient dosed in new phase 2 chronic hepatitis B virus trial evaluating combinations of VIR-2218, VIR-3434, PEG-IFNα and an NRTI [press release]. 2023.
Vaillant A. Oligonucleotide-based therapies for chronic HBV infection: a primer on biochemistry, mechanisms and antiviral effects. Viruses. 2022;14(9).
Yuen MF, Schiefke I, Yoon JH, Ahn SH, Heo J, Kim JH, et al. RNA interference therapy with ARC-520 results in prolonged hepatitis B surface antigen response in patients with chronic hepatitis B infection. Hepatology. 2020;72(1):19–31.
Gane E, Lim YS, Cloutier D, Shen L, Cathcart A, Ding X, et al. Safety and antiviral activity of VIR-2218, an X-targeting RNAi therapeutic, in participants with chronic hepatitis B infection: week 48 follow-up results. J Hepatol. 2021;75:S287–8.
Yuen MF, Berliba E, Kim YJ, Holmes JA, Lim YS, Strasser SI, et al. Safety and pharmacodynamics of the GalNAc-siRNA AB-729 in subjects with chronic hepatitis B infection. Hepatology. 2020;72:62a-a63.
Yuen MFHJ, Strasser SI, Leerapun A, et al. Hepatitis B viral control maintained during extended follow up of HBeAg- chronic hepatitis B (CHB) subjects who discontinued nucleos(t)ide analogue (NA) therapy after completion of AB-729 treatment, and in HBeAg+ subjects still on NA therapy. AASLD 2022; Washington. 2022.
Gane E, Jucov A, Dobryanska M, Yoon KT, Lim TH, Arizpe A, et al. Safety, tolerability, and antiviral activity of the siRNA VIR-2218 in combination with investigational Ineutralizing monoclonal antibody VIR-3434 for the treatment of chronic hepatitis B virus infection: preliminary results from the phase 2 March trial. Hepatology. 2022;76:S18–9.
Yuen MF, Asselah T, Jacobson IM, Brunetto MR, Janssen HLA, Takehara T, et al. Efficacy and safety of the siRNA JNJ-73763989 and the capsid assembly modulator JNJ-56136379 (bersacapavir) with nucleos(t)ide analogues for the treatment of chronic hepatitis B virus infection (REEF-1): a multicentre, double-blind, active-controlled, randomised, phase 2b trial. Lancet Gastroenterol Hepatol. 2023;8(9):790–802.
Agarwal K, Lok J, Gane E. Antisense oligonucleotides (ASOs) in chronic hepatitis B infection: opportunities and challenging the orthodoxy. J Hepatol. 2022;77(4):906–8.
Gane E, Yuen MF, Kim DJ, Chan HL, Surujbally B, Pavlovic V, et al. Clinical study of single-stranded oligonucleotide RO7062931 in healthy volunteers and patients with chronic hepatitis B. Hepatology. 2021;74(4):1795–808.
Yuen MF, Heo J, Kumada H, Suzuki F, Suzuki Y, Xie Q, et al. Phase IIa, randomised, double-blind study of GSK3389404 in patients with chronic hepatitis B on stable nucleos(t)ide therapy. J Hepatol. 2022;77(4):967–77.
Yan Z, Wu D, Hu H, Zeng J, Yu X, Xu Z, et al. Direct inhibition of hepatitis B e antigen by core protein allosteric modulator. Hepatology. 2019;70(1):11–24.
Bartoli A, Gabrielli F, Tassi A, Cursaro C, Pinelli A, Andreone P. Treatments for HBV: A Glimpse into the Future. Viruses. 2021;13(9):1767.
Yuen MF, Zhou X, Gane E, Schwabe C, Tanwandee T, Feng S, et al. Safety, pharmacokinetics, and antiviral activity of RO7049389, a core protein allosteric modulator, in patients with chronic hepatitis B virus infection: a multicentre, randomised, placebo-controlled, phase 1 trial. Lancet Gastroenterol Hepatol. 2021;6(9):723–32.
Gane EJ, Schwabe C, Berliba E, Tangkijvanich P, Jucov A, Ghicavii N, et al. Safety, antiviral activity and pharmacokinetics of JNJ-64530440, a novel capsid assembly modulator, as 4 week monotherapy in treatment-naive patients with chronic hepatitis B virus infection. J Antimicrob Chemother. 2022;77(4):1102–10.
Yuen MF, Agarwal K, Gane EJ, Schwabe C, Ahn SH, Kim DJ, et al. Safety, pharmacokinetics, and antiviral effects of ABI-H0731, a hepatitis B virus core inhibitor: a randomised, placebo-controlled phase 1 trial. Lancet Gastroenterol Hepatol. 2020;5(2):152–66.
Sulkowski MS, Agarwal K, Ma X, Nguyen TT, Schiff ER, Hann HL, et al. Safety and efficacy of vebicorvir administered with entecavir in treatment-naïve patients with chronic hepatitis B virus infection. J Hepatol. 2022;77(5):1265–75.
Yuen MF, Gane EJ, Kim DJ, Weilert F, Yuen Chan HL, Lalezari J, et al. Antiviral activity, safety, and pharmacokinetics of capsid assembly modulator NVR 3–778 in patients with chronic HBV infection. Gastroenterology. 2019;156(5):1392-403.e7.
Gane EJ, Yuen M, Jucov A, Le K, Westland C, Schwabe C, et al. Safety, pharmacokinetics (pk), and antiviral activity of the capsid assembly modulator (Cam) Alg-000184 in subjects with chronic hepatitis B (Chb). Hepatology. 2021;74:516a-a517.
Cai DW, Evanhcik M, Yan R, Kumar R, Zong YH, Huang Q, et al. Second generation hepatitis B virus core inhibitors ABI-H2158 and ABI-H3733 have enhanced potency and target coverage for both antiviral inhibition and covalently closed circular DNA establishment activities. J Hepatol. 2021;75:S290-S.
Gane EJ, Jucov A, Kolomiichuk L, Eley T, Brown J, King Y, et al. Safety, tolerability, pharmacokinetics (PK), and antiviral activity of the 3rd generation capsid inhibitor AB-836 in healthy subjects (HS) and subjects with chronic hepatitis B (CHB). J Hepatol. 2022;77:S848–9.
Efficacy and safety of the siRNA JNJ-3989 and/or the capsid assembly modulator JNJ-6379 for the treatment of chronic hepatitis B virus infection: results from the phase 2b REEF-1 study [Internet]. 2021. Available from: https://www.natap.org/2021/AASLD/AASLD_152.htm. Accessed 26 Oct 2023.
Agarwal K, Buti M, van Bömmel F, et al. Efficacy and safety of combination treatment with siRNA JNJ-73763989 and capsid assembly modulator JNJ-56136379 (bersacapavir) in HBeAg negative virologically suppressed chronic hepatitis B patients: Follow-up week 48 end of study results from REEF-2. American Association for the Study of Liver Diseases (AASLD) Liver Meet. 2022;4–8 (abstr 5012).
Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64(12):1972–84.
Yardeni D, Ghany MG. Review article: hepatitis B—current and emerging therapies. Aliment Pharmacol Ther. 2022;55(7):805–19.
Cai DW, Mills C, Yu WQ, Yan R, Aldrich CE, Saputelli JR, et al. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob Agents Chemother. 2012;56(8):4277–88.
Wang L, Zhu Q, Zhang JD, Zhang Y, Ni X, Xiang K, et al. Discovery of a first-in-class orally available HBV cccDNA inhibitor. J Hepatol. 2023;78(4):742–53.
Mouzannar K, Fusil F, Lacombe B, Ollivier A, Menard C, Lotteau V, et al. Farnesoid X receptor-alpha is a proviral host factor for hepatitis B virus that is inhibited by ligands in vitro and in vivo. FASEB J. 2019;33(2):2472–83.
Scalfaro P, Heo J, Liu CJ, Hsu CW, Chuang WL, Hu TH, et al. A phase 2 study testing FXR agonist vonafexor in treatment naive patients with chronic hepatitis B (CHB): preliminary week 16 results. J Hepatol. 2021;75:S761–2.
Kostyushev D, Brezgin S, Kostyusheva A, Zarifyan D, Goptar I, Chulanov V. Orthologous CRISPR/Cas9 systems for specific and efficient degradation of covalently closed circular DNA of hepatitis B virus. Cell Mol Life Sci. 2019;76(9):1779–94.
Ely A, Moyo B, Arbuthnot P. Progress with developing use of gene editing to cure chronic infection with hepatitis B virus. Mol Ther. 2016;24(4):671–7.
Sekiba K, Otsuka M, Ohno M, Yamagami M, Kishikawa T, Seimiya T, et al. Pevonedistat, a neuronal precursor cell-expressed developmentally down-regulated protein 8-activating enzyme inhibitor, is a potent inhibitor of hepatitis B virus. Hepatology. 2019;69(5):1903–15.
Kao CC, Nie Y, Ren S, De Costa NTS, Pandey RK, Hong J, et al. Mechanism of action of hepatitis B virus S antigen transport-inhibiting oligonucleotide polymer, STOPS, molecules. Mol Ther - Nucleic Acids. 2022;27:335–48.
Bazinet M, Pântea V, Placinta G, Moscalu I, Cebotarescu V, Cojuhari L, et al. Benefit of transaminase elevations in establishing functional cure of HBV infection during nap-based combination therapy. J Viral Hepat. 2021;28(5):817–25.
Vaillant A. Targeting subviral particles: a critical step in achieving HBV functional cure but where are we with current agents in clinical development? Viruses. 2022;14(6).
Khanam A, Chua JV, Kottilil S. Immunopathology of chronic hepatitis B infection: role of innate and adaptive immune response in disease progression. Int J Mol Sci. 2021;22(11).
Lang-Meli J, Neumann-Haefelin C, Thimme R. Immunotherapy and therapeutic vaccines for chronic HBV infection. Curr Opin Virol. 2021;51:149–57.
Janssen HLA, Brunetto MR, Kim YJ, Ferrari C, Massetto B, Nguyen AH, et al. Safety, efficacy and pharmacodynamics of vesatolimod (GS-9620) in virally suppressed patients with chronic hepatitis B. J Hepatol. 2018;68(3):431–40.
Menne S, Tumas DB, Liu KH, Thampi L, AlDeghaither D, Baldwin BH, et al. Sustained efficacy and seroconversion with the Toll-like receptor 7 agonist GS-9620 in the Woodchuck model of chronic hepatitis B. J Hepatol. 2015;62(6):1237–45.
Daffis S, Balsitis S, Chamberlain J, Zheng J, Santos R, Rowe W, et al. Toll-like receptor 8 agonist GS-9688 induces sustained efficacy in the Woodchuck model of chronic hepatitis B. Hepatology. 2021;73(1):53–67.
Agarwal K, Afdhal N, Coffin C, Fung S, Dusheiko G, Foster G, Elkhashab M, Tam E, Ramji A, Iyer R, et al. Liver toxicity in the Phase 2 Catalyst 206 trial of Inarigivir 400 mg daily added to a nucleoside in HBV EAg negative patients. J Hepatol. 2020;73:S125.
Yuen MF, Chen CY, Liu CJ, Jeng WJ, Elkhashab M, Coffin CS, Kim W, Greenbloom S, Ramji A, Lim YS, Kim YJ, Fung SK, Kim DJ, Jang JW, Lee KS, Iyer RP, Macfarlane C, Jackson K, Locarnini SA, Chan HLY, Afdhal NH. A phase 2, open-label, randomized, multiple-dose study evaluating Inarigivir in treatment-naïve patients with chronic hepatitis B. Liver Int. 2023;43(1):77–89.
Fisicaro P, Valdatta C, Massari M, Loggi E, Biasini E, Sacchelli L, et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology. 2010;138(2):682-U348.
Gane E, Verdon DJ, Brooks AE, Gaggar A, Nguyen AH, Subramanian GM, et al. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: a pilot study. J Hepatol. 2019;71(5):900–7.
Bertoletti A, Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Postgrad Med J. 2013;89(1051):294–304.
Bohne F, Chmielewski M, Ebert G, Wiegmann K, Kurschner T, Schulze A, et al. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology. 2008;134(1):239–47.
Krebs K, Bottinger N, Huang LR, Chmielewski M, Arzberger S, Gasteiger G, et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology. 2013;145(2):456–65.
Meng F, Zhao J, Tan AT, Hu W, Wang SY, Jin J, et al. Immunotherapy of HBV-related advanced hepatocellular carcinoma with short-term HBV-specific TCR expressed T cells: results of dose escalation, phase I trial. Hepatol Int. 2021;15(6):1402–12.
Cargill T, Barnes E. Therapeutic vaccination for treatment of chronic hepatitis B. Clin Exp Immunol. 2021;205(2):106–18.
Ura T, Okuda K, Shimada M. Developments in viral vector-based vaccines. Vaccines. 2014;2(3):624–41.
Zoulim F, Fournier C, Habersetzer F, Sprinzl M, Pol S, Coffin CS, et al. Safety and immunogenicity of the therapeutic vaccine TG1050 in chronic hepatitis B patients: a phase 1b placebo-controlled trial. Hum Vaccin Immunother. 2020;16(2):388–99.
Boni C, Janssen HLA, Rossi M, Yoon SK, Vecchi A, Barili V, et al. Combined GS-4774 and tenofovir therapy can improve HBV-specific T-cell responses in patients with chronic hepatitis. Gastroenterology. 2019;157(1):227-+.
Yoshida O, Akbar SMF, Imai Y, Sanada T, Tsukiyama-Kohara K, Miyazaki T, et al. Intranasal therapeutic vaccine containing HBsAg and HBcAg for patients with chronic hepatitis B; 18 months follow-up results of phase IIa clinical study. Hepatol Res. 2023;53(3):196–207.
Michler T, Kosinska AD, Festag J, Bunse T, Su J, Ringelhan M, et al. Knockdown of virus antigen expression increases therapeutic vaccine efficacy in high-titer hepatitis B virus carrier mice. Gastroenterology. 2020;158(6):1762-75 e9.
Acknowledgements
The authors acknowledge the assistance of Erina He, Medical Illustrator, National Institutes of Health Medical Arts, for her assistance with creating the figures.
Author information
Authors and Affiliations
Contributions
H.G. and M.G.G conducted the review and wrote the manuscript. H.G. and M.G.G. approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gopalakrishna, H., Ghany, M.G. Perspective on Emerging Therapies to Achieve Functional Cure of Chronic Hepatitis B. Curr Hepatology Rep 23, 241–252 (2024). https://doi.org/10.1007/s11901-024-00652-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11901-024-00652-9