
Abstract
Purpose of Review
Hepatitis D Virus (HDV), although a small defective virus, poses a substantial public health challenge due to lack of awareness, underrecognized prevalence, and limited treatment options. Universal HDV screening within hepatitis B virus (HBV) cohorts is essential to address this issue. Despite its aggressive nature, effective HDV therapies have remained elusive for over four decades.
Recent Findings
Advances in understanding HDV’s biology and clinical behavior offer potential therapeutic breakthroughs, fostering optimism. As insights grow, effective and targeted therapies are being developed to improve HDV management.
Summary
This review delves into HDV’s intricate structure and biology, highlighting formidable hurdles in antiviral development. It emphasizes the importance of widespread screening, exploring noninvasive diagnostics, and examining current and emerging innovative therapeutic strategies. Moreover, the review explores models for monitoring treatment response. In essence, this review simplifies the complexities of effectively combating HDV.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatitis D Virus (HDV), also known as hepatitis delta virus, is the smallest yet one of the most potent pathogens, presenting a formidable global public health threat. HDV infection occurs in individuals who are co-infected with hepatitis B virus (HBV), resulting in an aggressive form of viral hepatitis [1]. The combination of HDV and HBV accelerates disease progression, with approximately 70–80% of patients developing cirrhosis within few decades of infection [2].
Recent estimates reveal that HDV impacts 5% of HBV-infected individuals, affecting around 12 million people globally [3•]. However, true prevalence is likely higher due to underutilized screening methods [4, 5]. Despite its substantial impact, clear monitoring guidelines for diagnosed cases are lacking. Advances in serological testing and non-invasive screening strategies are revolutionizing HDV management, reshaping the field [6].
Despite its discovery over four decades ago, there are no Food and Drug Administration (FDA)–approved therapies specifically targeting HDV, leaving patients with limited treatment options. Interferon-alpha is often used based on professional liver societies’ recommendations, but its success rates are generally modest [7]. Though bulevirtide, authorized in Europe in July 2020, marked a breakthrough cure for HDV, a potential gateway to HDV resolution remains elusive [8••].
With complex epidemiology, a rapid disease trajectory, and scarce treatment options, precise diagnosis and disease stratification assume paramount importance. This review aims to shed light on key aspects of managing HDV. It discusses about the intricate structure and biology of HDV, uncovering substantial challenges in antiviral development. This review highlights the importance of widespread screening, explores noninvasive diagnostics and current and emerging innovative therapeutic strategies.
Hepatitis D Virus: a Unique and Complex Pathogen
HDV, a satellite virus reliant on HBV for its life cycle (Fig. 1), is categorized as a “defective,” or “incomplete virus” [9]. The structure of HDV consists of an enveloped particle, 35–37 nm in size, harboring the delta antigen and a circular single-stranded RNA genome covered by the hepatitis B surface antigen (HBsAg) lipoprotein from HBV [10]. This resemblance enables HDV to exploit the same entry receptor, sodium taurocholate co-transporting polypeptide (NTCP), as HBV, facilitating its entry into hepatocytes [11].

HDV life cycle and sites of drug targets. HDV virion attaches to the hepatocyte via the sodium taurocholate cotransporting peptide (NTCP). HDV ribonucleoprotein (RNP) is translocated to nucleus. In the nucleus, HDV genome replication occurs by rolling-circle mechanism. HDV antigenome is transported out of the nucleus to the endoplasmic reticulum (ER). New HDV RNP associates with HBV envelop proteins and assemble into HDV virions. Completed HDV virions are released from the hepatocyte. Drugs approved for usage—as per expert guideline: interferon alpha (IFN-α), conditional approval in Europe: bulevirtide. Drugs under investigation—nucleic acid polymer (NAP), lonafarnib and interferon lambda (IFN-λ)
Once inside hepatocytes, the uncoated HDV genome moves to the nucleus for ribonucleic acid (RNA)–dependent replication, relying on host deoxyribonucleic acid (DNA)–dependent RNA polymerases I and II [12]. Encoded by the HDV genome, the delta antigen exists in two forms: small (S-HDAg) and large (L-HDAg) delta antigen [13]. RNA editing adds 19 amino acids to the C-terminus of L-HDAg, influencing the functions of both forms. S-HDAg serves as a replication co-factor, while L-HDAg inhibits replication [14]. The farnesylation of L-HDAg is essential for its interaction with HBsAg, anchoring it to the viral envelope and forming a ribonucleoprotein complex critical for the assembly and release of infectious HDV particles [15]. Cells infected with HBV often produce an excess of HBsAg beyond what is required for HBV virion assembly. HDV takes advantage of these surplus empty HBsAg envelopes to coat its ribonucleoprotein complexes [16].
HDV can propagate via cell division, generating clusters of infected cells [17]. Interferon-stimulated genes (ISGs) curtail spread, and lack of interferon (IFN) response enhances HDV propagation [18]. Therapeutic IFN treatment inhibits the mitosis-mediated intrahepatic spread of replicative HDV RNA in HBsAg-positive cells, showing potential therapeutic effects. However, IFN’s limited effects on HDV replication in resting hepatocytes explain its limited clinical efficacy [19].
Targeting HDV is challenging due to its intricate life cycle, HBV dependency, and host polymerase reliance. Still, enhanced HDV virology understanding has unveiled promising therapeutic avenues for control and potential cure, as shown in Fig. 1.
Natural History of the Disease
Transmission
HDV primarily spreads through the parenteral route, via infected body fluids (BF), similar to HBV. Intravenous users (IVDUs) are at highest risk for HDV transmission, but needle exchange programs and increasing vaccination efforts have led to a decline in HDV prevalence among them [20, 21]. Although rare, vertical transmission from an infected mother to the fetus can occur during pregnancy or childbirth [22], but breastfeeding-related transmission has not been reported.
Co-infection vs Superinfection
Co-infection refers to simultaneous HDV and HBV infection in individuals with no prior HBV exposure. It can lead to acute self-limiting hepatitis, with most individuals recovering fully [23].
Superinfection occurs when HDV infects an individual with a pre-existing HBV infection. Approximately 90% of superinfection cases are associated with a chronic aggressive disease course leading to increased risk of disease progression, including cirrhosis and hepatocellular carcinoma (HCC) [23]. Serological testing as shown in Table 1 can aid in differentiating acute co-infection and superinfection.
Genotypes and Regional Prevalence
Genetically diverse, HDV has eight genotypes and two to four subtypes each [24]. Genotype 1, the most prevalent globally, is associated with aggressive disease and found predominantly in Europe and North America. Genotype 2 is prevalent in Asia, genotype 3 is the most pathogenic and caused fulminant HDV outbreak in the Amazon basin. Genotype 4 (Japan, Taiwan) and genotypes 5–8 (Africa) differ in outcomes, with genotype 5 showing better IFN response [25, 26].
Underdiagnosis and Impact
Understanding the natural history of HDV has been challenging due to limited prospective studies and data heterogeneity. Prevalence estimates vary, but around 5–14% of HBV patients may have HDV co-infection [3•, 27]. Certain regions are HDV “hotspots,” like Mongolia, Pakistan, Afghanistan, sub-Saharan Africa, Mediterranean, and eastern European countries. Conversely, India, China, Iran, and Israel have declining HDV prevalence (1, 3).
Despite its impact, HDV remains underestimated, particularly in the USA. In a study of 2175 veterans with HBV, merely 8.5% were screened for HDV, and of those tested, 3.4% were HDV-positive. Notably, HDV-positive patients had a 2.9 times higher incidence of HCC compared to those without HDV testing (4). Another study involving 1191 HBV patients revealed that only 8% were tested for HDV, with 73% of the coinfected patients showing cirrhosis, compared to 22% in HBV mono-infection.(5).
Enhancing HDV Screening
Screening for HDV is crucial for early detection and understanding its natural history. Historically, global HDV screening rates have been low, leading to underdiagnosis, despite its severe health ramifications. In the USA, only 6.7% of HBV patients undergo HDV screening [28]. National Health and Nutrition Examination Survey (NHANES) data revealed about 42% of HBsAg carriers tested positive for anti-HDV, emphasizing the importance of routine HDV screening [29]. This urgency was further emphasized by a Spanish study, which showcased a fivefold increase in HDV diagnoses through universal screening among all HBV patients, advocating for broader screening initiatives [30].
High-risk groups, such as HBsAg-positive individuals, those with human immunodeficiency virus (HIV), IVDU, men who have sex with men, and immigrants from HDV-endemic areas, should be screened according to the American Association for the Study of Liver Diseases (AASLD) (7). Conversely, European and Asian associations advocate for universal HDV testing in all chronic HBV patients due to severity and rising prevalence [31, 32]. A systematic review revealed a pooled HDV co-infection prevalence of 14.57%, with a seroprevalence of 10.58% in individuals lacking risk factors, further endorsing the significance of routine screening over a risk-based approach [33]. Globally, HDV infection risks differ, with higher-income countries experiencing risks from immigration and IVDU [34], while lower-income countries face risks from sexual transmission, intra-household transmission, and cultural practices like tattoos and circumcision (1). Consequently, embracing universal screening emerges as the most effective strategy to gauge HDV’s global impact [35].
Diagnosis and Staging
Diagnostic Algorithm and Challenges in HDV Testing
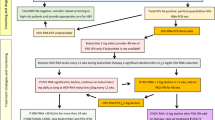
The diagnostic algorithm for HDV starts with screening for the presence of anti-HDV antibodies in patients who are HBsAg-positive [6, 7, 31]. A positive result could indicate prior HDV exposure, necessitating further testing to determine whether the infection is active or resolved (Fig. 2). To confirm active HDV infection, sensitive HDV RNA testing using reverse transcriptase-polymerase chain reaction (RT-PCR) assays is recommended [36]. Several assays have evolved with heightened sensitivity, detecting HDV RNA levels as low as 6 international units (IU) per milliliter. However, it is crucial to emphasize the utilization of validated commercial assays due to variations in detection among different assays and the need for standardized HDV RNA measurement across laboratories [37, 38].

Algorithm for the diagnosis and management of hepatitis D virus
The first World Health Organization international standard for HDV RNA was established in 2013, based on a genotype 1 HDV-infected patient. Despite its availability, in-house nucleic acid amplification testing protocols are commonly used for HDV RNA quantification. Automated isolation may underestimate viral load, and certain genotypes exhibit quantification errors [39, 40]. One other challenge that exists is diagnostic variability in anti-HDV serology assays due to factors like assay setup, antigen type (recombinant HDAg, peptides), and interference from other materials (serum proteins). Improved HDV diagnostic serology requires automated, reproducible assays with high diagnostic accuracy and quantification capability for better reliability [41]. Newly developed droplet digital PCR and cobas6800 offer improved accuracy with low limits of detection, promising enhanced HDV RNA quantification [42].
Staging Methods
Once HDV is confirmed, grading and staging become pivotal for assessing disease severity and progression. While liver biopsy has historically been the gold standard for staging, noninvasive alternatives have emerged [6, 43].
As shown in Fig. 2, serum fibrosis markers including fibrosis-4 score (FIB-4), aspartate aminotransferase (AST)-to-alanine aminotransferase (ALT) ratio, age-platelet index, and AST-to-platelet-ratio-index can help in predicting fibrosis and cirrhosis progression. Among these, FIB-4 stands out for its performance with AUROC of 0.7 for advanced fibrosis and 0.83 for cirrhosis in HDV, but its performance is relatively lower compared to HCV and HBV [43]. Despite certain limitations, these cost-effective markers hold practical applicability, particularly in resource-limited settings burdened by HDV (Fig. 2).
Vibration-controlled transient elastography (VCTE) and shear wave elastography (SWE) are promising non-invasive imaging modalities for fibrosis assessment in chronic hepatitis D [44]. VCTE is a notable noninvasive method that has demonstrated promising accuracy in cirrhosis detection, notably with a cutoff of 14 kPa [45]. However, further validation through larger studies is warranted to solidify its clinical utility. VCTE, although effective, faces limitations in accessibility due to cost and availability. In contrast, SWE’s global application potential is augmented by its compatibility with existing ultrasound machines through software additions.
Novel Fibrosis Scoring System
The Delta Fibrosis Score (DFS) and Delta-4 Fibrosis Score (D4FS) have been developed as non-invasive fibrosis scores for HDV. DFS incorporates patient age, albumin, gamma-glutamyl transferase, and cholinesterase (CHE) levels to predict advanced fibrosis, exhibiting good performance with an AUROC of 0.87 [46]. D4FS, which integrates serum fibrosis markers and VCTE, is a better predictor of cirrhosis with an impressive AUROC of 0.94 [47]. Nevertheless, the clinical application of DFS score might be hampered by marker availability, as seen in DFS with CHE. Larger prospective studies are needed to validate and establish the clinical utility of these scores in managing HDV patients.
Treatment
Who Needs to be Treated?
Ideally, every HDV-infected individual should receive treatment, particularly considering the deadly nature of the infection. Nonetheless, due to the clear link between HDV viremia and worsened outcomes, highlighted by a Swedish study indicating a 3.8-fold higher risk of liver-related complications due to HDV RNA viremia over 6.5 years, strategic measures are advised [48]. Accordingly, the AASLD prioritizes treatment for those with elevated HDV RNA levels and ALT elevation (7). Elevated HDV RNA levels frequently correspond to more severe disease manifestations [49]. Conversely, the absence or substantial reduction of HDV RNA aligns with improved clinical outcomes. Emphasizing the pressing requirement for effective treatment strategies, the objective is to manage HDV infection and alleviate its adverse impact.
Definition of Treatment Response: Ideal vs Reality
Defining effective endpoints in HDV therapy is complex due to the dual challenge of suppressing both HBV and HDV. Proposed surrogate endpoints, like HBsAg clearance and HDV RNA below the lower limit of quantification (LLOQ), correlate with improved clinical outcomes. Sustained virological response (SVR), characterized by undetectable HDV RNA for 24 weeks post-therapy, reduces adverse events during long-term follow-up [50]. However, achieving SVR at 24 weeks remains challenging, as evidenced by relapses observed even after its attainment [51].
An alternative endpoint, proposed by Yurdaydin et al., suggests a ≥ 2 log10 HDV RNA decline at the end of treatment, though this should be labeled a partial response [52, 53••]. Regulatory agencies in the US and Europe have accepted this proposal, acknowledging its potential significance. However, any presence of HDV is associated with increased risks compared to undetectable HDV RNA, casting doubt on the current endpoint’s validity [50, 54]. A study from Spain further questions the endpoint’s relevance by revealing no significant clinical outcome differences between those with ≥ 2 log10 HDV-RNA decline and those without [55]. But patients in this study achieved SVR spontaneously.
As per AASLD-EASL guidelines, for finite therapies, the ideal endpoint involves HBsAg loss, anti-HB seroconversion, and HDV RNA < LLOQ. An alternate goal is HDV RNA < LLOQ at 24 weeks post-treatment, with prolonged follow-up. For maintenance therapy, endpoint is HDV RNA < LLOQ at 48 weeks on-treatment, although optimal duration remains uncertain. Reminiscent of predicting duration of anti-viral treatment for hepatitis C needed to reach < 1 virus copy in a patient’s total extracellular BF [56], Shekhtman et al. [57] predicted time to reach < 1 HDV copy per BF in two patients who were treated with bulevirtide monotherapy for 144 weeks [58]. One of these two patients remained HDV RNA undetectable 72 weeks after bulevirtide discontinuation, suggesting HDV viral cure [59]. If HDV RNA < LLOQ is unattainable, an alternative endpoint is a combined response: > 2 log HDV RNA reduction and normal ALT at 48 weeks, with subsequent follow-up [53••]. These evolving endpoints reflect ongoing efforts to effectively manage HDV infection’s intricate dynamics.
Treatment Strategies
Interferon Alpha
Current guidelines recommend pegylated formulations of interferon alpha (PEG-IFNα) as a primary treatment for HDV [60]. While PEG-IFNα’s 48-week regimen offers convenience and acceptable response rates, complete HDV eradication remains modest [61]. Post-treatment, only a subset of patients (ranging from 23 to 57%) achieve undetectable HDV RNA levels at 24 weeks, often accompanied by high relapse rates [51], especially in HBsAg-positive cases. Undetectable HDV RNA level at 24 weeks off treatment does not universally correlate with HDV clearance, necessitating extended follow-up [51].
A Belgian long-term follow-up study demonstrated SVR in 52% of interferon alpha (IFNα)–treated patients at 24 weeks, with 33% patients relapsing over 3 years. Prolonging treatment beyond 48 weeks minimally impacted durable off-treatment virological response [62]. Importantly, responders to interferon exhibit reduced mortality rates compared to non-responders, emphasizing its clinical significance. A German study with mean follow up of 5.2 years demonstrated that IFNα treatment yields better long-term outcomes than untreated or nucleotide analog-receiving patients [63].
Interferon Lambda
Interferon lambda (IFN-λ) offers a promising avenue for HDV treatment, thanks to its selective hepatocyte receptor expression and reduced side effect profile. A phase II trial explored two doses (120 g and 180 g) of IFN-λ for 48 weeks and resulted in viral load below limit of quantification (BLQ) in 16% and 36% of patients in the respective arms at 24 weeks post-treatment. The 180-g arm demonstrated comparable SVR to PEG-IFNα, with fewer side effects [64].
Bulevirtide
HDV exploits the NTCP receptor, similar to HBV, for hepatocyte entry. Bulevirtide (myrcludex B), is a synthetic myristoylated peptide that binds to NTCP receptors, disrupting viral entry. In the MYR-202 trial with 120 HDV patients, different bulevirtide doses with tenofovir combinations showed significant HDV RNA reduction (50–77%) compared to tenofovir alone (4%). However, bulevirtide had minimal impact on HBsAg levels, and discontinuation led to HDV RNA rebound [65]. In a phase 3 MYR-301 trial, patients received subcutaneous bulevirtide (2 mg or 10 mg daily) or no treatment for 144 weeks. The 48-week interim analysis showed a higher response rate (45% in 2 mg, 48% in 10 mg) than controls (2%), meeting the primary endpoint of undetectable HDV RNA or significant decrease with ALT normalization. Yet only 12% in 2 mg and 20% in 10 mg groups achieved undetectable HDV RNA at week 48. HBsAg levels did not significantly decrease in either bulevirtide group (8). Real-world data demonstrated a 76% virologic response (≥ 2 log HDV RNA decline or undetectable) with bulevirtide, but relapse occurred in some cases, and no patients lost HBsAg [66].
Combining Bulevirtide with Peginterferon
The MYR-203 trial examined bulevirtide alone or with PEG-IFNα or tenofovir in 60 patients across four arms for 48 weeks. Low-dose bulevirtide with PEG-IFNα and high-dose bulevirtide with PEG-IFNα maintained undetectable HDV RNA levels in 8/15 and 4/15 patients, respectively [67]. MYR-204, a phase 2b study, demonstrated higher HDV-RNA decline rates in combination arms compared to PEG-IFNα monotherapy [68].
Despite bulevirtide showing promise, its long-term use, discontinuation viability, safety in decompensated cirrhotic patients, and potential dose-related bile acid increase will require further exploration.
Nucleic Acid Polymer
Nucleic acid polymers targeting host chaperones have shown potential in disrupting HBsAg assembly and release. One promising candidate is REP 2139-Ca. In a small study, patients received REP 2139-Ca monotherapy for 15 weeks, followed by PEG-IFNα for an additional 15 weeks, and then PEG-IFNα monotherapy for 33 weeks. Astonishingly, seven of 12 patients achieved undetectable HDV RNA levels, and five had HBsAg loss. Impressively, a 3.5-year follow-up demonstrated sustained outcomes, with seven of 11 patients maintaining undetectable HDV RNA levels and four achieving HBsAg loss [69, 70]. Mathematical modeling indicated that the median efficacy of REP 2139-Ca monotherapy in blocking HBsAg and HDV production was 98.2% and 99.7%, respectively. In addition, a short HBsAg half-life of 1.3 days was estimated, suggesting a rapid turnover of HBsAg in HBV/HDV co-infected patient [71•]. To validate these findings, large-scale studies are imperative.
Lonafarnib
Lonafarnib, a farnesyl-transferase inhibitor, interferes with HDV assembly and secretion of HDV virions [72]. Mathematical modeling indicated high efficacy of lonafarnib in blocking HDV production (95%) along with an estimated HDV half-life of 1.6 days. While initial lonafarnib monotherapy studies reduced HDV viral load, they resulted in elevated gastrointestinal adverse events. However, combining low-dose lonafarnib (25 or 50 mg twice daily) with the cytochrome P450 3A4 inhibitor ritonavir proved beneficial, yielding favorable antiviral effects with reduced side effects. Notably, lonafarnib 25 mg with ritonavir + PEG-IFNα achieved a remarkable mean log decline of − 5.57 log10 U/ml in HDV RNA levels, with 60% of patients achieving undetectable HDV-RNA and ALT normalization [73].
Combining Lonafarnib with Peginterferon
The preliminary data from recently concluded LIFT study explored triple therapy with peginterferon lambda (PEG-IFN-λ), lonafarnib, and ritonavir in HDV patients for 24 weeks. After 12 weeks, mean HDV RNA levels dropped by 3.4 log IU/mL. At therapy conclusion, the mean decline was 3.2 log IU/mL (50% undetectable or BLQ). Impressively, 96% achieved a > 2 log decline during the 24-week treatment period, accompanied by mostly mild to moderate adverse events [74•]. The encouraging LIFT study outcomes set the stage for an upcoming 48-week trial, exploring the extended efficacy of triple combination therapy.(ClinicalTrials.gov NCT05953545). This extension holds potential for further enhancing positive results.
In the phase 3 D-LIVR study with 407 patients, ritonavir-boosted lonafarnib alone or combined with PEG-IFNα was evaluated for 48 weeks. Combination therapy (19.2%) showed superior response rates compared to lonafarnib/ritonavir (10.1%) or PEG-IFNα alone (9.6%) in achieving the primary endpoint of ≥ 2 log HDV RNA reduction with ALT normalization [75].
Role of Nucleos(t)Ide Analogues in HDV Management
While nucleos(t)ide analogues (NAs) alone may not achieve HDV cure [76], they still might have a role to play and are frequently used alongside HDV treatment due to potential HBV reactivation after HDV control [72]. This requires further clarification. Additionally, the role of NA in compensated HBV/HDV cirrhosis with low HBV DNA levels needs investigation.
Monitoring Response to Treatment and Disease Progression
The study conducted by Guedj et al. has yielded a significant breakthrough, shedding light on the intricate dynamics of early HDV and HBsAg kinetics during PEG-IFNα therapy. Through a novel mathematical model, this research offers crucial insights into the complex interplay among HDV, the host, interferon treatment impact, and treatment response predictors. Analyzing comprehensive HDV and HBsAg kinetic data over 28 weeks from 10 PEG-IFNα-treated patients, the study revealed a biphasic decline in HDV levels—a rapid initial phase followed by a slower or plateau phase. Impressively, PEG-IFNα’s efficacy in suppressing HDV production was high, with a median effectiveness of 96%. The correlation between HBsAg kinetics and the latter phase of HDV decline highlighted the role of HBsAg-productive–infected cells in HDV production. Clinically, this finding is notable, particularly in identifying a flat second phase in HDV and HBsAg kinetics as a potential predictive marker for incomplete virological response during PEG-IFNα therapy. This discovery holds promise for shaping early discontinuation criteria, optimizing decision-making in HDV management [77].
Another study explored the role of HBsAg in HDV-RNA clearance during PEG-IFNα treatment. A reduction in HBsAg below 1000 IU/mL at the 6 months indicated favorable outcomes. A 0.105 log HBsAg reduction correlated with 1.610 log HDV-RNA reduction at 6 months on treatment, predicting HDV clearance [78]. Hence, quantitative HBsAg could guide therapy decisions. Hepatitis B core-related antigen (HBcrAg) linked to intrahepatic covalently closed circular DNA levels can aid in predicting treatment response. In a study which investigated this, baseline HBcrAg < 4.5 log IU/ml increased chances of achieving undetectable HDV RNA at 24-week follow-up (NPV 81.8%), with higher end-of-treatment HBcrAg linked to a higher relapse rate [79].
Screening for Hepatocellular Carcinoma
The intricate relationship between HDV and HBV, where HDV relies on HBV for transmission, adds complexity to understanding HDV’s role in hepatocarcinogenesis. In Italy, a long-term study estimated annual incidences of liver decompensation in HDV cirrhosis at 4%, with HCC incidence at 2.8% [54]. Patients with HDV cirrhosis are at threefold higher risk of developing HCC than those with HBV mono-infection associated cirrhosis [80], emphasizing importance of strict screening in those with HDV infection.
Interestingly, in non-cirrhotic patients, HDV replication increased the likelihood of progression to HCC even in absence of cirrhosis [81]. However, uncertainties persist regarding whether individuals without cirrhosis should undergo HCC screening and the appropriate age to initiate screening for those without cirrhosis. A study from Mongolia showed that HBV-HDV coinfection contributes to HCC development at a comparatively younger age in contrast to those infected with hepatitis C virus (mean age 51 vs 59). However, this study did not differentiate between cirrhotic and non-cirrhotic patients [82].
These findings emphasize the relationship between HDV and HCC, suggesting the need for vigilant HCC screening strategies, especially in HDV-infected cirrhotic. Further research is required to delineate optimal screening protocols and the age at which to start HCC surveillance in non-cirrhotic HDV patients.
Liver Transplantation
In HDV patients with decompensated cirrhosis or fulminant hepatitis, the use of interferon is restricted due to adverse effects and limited data. Consequently, liver transplantation (LT) remains the primary treatment avenue [83]. In one of the most extensive studies involving 76 LT in HDV patients, a remarkable 88% survival rate was observed for 5 years follow-up [84].
Historically, LT for HDV-induced cirrhosis has yielded favorable clinical outcomes, although accompanied by significant graft reinfection rates (70–80%) [85]. As expected, the presence of HBV viremia proved pivotal for HDV recurrence (1). The introduction of prolonged hepatitis B immunoglobulin (HBIg) prophylaxis successfully lowered HDV reinfection rates and further improved through HBIg and NA combinations [86]. A comprehensive European cohort study revealed a mere 3.5% HBV recurrence post-LT, with negligible HDV recurrences [87].
Recent studies investigate stopping HBIg with ongoing NAs after HDV-related transplants, noting minimal HDV reinfections, validating efficacy of NAs alone in controlling relapse [88, 89]. Collectively, prophylactic approaches have substantially diminished HDV recurrence rates post-LT, offering optimism for enhanced patient outcomes.
HBV Vaccination to Reduce HDV
The absence of an HDV vaccine underscores the significance of HBV vaccination as a key strategy for preventing HBV and thus potentially curtailing HDV transmission. The implementation of HBV vaccination has led to a decline in HBV infections, consequently protecting high-risk groups from acquiring HDV infection [90]. This is evidenced by the age-related shift in HDV prevalence towards older individuals, reflecting the success of HBV vaccination programs in preventing early HBV infections [91]. By effectively preventing HBV, vaccination indirectly contributes to the reduction of HDV transmission, offering long-term protection against these viral infections.
Critical Questions
In the realm of HDV infection management, several pivotal questions arise. What is the true prevalence of HDV? What drives the swift advancement to cirrhosis and hepatocellular carcinoma in HDV-infected individuals? As we seek meaningful treatment endpoints, does a 2-log decline in HDV RNA levels truly correlate with improved clinical outcomes, and are there alternative endpoints which offer valuable insights? The optimal treatment duration for each HDV agent warrants consideration, as does the potential synergy of combination therapies. Amid these complexities, the feasibility of treating individuals with decompensated cirrhosis remains a critical query. Shifting focus, determining the appropriate age to initiate HCC screening in HDV-infected patients and investigating possible associations between genotypes and disease progression add further layers of inquiry. Lastly, the question of reactivation or relapse in those who have cleared HDV infection looms, demanding vigilant understanding. Addressing these questions are important in advancing comprehension of HDV infection management.
Conclusions
Addressing the complex challenges posed by HDV infection requires a multi-faceted approach. Raising awareness among healthcare professionals is imperative, advocating for reflex screening of HBV patients for anti-HDV antibodies. In cases of positivity, a subsequent step involves using a reliable and validated quantitative HDV RNA assays to confirm active infection. Timely and accurate assessment, facilitated by validated serological biomarkers and non-invasive tests, is crucial for effective diagnosis and risk stratification. Immunization against HBV stands as a potent preventive measure. While financial constraints and awareness gaps persist, ongoing research and innovative therapies fuel hope for better outcomes. Understanding viral-host-drug dynamics may help to develop and optimize response-guided therapies for HDV patients. This dedication to understanding and treating HDV brings hope for millions suffering from this deadly infection, promising enhanced global health in the face of its enduring challenges.
Abbreviations
- AAR:
-
AST-to-ALT ratio
- AASLD:
-
American Association for the Study of Liver Disease
- ALT:
-
Alanine aminotransferase
- API:
-
Age platelet index
- APRI:
-
AST-to-platelet ratio index
- AST:
-
Aspartate aminotransferase
- AUROC:
-
Area under the receiver operating characteristic curve
- BF:
-
Body fluid
- CHB:
-
Chronic hepatitis B
- CHC:
-
Chronic hepatitis C
- CHD:
-
Chronic hepatitis D
- CHE:
-
Cholinesterase
- CSPH:
-
Clinically significant portal hypertension
- D4FS:
-
Delta-4 fibrosis score
- DFS:
-
Delta fibrosis score
- DNA:
-
Deoxyribonucleic acid
- EASL:
-
European Association for the Study of the Liver
- ELISA:
-
Enzyme-linked immunosorbent assay
- FIB-4:
-
Fibrosis-4
- FDA:
-
Food and Drug Administration
- GGT:
-
Gamma glutamyl transferase
- HBcrAg:
-
Hepatitis B core antigen
- HBeAg:
-
Hepatitis B e antigen
- HBsAg:
-
Hepatitis B surface antigen
- HBV:
-
Hepatitis B virus
- HBIg:
-
Hepatitis B immunoglobulin
- HDAg:
-
Hepatitis D antigen
- HDV:
-
Hepatitis delta virus
- HCC:
-
Hepatocellular carcinoma
- HIV:
-
Human immunodeficiency virus
- IU:
-
International units
- IVDU:
-
Intravenous drug users
- kPa:
-
Kilopascals
- LLOQ:
-
Lower limit of quantification
- LT:
-
Liver transplantation
- NHANES:
-
National Health and Nutrition Examination Survey
- NPV:
-
Negative predictive value
- NTCP:
-
Sodium taurocholate cotransporting polypeptide
- RT-PCR:
-
Reverse transcriptase-polymerase chain reaction
- PEG-IFN:
-
Pegylated interferon
- PCR:
-
Polymerase chain reaction
- RNA:
-
Ribonucleic acid
- SWE:
-
Shear wave elastography
- VCTE:
-
Vibration-controlled transient elastography
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Rizzetto M, Hamid S, Negro F. The changing context of hepatitis D. J Hepatol. 2021;74(5):1200–11.
Negro F, Baldi M, Bonino F, Rocca G, Demartini A, Passarino G, et al. Chronic Hdv (hepatitis delta-virus) hepatitis - intrahepatic expression of delta antigen, histologic activity and outcome of liver-disease. J Hepatol. 1988;6(1):8–14.
• Stockdale AJ, Kreuels B, Henrion MYR, Giorgi E, Kyomuhangi I, de Martel C, et al. The global prevalence of hepatitis D virus infection: systematic review and meta-analysis. J Hepatol. 2020;73(3):523–32.Systematic review showing high prevalance of HDV globally.
Kushner T, Serper M, Kaplan DE. Delta hepatitis within the Veterans Affairs medical system in the United States: prevalence, risk factors, and outcomes. J Hepatol. 2015;63(3):586–92.
Gish RG, Yi DH, Kane S, Clark M, Mangahas M, Baqai S, et al. Coinfection with hepatitis B and D: epidemiology, prevalence and disease in patients in Northern California. J Gastroen Hepatol. 2013;28(9):1521–5.
Majeed NA, Hitawala AA, Heller T, Koh C. Diagnosis of HDV: from virology to non-invasive markers of fibrosis. Liver Int. 2023;1:31–46. https://doi.org/10.1111/liv.15515.
Terrault NA, Lok ASF, McMahon BJ, Chang KM, Hwang JP, Jonas MM, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67(4):1560–99.
•• Wedemeyer H, Aleman S, Brunetto MR, Blank A, Andreone P, Bogomolov P, et al. A phase 3, randomized trial of bulevirtide in chronic hepatitis D. N Engl J Med. 2023.Phase 3 trial of Bulevirtide showing its effectiveness in HDV management.
Rizzetto M, Hoyer B, Canese MG, Shih JWK, Purcell RH, Gerin JL. Delta-agent - association of delta-antigen with hepatitis-b surface-antigen and RNA in serum of delta-infected chimpanzees. P Natl Acad Sci-Biol. 1980;77(10):6124–8.
Bonino F, Heermann KH, Rizzetto M, Gerlich WH. Hepatitis delta virus - protein-composition of delta-antigen and its hepatitis-b virus derived envelope. Ital J Gastroenterol. 1986;18(5):297.
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;13;1:e00049. https://doi.org/10.7554/eLife.00049.
Greco-Stewart VS, Miron P, Abrahem A, Pelchat M. The human RNA polymerase II interacts with the terminal stem-loop regions of the hepatitis delta virus RNA genome. Virology. 2007;357(1):68–78.
Gudima S, Chang JH, Moraleda G, Azvolinsky A, Taylor J. Parameters of human hepatitis delta virus genome replication: the quantity, quality, and intracellular distribution of viral proteins and RNA. J Virol. 2002;76(8):3709–19.
Chao M, Hsieh SY, Taylor J. Role of two forms of hepatitis delta virus antigen: evidence for a mechanism of self-limiting genome replication. J Virol. 1990;64(10):5066–9.
O’Malley B, Lazinski DW. Roles of carboxyl-terminal and farnesylated residues in the functions of the large hepatitis delta antigen. J Virol. 2005;79(2):1142–53.
Giersch K, Bhadra OD, Volz T, Allweiss L, Riecken K, Fehse B, et al. Hepatitis delta virus persists during liver regeneration and is amplified through cell division both in vitro and in vivo. Gut. 2019;68(1):150–7.
Zhang ZF, Ni Y, Lempp FA, Walter L, Mutz P, Bartenschlager R, et al. Hepatitis D virus-induced interferon response and administered interferons control cell division-mediated virus spread. J Hepatol. 2022;77(4):957–66.
Alfaiate D, Lucifora J, Abeywickrama-Samarakoon N, Michelet M, Testoni B, Cortay JC, et al. HDV RNA replication is associated with HBV repression and interferon-stimulated genes induction in super-infected hepatocytes. Antivir Res. 2016;136:19–31.
Zhang ZF, Urban S. New insights into HDV persistence: the role of interferon response and implications for upcoming novel therapies. J Hepatol. 2021;74(3):686–99.
Kucirka LM, Farzadegan H, Feld JJ, Mehta SH, Winters M, Glenn JS, et al. Prevalence, correlates, and viral dynamics of hepatitis delta among injection drug users. J Infect Dis. 2010;202(6):845–52.
Aguilera A, Trastoy R, Barreiro P, Costa JJ, de Mendoza C, Pena JM, et al. Decline and changing profile of hepatitis delta among injection drug users in Spain. Antivir Ther. 2018;23(1):87–90.
Sellier PO, Maylin S, Brichler S, Bercot B, Lopes A, Chopin D, et al. Hepatitis B virus-hepatitis D virus mother-to-child co-transmission: a retrospective study in a developed country. Liver Int. 2018;38(4):611–8.
Negro F. Hepatitis D virus coinfection and superinfection. Cold Spring Harb Perspect Med. 2014;4(11):a021550. https://doi.org/10.1101/cshperspect.a021550.
Le Gal F, Gault E, Ripault MP, Serpaggi J, Trinchet JC, Gordien E, et al. Eighth major clade for hepatitis delta virus. Emerg Infect Dis. 2006;12(9):1447–50.
Wranke A, Borzacov LMP, Parana R, Lobato C, Hamid S, Ceausu E, et al. Clinical and virological heterogeneity of hepatitis delta in different regions world-wide: the Hepatitis Delta International Network (HDIN). Liver Int. 2018;38(5):842–50.
Spaan M, Carey I, Bruce M, Shang DZ, Horner M, Dusheiko G, et al. Hepatitis delta genotype 5 is associated with favourable disease outcome and better response to treatment compared to genotype 1. J Hepatol. 2020;72(6):1097–104.
Miao ZJ, Zhang SS, Ou XM, Li S, Ma ZR, Wang WS, et al. Estimating the global prevalence, disease progression, and clinical outcome of hepatitis delta virus infection. J Infect Dis. 2020;221(10):1677–87.
Wong RJ, Kaufman HW, Niles JK, Chen C, Yang ZY, Kapoor H, et al. Low performance of hepatitis delta virus testing among 2 national cohorts of chronic hepatitis B patients in the United States. Am J Gastroenterol. 2022;117(12):2067–70.
Patel EU, Thio CL, Boon D, Thomas DL, Tobian AAR. Prevalence of hepatitis B and hepatitis D virus infections in the United States, 2011–2016. Clin Infect Dis. 2019;69(4):709–12.
Palom A, Rando-Segura A, Vico J, Pacin B, Vargas E, Barreira-Diaz A, et al. Implementation of anti-HDV reflex testing among HBsAg-positive individuals increases testing for hepatitis D. JHEP Rep. 2022;4(10):100547.
Sarin SK, Kumar M, Lau GK, Abbas Z, Chan HLY, Chen CJ, et al. Asian-Pacific clinical practice guidelines on the management of hepatitis B: a 2015 update. Hep Intl. 2016;10(1):1–98.
Liver EAS. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370–98.
Chen HY, Shen DT, Ji DZ, Han PC, Zhang WM, Ma JF, et al. Prevalence and burden of hepatitis D virus infection in the global population: a systematic review and meta-analysis. Gut. 2019;68(3):512–21.
Da BL, Rahman F, Lai WC, Kleiner DE, Heller T, Koh C. Risk factors for delta hepatitis in a North American cohort: who should be screened? Am J Gastroenterol. 2021;116(1):206–9.
Razavi HA, Buti M, Terrault NA, Zeuzem S, Yurdaydin C, Tanaka J, et al. Hepatitis D double reflex testing of all hepatitis B carriers in low-HBV- and high-HBV/HDV-prevalence countries. J Hepatol. 2023;79(2):576–80.
Stelzl E, Ciesek S, Cornberg M, Maasoumy B, Heim A, Chudy M, et al. Reliable quantification of plasma HDV RNA is of paramount importance for treatment monitoring: a European multicenter study. J Clin Virol. 2021;142:104932.
Le Gal F, Brichler S, Sahli R, Chevret S, Gordien E. First international external quality assessment for hepatitis delta virus RNA quantification in plasma. Hepatology. 2016;64(5):1483–94.
Le Gal F, Dziri S, Gerber A, Alloui C, Ben Abdesselam Z, Roulot D, et al. Performance characteristics of a new consensus commercial kit for hepatitis D virus RNA viral load quantification. J Clin Microbiol. 2017;55(2):431–41.
Bremer B, Anastasiou OE, Ciesek S, Weelemeyer H. Automated nucleic acid isolation methods for HDV viral load quantification can lead to viral load underestimation. Antivir Ther. 2019;24(2):117–23.
Brichler S, Le Gal F, Butt A, Chevret S, Gordien E. Commercial real-time reverse transcriptase PCR assays can underestimate or fail to quantify hepatitis delta virus viremia. Clin Gastroenterol H. 2013;11(6):734–40.
Lempp FA, Roggenbach I, Nkongolo S, Sakin V, Schlund F, Schnitzler P, et al. A Rapid point-of-care test for the serodiagnosis of hepatitis delta virus infection. Viruses. 2021;13(12):2371. https://doi.org/10.3390/v13122371.
Pfluger LS, Norz D, Volz T, Giersch K, Giese A, Goldmann N, et al. Clinical establishment of a laboratory developed quantitative HDV PCR assay on the cobas6800 high-throughput system. JHEP Rep. 2021;3(6):100356.
Takyar V, Surana P, Kleiner DE, Wilkins K, Hoofnagle JH, Liang TJ, et al. Noninvasive markers for staging fibrosis in chronic delta hepatitis. Aliment Pharm Ther. 2017;45(1):127–38.
Yang AH, Yardeni D, Hercun J, Kleiner DE, Ling A, Marko J, et al. Shear wave elastography: how well does it perform in chronic hepatitis D virus infection? J Viral Hepat. 2022;29(12):1127–33.
Da BL, Surana P, Takyar V, Kleiner DE, Heller T, Koh C. Vibration-controlled transient elastography for the detection of cirrhosis in chronic hepatitis D infection. J Viral Hepatitis. 2020;27(4):428–36.
Lutterkort GL, Wranke A, Yurdaydin C, Budde E, Westphal M, Lichtinghagen R, et al. Non-invasive fibrosis score for hepatitis delta. Liver Int. 2017;37(2):196–204.
Da BL, Surana P, Kleiner DE, Heller T, Koh C. The Delta-4 fibrosis score (D4FS): a novel fibrosis score in chronic hepatitis D. Antivir Res. 2020;174:104691. https://doi.org/10.1016/j.antiviral.2019.104691.
Kamal H, Westman G, Falconer K, Duberg AS, Weiland O, Haverinen S, et al. Long-term study of hepatitis delta virus infection at secondary care centers: the impact of viremia on liver-related outcomes. Hepatology. 2020;72(4):1177–90.
Palom A, Rodriguez-Tajes S, Navascues CA, Garcia-Samaniego J, Riveiro-Barciela M, Lens S, et al. Long-term clinical outcomes in patients with chronic hepatitis delta: the role of persistent viraemia. Aliment Pharm Ther. 2020;51(1):158–66.
Yurdaydin C, Keskin O, Kalkan C, Karakaya F, Caliskan A, Kabacam G, et al. Interferon treatment duration in patients with chronic delta hepatitis and its effect on the natural course of the disease. J Infect Dis. 2018;217(8):1184–92.
Heidrich B, Yurdaydin C, Kabacam G, Ratsch BA, Zachou K, Bremer B, et al. Late HDV RNA relapse after peginterferon alpha-based therapy of chronic hepatitis delta. Hepatology. 2014;60(1):87–97.
Farci P, Roskams T, Chessa L, Peddis G, Mazzoleni AP, Scioscia R, et al. Long-term benefit of interferon alpha therapy of chronic hepatitis D: regression of advanced hepatic fibrosis. Gastroenterology. 2004;126(7):1740–9.
•• Yurdaydin C, Abbas Z, Buti M, Cornberg M, Esteban R, Etzion O, et al. Treating chronic hepatitis delta: the need for surrogate markers of treatment efficacy. J Hepatol. 2019;70(5):1008–15.Seminal paper which resulted in implementation of surrogate markers assessing the effectiveness of HDV management.
Romeo R, Del Ninno E, Rumi M, Russo A, Sangiovanni A, De Franchis R, et al. A 28-year study of the course of hepatitis delta infection: a risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology. 2009;136(5):1629–38.
Palom A, Sopena S, Riveiro-Barciela M, Carvalho-Gomes A, Madejon A, Rodriguez-Tajes S, et al. One-quarter of chronic hepatitis D patients reach HDV-RNA decline or undetectability during the natural course of the disease. Aliment Pharmacol Ther. 2021;54(4):462–9.
Etzion O, Dahari H, Yardeni D, Issachar A, Nevo-Shor A, Cohen-Naftaly M, et al.Response guided therapy for reducing duration of direct acting antivirals in chronic hepatitis C infected patients: a Pilot study. Sci Rep. 2020 Oct 20;10(1):17820. https://doi.org/10.1038/s41598-020-74568-x.
Shekhtman L, Cotler SJ, Ploss A, Dahari H. Mathematical modeling suggests that entry-inhibitor bulevirtide may interfere with hepatitis D virus clearance from circulation. J Hepatol. 2022;76(5):1229–31.
Loglio A, Ferenci P, Uceda Renteria SC, Tham CYL, Scholtes C, Holzmann H, et al. Safety and effectiveness of up to 3 years’ bulevirtide monotherapy in patients with HDV-related cirrhosis. J Hepatol. 2022;76(2):464–9.
Anolli MP, Degasperi E, Allweiss L, Sangiovanni A, Maggioni M, Scholtes C, et al. A 3-year course of bulevirtide monotherapy may cure HDV infection in cirrhotics. J Hepatol. 2023;78(4):876–80.
Heller T, Rotman Y, Koh C, Clark S, Haynes-Williams V, Chang R, et al. Long-term therapy of chronic delta hepatitis with peginterferon alfa. Aliment Pharm Ther. 2014;40(1):93–104.
Hercun J, Kim GE, Da BL, Rotman Y, Kleiner DE, Chang R, et al. Durable virological response and functional cure of chronic hepatitis D after long-term peginterferon therapy. Aliment Pharm Ther. 2021;54(2):176–82.
Yurdaydin C, Bozkaya H, Karaaslan H, Onder FO, Erkan OE, Yalcin K, et al. A pilot study of 2 years of interferon treatment in patients with chronic delta hepatitis. J Viral Hepat. 2007;14(11):812–6.
Wranke A, Serrano BC, Heidrich B, Kirschner J, Bremer B, Lehmann P, et al. Antiviral treatment and liver- related complications in hepatitis delta. Hepatology. 2017;65(2):414–25.
Etzion O, Hamid S, Lurie Y, Gane EJ, Yardeni D, Duehren S, et al. Treatment of chronic hepatitis D with peginterferon lambda-the phase 2 LIMT-1 clinical trial. Hepatology. 2023;77(6):2093–103.
Wedemeyer H, Schoneweis K, Bogomolov P, Blank A, Voronkova N, Stepanova T, et al. Safety and efficacy of bulevirtide in combination with tenofovir disoproxil fumarate in patients with hepatitis B virus and hepatitis D virus coinfection (MYR202): a multicentre, randomised, parallel-group, open-label, phase 2 trial. Lancet Infect Dis. 2023;23(1):117–29.
Dietz-Fricke C, Tacke F, Zollner C, Demir M, Schmidt HH, Schramm C, et al. Treating hepatitis D with bulevirtide-real-world experience from 114 patients. JHEP Rep. 2023;5(4):100686. https://doi.org/10.1016/j.jhepr.2023.100686.
Wedemeyer H, Schoneweis K, Bogomolov PO, Voronkova N, Chulanov V, Stepanova T, et al. Final results of a multicenter, open-label phase 2 clinical trial (MYR203) to assess safety and efficacy of myrcludex B in cwith PEG-interferon alpha 2a in patients with chronic HBV/HDV co-infection. J Hepatol. 2019;70(1):E81.
Asselah T, Arama SS, Bogomolov P, Bourliere M, Fontaine H, Gherlan GS, et al. Safety and efficacy of bulevirtide monotherapy and in combination with Peginterferon alfa-2a in patients with chronic hepatitis delta: 24 weeks interim data of MYR204 Phase 2b study. J Hepatol. 2021;75:S291.
Bazinet M, Pantea V, Cebotarescu V, Cojuhari L, Jimbei P, Albrecht J, et al. Safety and efficacy of REP 2139 and pegylated interferon alfa-2a for treatment-naive patients with chronic hepatitis B virus and hepatitis D virus co-infection (REP 301 and REP 301-LTF): a non-randomised, open-label, phase 2 trial. Lancet Gastroenterol. 2017;2(12):877–89.
Bazinet M, Pantea V, Cebotarescu V, Cojuhari L, Jimbei P, Anderson M, et al. Persistent control of hepatitis B virus and hepatitis delta virus infection following REP 2139-Ca and pegylated interferon therapy in chronic hepatitis B virus/hepatitis delta virus coinfection. Hepatol Commun. 2021;5(2):189–202.
• Shekhtman L, Cotler SJ, Hershkovich L, Uprichard SL, Bazinet M, Pantea V, et al. Modelling hepatitis D virus RNA and HBsAg dynamics during nucleic acid polymer monotherapy suggest rapid turnover of HBsAg. Sci Rep. 2020;10(1):7837. Study showed the rapid turnover of HBsAg subviral particles in patients with HDV infection.
Koh C, Canini L, Dahari H, Zhao X, Uprichard SL, Haynes-Williams V, et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: a proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect Dis. 2015;15(10):1167–74.
Yurdaydin C, Idilman R, Keskin O, Kalkan C, Karakaya MF, Caliskan A, et al. A phase 2 dose-optimization study of lonafarnib with ritonavir for the treatment of chronic delta hepatitis-end of treatment results from the LOWR HDV-2 study. J Hepatol. 2017;66(1):S33–4.
• Koh C, Hercun J, Rahman F, Huang A, Da B, Surana P, et al. A phase 2 study of peginterferon lambda, lonafarnib and ritonavir for 24 weeks: end-of-treatment results from the LIFT HDV Study. J Hepatol. 2020;73:S130–S130.Important paper showing effectivess of combining lonafanib/ritonavir with peginterferon lambda in HDV management.
Etzion O et al. Week 48 results of the phase 3 D-LIVR study: a randomized double-blind, placebo-controlled trial, evaluating the safety and efficacy of lonafarnib-boosted with ritonavir with or without peginterferon alfa in patients with chronic hepatitis delta. The International Liver Congress™ EASL - European Association for the Study of the Liver June 21–24 2023 Vienna: J Hepatology.
Wedemeyer H, Yurdaydin C, Hardtke S, Caruntu FA, Curescu MG, Yalcin K, et al. Peginterferon alfa-2a plus tenofovir disoproxil fumarate for hepatitis D (HIDIT-II): a randomised, placebo controlled, phase 2 trial. Lancet Infect Dis. 2019;19(3):275–86.
Guedj J, Rotman Y, Cotler SJ, Koh C, Schmid P, Albrecht J, et al. Understanding early serum hepatitis D virus and hepatitis B surface antigen kinetics during pegylated interferon-alpha therapy via mathematical modeling. Hepatology. 2014;60(6):1902–10.
Niro GA, Smedile A, Fontana R, Olivero A, Ciancio A, Valvano MR, et al. HBsAg kinetics in chronic hepatitis D during interferon therapy: on-treatment prediction of response. Aliment Pharm Ther. 2016;44(6):620–8.
Sandmann L, Yurdaydin C, Deterding K, Heidrich B, Hardtke S, Lehmann P, et al. HBcrAg levels are associated with virological response to treatment with interferon in patients with hepatitis delta. Hepatol Commun. 2022;6(3):480–95.
Fattovich G, Giustina G, Christensen E, Pantalena M, Zagni I, Realdi G, et al. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep). Gut. 2000;46(3):420–6.
Romeo R, Foglieni B, Casazza G, Spreafico M, Colombo M, Prati D. High serum levels of HDV RNA are predictors of cirrhosis and liver cancer in patients with chronic hepatitis delta. Plos One. 2014;9(3).
Oyunsuren T, Kurbanov F, Tanaka Y, Elkady A, Sanduijav R, Khajidsuren O, et al. High frequency of hepatocellular carcinoma in Mongolia; association with mono-, or co-infection with hepatitis C, B, and delta viruses. J Med Virol. 2006;78(12):1688–95.
Kushner T, Da BL, Chan AA, Dieterich D, Sigel K, Saberi B. Liver transplantation for hepatitis D virus in the United States: a UNOS study on outcomes in the MELD era. Transplant Direct. 2022;8(1).
Samuel D, Zignego AL, Reynes M, Feray C, Arulnaden JL, David MF, et al. Long-term clinical and virological outcome after liver-transplantation for cirrhosis caused by chronic delta-hepatitis. Hepatology. 1995;21(2):333–9.
Ottobrelli A, Marzano A, Smedile A, Recchia S, Salizzoni M, Cornu C, et al. Patterns of hepatitis delta-virus reinfection and disease in liver-transplantation. Gastroenterology. 1991;101(6):1649–55.
Duvoux C, Belli LS, Fung J, Angelico M, Buti M, Coilly A, et al. 2020 position statement and recommendations of the European Liver and Intestine Transplantation Association (ELITA): management of hepatitis B virus-related infection before and after liver transplantation. Aliment Pharm Ther. 2021;54(5):583–605.
Beckebaum S, Herzer K, Bauhofer A, Gelson W, De Simone P, de Man R, et al. Recurrence of hepatitis B infection in liver transplant patients receiving long-term hepatitis b immunoglobulin prophylaxis. Ann Transpl. 2018;23:789–801.
Manini MA, Whitehouse G, Bruce M, Passerini M, Lim TY, Carey I, et al. Entecavir or tenofovir monotherapy prevents HBV recurrence in liver transplant recipients: a 5-year follow-up study after hepatitis B immunoglobulin withdrawal. Dig Liver Dis. 2018;50(9):944–53.
Cholongitas E, Goulis I, Antoniadis N, Fouzas I, Imvrios G, Giakoustidis D, et al. Nucleos(t)ide analog(s) prophylaxis after hepatitis B immunoglobulin withdrawal against hepatitis B and D recurrence after liver transplantation. Transpl Infect Dis. 2016;18(5):667–73.
Lin HH, Lee SS, Yu ML, Chang TT, Su CW, Hu BS, et al. Changing hepatitis D virus epidemiology in a hepatitis B virus endemic area with a national vaccination program. Hepatology. 2015;61(6):1870–9.
Stroffolini T, Sagnelli E, Sagnelli C, Russello M, De Luca M, Rosina F, et al. Hepatitis delta infection in Italian patients: towards the end of the story? Infection. 2017;45(3):277–81.
Acknowledgements
The authors acknowledge the assistance of Erina He, Medical Illustrator, National Institutes of Health Medical Arts, for her assistance with creating the figures.
Author information
Authors and Affiliations
Contributions
HG and TH interpreted the data and drafted the manuscript. MM, HD, and CK critically revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Conflict of Interest
None.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gopalakrishna, H., Mironova, M., Dahari, H. et al. Advances and Challenges in Managing Hepatitis D Virus: Evolving Strategies. Curr Hepatology Rep 23, 32–44 (2024). https://doi.org/10.1007/s11901-024-00643-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11901-024-00643-w