
Abstract
The work reports ultrasound-mediated greener synthesis of 11 novel 3-(4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-ylimino)indolin-2-one (7a–7k) derivatives. The synthesized derivatives were evaluated for their in vitro anticancer activity against a panel of selected human cancer cell lines of breast (MCF-7), cervix (HeLa), prostate (PC-3) and lung (A-549). Among the tested compounds, 7b exhibited most promising in vitro anticancer activity against HeLa, PC-3 and A-549 with GI50 value 15.38, 19.67 and 4.37 µM, respectively. The compounds (7a–7k) were also screened for induction of apoptosis and morphological changes in cancer cells at their GI50 concentration. The treatment of HeLa, PC-3 and A549 cancer cells with 7b and treatment of MCF-7 cancer cells with 7h showed apoptosis and morphological changes such as cell shrinkage, cell wall deformation and reduced number of viable cells. The compound 7b has shown almost 5.00 times more selectivity for PC-3 cancer cell lines in comparison to the RWPE-1 normal prostate epithelial cells. Molecular docking study has been carried out, which replicates results of biological activity in cases of initial hits 7b, 7c and 7d, suggesting that these compounds have a potential to become lead molecules in the drug discovery process. In silico ADMET study was performed for predicting pharmacokinetic properties and toxicity profile of the synthesized compounds and expressed good oral drug-like behaviour. An in vivo acute oral toxicity study was performed using Swiss albino mice for the most active compounds 7b and 7c, and results indicate that the compounds are non-toxic in nature.
Similar content being viewed by others
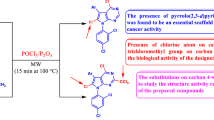

Introduction
Cancer is a life threatening disease characterized by uncontrolled growth of cells, leading to invasion of surrounding tissue and often spreading to other parts of the body; cancer affects millions of people across the globe [1, 2]. Therefore, the development of new anticancer agents is the need of the hour, and scientists all over the world are in search of an active molecule that can save the lives of patients with cancer.
During carcinogenesis, an angiogenic switch occurs, and several angiogenic growth factors stimulate a cell's receptor tyrosine kinases (RTKs) to initiate multiple pro-angiogenic events [3]. A therapeutic strategy to inhibit these key angiogenic proteins or their RTKs was envisioned [4, 5], and multiple inhibitors targeting epidermal growth factor receptors (EGFR), vascular endothelial growth factors (VEGF) and/or platelet-derived growth factor receptors (PDGFR-2), among others, are now used clinically. These RTKs are noted to have multi-kinase effects [6], and this appears to be important for enhanced anticancer activity.
Turning to cytotoxic chemotherapy, tubulin binding agents, such as vincristine, vinblastine and vindesine, are among the most successful anticancer drugs in clinical use [7]. These compounds can be classified as microtubule stabilizers that stimulate tubulin polymerization or destabilizers that inhibit tubulin polymerization. The destabilizing agents bind to tubulin at different binding sites, including the vinca domain and the colchicines site [7].
Combination cancer chemotherapy is not a new idea. Recent studies indicate that the combination of antiangiogenic agents with cytotoxic agents is more effective in cancer treatment [8]. Combination chemotherapy with RTK inhibitors as the anti-angiogenic component along with cytotoxic chemotherapeutic agents are in clinical trials [9, 10]. Examples of such combinations currently in clinical trials include the combination of lapatinib with carboplatin, paclitaxel and trastuzumab in metastatic breast cancer [11, 12] and docetaxel, gemcitabine and pazopanib as treatment for soft tissue sarcoma [13], among others [11]. The advantages of combination chemotherapy, particularly with RTK inhibitors, addresses pathway redundancy [10], as well as tumor heterogeneity among other resistance mechanisms and is beneficial when RTK inhibitors are combined with conventional cancer therapeutics [9, 10]. RTK inhibitors are cytostatic in nature, i.e. they inhibit the growth of tumor cells. The tubulin inhibitors are cytotoxic in nature, i.e. they kill the tumor cells. If the designed molecule contains pharmacophores, making it cytostatic plus cytotoxic, then such a molecule could be very helpful in treating cancer patients. In keeping with the principles of combination chemotherapy [9, 10], such single entities would act simultaneously at two or more distinct targets and prevent or delay the emergence of resistance, avoid drug–drug interactions, circumvent pharmacokinetic problems and overlapping toxicities that plague combination chemotherapy with two or more separate agents. Therefore, we sought to combine inhibitory and cytotoxic activities of RTKs in a single molecule to afford combination chemotherapeutics via a single agent [14].
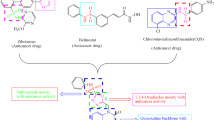
In the search of novel future anticancer agents, considerable literature has been made on the development of heterocyclic motifs based on their structural design. It is worth noting that isatin (indolin-2,3-dione) a “privileged scaffold” has been found to be an important class of heterocyclic compounds endowed of interesting pharmacological [15, 16] and biological activities such as antimicrobial [17], cholinesterases [18] and anticancer properties [19]. As exemplified by the clinically approved sunitinib, an indole-2-one-based kinase inhibitor paved the way for the design and synthesis of various indole-2-one-based molecules [20]. Moreover, orantinib [21], toceranib [22], SU5614 [23] and semaxanib [24] are some clinical drug candidates of “indole-2-one” class molecules known to exhibit potential anticancer activity. Thus, a profusion of pharmacologically and biologically active “C-3 position” of indole-2-one ring has lured researchers all over the world to develop novel molecules of this class for the treatment of cancer. Moreover, the introduction of N-4-chlorobenzyl moiety with isatin as in NSC635473 significantly increases the hydrophobicity and anticancer activity towards cancer cells.
On the other hand, pyrimidine moiety has been found to be an eminent pharmacophore in medicinal chemistry [25]. The meridianin alkaloids and hyrtinadine alkaloids contain indole and pyrimidine scaffolds and are found to posses good anticancer activity [26]. The design protocol for the target compound is presented in Fig. 1.

The design protocol of the target compounds (7a–7k)
The amalgamation of the two dissimilar bioactive pharmacophores made into a single molecule using ultrasound technique is a green approach applied by many researchers all over the world [27,28,29]. Using prominence of indole-2-one, N-4-chlorobenzyl moiety and pyrimidine moiety along with our interest in the synthesis of biologically imperative frameworks with medicinal potential [27, 28], the present report presents the synthesis of a suite of 3-(4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-ylimino)indolin-2-one (7a–7k) derivatives using ultrasound irradiation with a view to engender the promising anticancer agents.
Molecular docking has given brief insight about strength of molecular complexes, suggesting synthesised derivatives have a strong potential to inhibit tubulin and vascular endothelial growth factor receptors (VEGFR2). Vascular endothelial growth factor receptors and related family of enzymes are inhibited by the number of small molecule anticancer agents such as erlotinib, semaxanib, sunitinib, afatinib, axitinib and cabozanitinb that have significant potential as targeted cancer chemotherapeutic agents [30].
The microtubule’s dynamic instability is mostly elicited by compounds such as polypeptide stathmin and ligands such as colchicines. Molecular interactions between colchicines- stathmin and with two tubulin α/β heterodimers in a complex by stathmin lid uped by the stathmin amino-terminal domain, which prevents the complex formation of tubulin into microtubules. The microtubules' structure conformation changes once tubulin in the protofilament becomes curved. Change in the form of tubulin leads to failure in sideways contacts and provides a basis for spontaneous microtubule depolymerization, which is characteristic of dynamic instability. The tubulin–colchicine complex sheds light on the possible mechanism of colchicine activity. It has been shown that colchicine binds at a position where it prevents lid-up tubulin from forming a straight structure, which inhibits assembly [31].
In addition, we have also explored the physicochemical parameters. The synthesized compounds that showed promising in vitro anticancer activity were further tested for their in vivo acute oral toxicity study and gross behavioral studies using Swiss albino mice.
Materials and methods
Chemistry
All chemicals, unless otherwise specified, were purchased from commercial sources and were used without further purification. The major chemicals were purchased from Sigma Aldrich and Avra labs. The progress of the reactions was monitored by thin layer chromatography (TLC) analysis on Merck pre-coated silica gel 60 F254 aluminum sheets, visualized by UV light. An ultrasound (Sonics Vibra-cell, Model no. VCX 500) equipped with solid synthetic probe, 13 mm in tip diameter, operating at 20 kHz with a maximum power output of 500 W, was used for synthesis of final title compounds. Infrared (IR) spectra were recorded on JASCO FTIR (PS 4000) (Japan) using KBr pallet. Melting points were determined in open capillary tubes and are uncorrected. The 1H-NMR and 13C-NMR spectra of synthesized compounds were recorded on Bruker Advance II 400 NMR Spectrometer (Billerica, MA, USA) at 400 MHz frequency in deuterated DMSO. Tetramethylsilane (TMS) was used as an internal standard. Chemical shift values are given in ppm relative to TMS as internal reference and coupling constant (J) in Hertz. The chemical shifts are reported as NMR spectra δppm units. The following abbreviations are used; singlet (s), doublet (d), multiplet (m). Mass spectra were taken with WATERS, Q-TOF MICROMASS (E SI-MS). Elemental analyses were done with a FLASHEA 112 Shimadzu analyzer (Mumbai, Maharashtra, India), and all analyses were consistent (within 0.4%) with theoretical values.
In the present work synthesis of 3-(4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-ylimino)indolin-2-one (7a–7k) derivatives using ultrasound irradiation was carried out in three steps as shown in Scheme 1. The structures of the synthesized compounds were confirmed by spectral studies and elemental analyses.

Synthesis of the target compounds (7a–7k)
General procedure for synthesis of 1-(4-chlorophenyl)-3-(substituted phenyl/heteryl)prop-2-en-1-one (3a–3k)
Classical synthesis of 1-(4-chlorophenyl)-3-(substituted phenyl/heteryl)prop-2-en-1-one (3a–3k)
A mixture of 4-chloroacetophenone (1) (0.5 mmol) and suitable aldehyde (2a–2k) (0.5 mmol) in ethanol (5–8 mL) was taken. Then cold 40% potassium hydroxide (KOH) solution was added dropwise to the reaction mixture on vigorous stirring until the solution became turbid. Then the mixture was stirred at room temperature for the period indicated in Table 1. After completion of the reaction (monitored by TLC), the mixture was poured into ice cold water and was neutralized by dilute acetic acid. The resultant solid was filtered, dried and purified by recrystallization.
Ultrasonic mediated synthesis of 1-(4-chlorophenyl)-3-(substituted phenyl/heteryl)prop-2-en-1-one (3a–3k)
In a 50 mL borosil beaker, 4-chloroacetophenone (1) (0.5 mmol) and a suitable aldehyde (2a–2k) (0.5 mmol) were mixed in ethanol (10 mL) and 40% of KOH cold solution was added dropwise to the reaction mixture with vigorous stirring until the solution became turbid. The ultrasound probe was immersed directly into the reaction mixture. The ultrasound probe emits a sonic vibration into the reaction mixture for the period indicated in Table 1. Sonication was achieved at frequencies of 20 kHz (amplitude of 50%). The reaction was carried out at room temperature. After completion of the reaction (monitored by TLC), the mixture was poured into ice cold water and was neutralized by dilute acetic acid. The resultant solid was filtered, dried and purified by recrystallisation.
(E)-1-(4-chlorophenyl)-3-phenylprop-2-en-1-one ( 3a )
Light yellow solid; Yield 90%; m.p.: 113–115 °C; IR: (KBr vmax in cm−1): 3050 (C–H of Aromatic), 2900 (C–H of alkyl), 1649 (C=O), 1557 (C=C); 1H NMR (400 MHz, CDCl3, δH ppm): 6.75 (d, 1H, CH=CH), 7.33–7.44 (m, 3H, Aromatic), 7.56 (d, 1H, CH=CH), 7.68–7.99 (m, 6H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 121.32, 127.92, 128.64, 125.53, 129.33, 130.31, 135.24, 136.27, 141.27, 145.14, 189.77; MS m/z 244.70 [M + 2]+; Anal. Calcd. for C15H11ClO: C, 74.23; H, 4.57. Found: C, 74.25; H, 4.55.
(E)-1,3-bis(4-chlorophenyl)prop-2-en-1-one ( 3b )
Yellow solid; Yield 90%; m.p.: 157–159 °C; IR: (KBr vmax in cm−1): 3055 (C–H of Aromatic), 2900 (C–H of alkyl), 1650 (C=O), 1557 (C=C), 760 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 6.75 (d, 1H,CH=CH), 7.24 (dd, 2H, Aromatic), 7.51 (d, 1H, CH=CH), 7.58–7.88 (m, 6H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 121.50, 128.77, 129.15, 129.48, 130.51, 133.54, 133.97, 136.36, 140.72, 145.63, 189.91; MS m/z 281.15 [M + 4]+; Anal. Calcd. for C15H10Cl2O: C, 65.01; H, 3.64. Found: C, 65.06; H, 3.62.
(E)-1-(4-chlorophenyl)-3-(4-fluorophenyl)prop-2-en-1-one ( 3c )
Cream solid; Yield 90%; m.p.: 130–132 °C; IR: (KBr vmax in cm−1): 3045 (C–H of Aromatic), 2900 (C–H of alkyl), 1650 (C=O), 1560 (C=C), 1350 (C–F), 765 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 6.68 (d, 1H, CH=CH), 7.17 (dd, 2H, Aromatic), 7.47 (d, 1H, CH=CH), 7.55–7.72 (m, 6H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 115.71, 121.41, 129.45, 130.52, 130.97, 131.11, 136.29, 140.73, 145.56, 162.36, 189.91; MS m/z 262.69 [M + 2]+; Anal. Calcd. for C15H10ClFO: C, 69.11; H, 3.87. Found: C, 69.14; H, 3.85.
(E)-3-(4-bromophenyl)-1-(4-chlorophenyl)prop-2-en-1-one ( 3d )
Light yellow solid; Yield 90%; m.p.: 167–168 °C; IR: (KBr vmax in cm−1): 3050 (C–H of Aromatic), 2900 (C–H of alkyl), 1650 (C=O), 1562 (C=C), 765 (C–Cl), 545 (C–Br); 1H NMR (400 MHz, CDCl3, δH ppm): 6.66 (d, 1H, CH=CH), 7.46 (d, 1H, CH=CH), 7.50–7.81 (m, 8H, Aromaic); 13C NMR (100 MHz, CDCl3, δC ppm): 121.46, 122.65, 129.41, 130.47, 130.90, 131.71, 134.51, 136.57, 140.70, 145.10, 189.91; MS m/z 324.96 [M + 4]+; Anal. Calcd. for C15H10BrClO: C, 56.02; H, 3.13. Found: C, 56.05; H, 3.10.
(E)-1-(4-chlorophenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one ( 3e )
White solid; Yield 90%; m.p.: 177–179 °C; IR: (KBr vmax in cm−1): 3500 (–OH), 3050 (C–H of Aromatic), 2890 (C–H of alkyl), 1650 (C=O), 1562 (C=C), 1350 (C–O), 765 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 5.35 (s, 1H, OH), 6.66 (d, 1H, CH=CH), 6.89 (dd, 2H, Aromatic), 7.51–7.78 (m, 7H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 117.51, 121.51, 127.85, 129.43, 130.33, 130.92, 136.46, 140.17, 145.13, 157.74, 189.79; MS m/z 260.70 [M + 2]+; Anal. Calcd. for C15H11ClO2: C, 69.64; H, 4.29. Found: C, 69.67; H, 4.27.
(E)-1-(4-chlorophenyl)-3-(4-methoxyphenyl)prop-2-en-1-one ( 3f )
Yellow solid; Yield 90%; m.p.: 128–130 °C; IR: (KBr vmax in cm−1): 3050 (C–H of Aromatic), 2890 (C–H of alkyl), 1645 (C=O), 1560 (C=C), 1230 (C–OCH3 of aromatic rings), 1025 (–O–), 762 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 3.77 (s, 3H, OCH3), 6.66 (d, 1H, CH=CH), 7.15–7.51 (m, 6H, Aromatic), 7.58 (d, 1H, CH=CH), 7.77 (dd, 2H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 55.82, 115.26, 121.34, 127.57, 129.63, 130.49, 130.91, 136.30, 140.22, 145.46, 159.53, 189.59; MS m/z 274.72 [M + 2]+; Anal. Calcd. for C16H13ClO2: C, 70.46; H, 4.80. Found: C, 70.49; H, 4.78.
1-(4-Chlorophenyl)-3-(2,4-dimethoxyphenyl)prop-2-en-1-one ( 3g )
Yellow solid; Yield 90%; m.p.: 124–126 °C; IR: (KBr vmax in cm−1): 3050 (C–H of Aromatic), 2900 (C–H of alkyl), 1650 (C=O), 1555 (C=C), 1230 (C–OCH3 of aromatic rings), 1002 (–O–), 765 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 3.78 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 6.47 (d, 1H, CH=CH), 6.53–6.60 (m, 2H, Aromatic), 7.46 (d, 1H, CH=CH), 7.51–7.74 (m, 5H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 516.11, 115.77, 117.54, 121.57, 122.72, 127.49, 129.39, 130.42, 136.17, 140.55, 149.63, 149.91, 189.79; MS m/z 304.72 [M + 2]+; Anal. Calcd. for C17H15ClO3: C, 67.44; H, 4.99. Found: C, 67.46; H, 4.96.
(E)-1-(4-chlorophenyl)-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one ( 3h )
Yellow solid; Yield 90%; m.p.: 108–110 °C; IR: (KBr vmax in cm−1): 3052 (C–H of Aromatic), 2890 (C–H of alkyl), 1648 (C=O), 1550 (C=C), 1230 (C–OCH3 of aromatic rings), 1020 (–O–), 760 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 3.84 (s, 3H, OCH3), 3.91 (s, 6H, OCH3), 6.44 (d, 1H, CH=CH), 6.56 (d, 1H, Aromatic), 7.04 (d, 1H, Aromatic), 7.58–7.78 (m, 5H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 56.14, 60.82, 104.21, 121.33, 126.42, 129.47, 130.31, 136.34, 138.47, 140.11, 145.21, 153.67, 189.79; MS m/z 334.78 [M + 2]+; Anal. Calcd. for C18H17ClO4: C, 64.97; H, 5.15. Found: C, 64.99; H, 5.14.
(E)-1-(4-chlorophenyl)-3-(pyridin-2-yl)prop-2-en-1-one ( 3i )
Yellow solid; Yield 90%; m.p.: 124–126 °C; IR: (KBr vmax in cm−1): 3050 (C–H of Aromatic), 2900 (C–H of alkyl), 1645 (C=O), 1550 (C=C), 765 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 6.81 (d, 1H, CH=CH), 7.41(d, 1H, CH=CH), 7.52–8.73 (m, 8H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 122.71, 124.44, 127.58, 129.58, 130.30, 136.41, 137.32, 140.45, 143.43, 148.86, 154.77, 189.77; MS m/z 245.69 [M + 2]+; Anal. Calcd. for C14H10ClNO: C, 69.00; H, 4.14; N, 5.75. Found: C, 69.03; H, 4.12; N, 5.77.
(E)-1-(4-chlorophenyl)-3-(furan-2-yl)prop-2-en-1-one ( 3j )
Brown solid; Yield 90%; m.p.: 88–90 °C; IR: (KBr vmax in cm−1): 3050 (C–H of Aromatic), 2900 (C–H of alkyl), 1645 (C=O), 1550 (C=C), 760 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 6.33 (dd, 1H, furan ring), 6,71 (d, 1H, CH=CH), 7.02 (d, 1H, CH=CH), 7.23–7.77 (m, 5H, Aromatic), 7.80 (dd, 1H, furan ring); 13C NMR (100 MHz, CDCl3, δC ppm): 112.77, 113.63, 120.92, 127.41, 129.72, 130.44, 136.44, 140.38, 143.79, 151.99, 189.91; MS m/z 234.62 [M + 2]+; Anal. Calcd. for C13H9ClO2: C, 67.11; H, 3.90. Found: C, 67.14; H, 3.88.
(E)-1-(4-chlorophenyl)-3-(thiophen-2-yl)prop-2-en-1-one ( 3 k )
Yellow solid; Yield 90%; m.p.: 100–102 °C; IR: (KBr vmax in cm−1): 3048 (C–H of Aromatic), 2900 (C–H of alkyl), 1648 (C=O), 1550 (C=C), 763 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 6.78 (d, 1H, CH=CH), 7.12–7.41 (m, 2H, thiophene ring), 7.51 (dd, 2H, Aromatic), 7.55 (d, 1H, CH=CH), 7.56 (dd, 1H, thioaphene ring), 7.73 (dd, 2H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 127.32, 128.39, 129.19, 129.32, 130.46, 130.94, 134.57, 136.77, 140.43, 140.91, 189.78; MS m/z 250.73 [M + 2]+; Anal. Calcd. for C13H9ClOS: C, 62.78; H, 3.65. Found: C, 62.79; H, 3.63.
General procedure for the synthesis of 4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-amine (5a–5k)
Classical synthesis of 4-(4-chlorophenyl)-6-(substituted phenyl)pyrimidin-2-amine (5a–5k)
A mixture of 1-(4-chlorophenyl)-3-(substituted phenyl/heteryl)prop-2-en-1-one (3a–3k) (0.5 mmol), guanidine hydrochloride (4) (0.5 mmol) and 40% of KOH solution in ethanol (5–8 mL) was stirred at reflux. The reaction mixture was refluxed for the appropriate time as indicated in Table 2. After completion of the reaction (monitored by TLC), the mixture was poured into ice cold water and was neutralized by dilute acetic acid. The resultant solid was filtered, dried and purified by recrystallization.
Ultrasonic mediated synthesis of 4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-amine (5a–5k)
In a 50 mL borosil beaker a mixture of 1-(4-chlorophenyl)-3-(substituted phenyl/heteryl)prop-2-en-1-one (3a–3k) (0.5 mmol), guanidine hydrochloride (4) (0.5 mmol) and 40% of KOH solution in ethanol (5–8 mL) was added. The ultrasound probe was immersed directly into the reaction mixture. The ultrasound probe emits a sonic vibration into the reaction mixture at frequencies of 20 kHz (amplitude of 50%) at 25–30 °C for the appropriate time as indicated in Table 2. After completion of the reaction (monitored by TLC), the mixture was poured into ice cold water and was neutralized by dilute acetic acid. The resultant solid was filtered, dried and purified by recrystallization.
4-(4-chlorophenyl)-6-phenylpyrimidin-2-amine ( 5a )
Light yellow solid; Yield 80%; m.p.: 160–162 °C; IR: (KBr vmax in cm−1): 3450 (NH2), 3010 (CH of Aromatic ring), 2400 (C=N), 760 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 6.99 (s, 2H, NH2), 7.41–8.05 (m, 10H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 101.31, 127.55, 128.72, 128.95, 129.31, 133.92, 134.37, 135.84, 138.51, 156.39, 156.76, 160.99; MS m/z 283.71 [M + 2]+; Anal. Calcd. for C16H12ClN3: C, 68.21; H, 4.29; N, 14.91; Found: C, 68.24; H, 4.26; N, 14.94.
4,6-bis(4-chlorophenyl)pyrimidin-2-amine ( 5b )
Light yellow solid; Yield 88%; m.p.: 203–204 °C; IR: (KBr vmax in cm−1): 3450 (NH2), 3015 (CH of Aromatic ring), 2420 (C=N), 765 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 7.09 (s, 2H, NH2), 7.55–8.11 (m, 9H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm):112.32, 128.70, 129.35, 131.96, 138.16, 156.38, 160.34; MS m/z 320.03 [M + 4]+; Anal. Calcd. for C16H11Cl2N3: C, 60.78; H, 3.51; N, 13.29; Found: C, 60.79; H, 3.50; N, 13.31.
4-(4-chlorophenyl)-6-(4-fluorophenyl)pyrimidin-2-amine ( 5c )
Light yellow solid; Yield 88%; m.p.: 188–190 °C; IR: (KBr vmax in cm−1): 3450 (NH2), 3015 (CH of Aromatic ring), 2420 (C=N), 1350 (C-F), 765 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 7.09 (s, 2H, NH2), 7.33–8.19 (m, 9H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 112.77, 116.01, 116.21, 128.70, 129.16, 129.24, 129.35, 130.29, 130.32, 131.96, 138.16, 156.38, 156.48, 160.34, 163.22, 165.74; MS m/z 301.73 [M + 2]+; Anal. Calcd. for C16H11ClFN3: C, 64.11; H, 3.70; N, 14.02; Found: C, 64.13; H, 3.68; N, 14.05.
4-(4-bromophenyl)-6-(4-chlorophenyl)pyrimidin-2-amine ( 5d )
Light orange solid; Yield 86%; m.p.: 168–170 °C; IR: (KBr vmax in cm−1): 3450 (NH2), 3015 (CH of Aromatic ring), 2415 (C=N), 765 (C–Cl), 545 (C–Br); 1H NMR (400 MHz, CDCl3, δH ppm): 7.09 (s, 2H, NH2), 7.55–8.09 (m, 9H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 112.77, 121.18, 128.70, 129.12, 129.35, 131.96, 132.24, 132.65, 138.16, 156.38, 156.44, 160.34; MS m/z 364.98 [M + 4]+; Anal. Calcd. for C16H11BrClN3: C, 53.29; H, 3.07; N, 11.65; Found: C, 53.32; H, 3.02; N, 11.69.
4-(2-amino-6-(4-chlorophenyl)pyrimidin-4-yl)phenol ( 5e )
Yellow solid; Yield 84%; m.p.: 170–172 °C; IR: (KBr vmax in cm−1): 3500 (OH), 3450 (NH2), 3015 (CH of Aromatic ring), 2415 (C=N), 765 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 5.35 (s, 1H, OH), 6.99 (s, 2H, NH2), 7.00–8.09 (m, 9H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 112.77, 115.71, 127.75, 128.70, 129.35, 129.47, 131.96, 138.16, 156.38, 156.48, 159.01, 160.34; MS m/z 299.98 [M + 2]+; Anal. Calcd. for C16H12ClN3O: C, 64.54; H, 4.06; N, 14.11; Found: C, 64.56; H, 4.02; N, 14.13.
4-(4-chlorophenyl)-6-(4-methoxyphenyl)pyrimidin-2-amine [ 5f ]
Light yellow solid; Yield 84%; m.p.: 148–150 °C; IR: (KBr vmax in cm−1): 3450 (NH2), 3010 (CH of Aromatic ring), 2415 (C=N), 1230 (C–OCH3 of aromatic rings), 760 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 3.35 (s, 3H, OCH3), 6.99 (s, 2H, NH2), 7.11–8.19 (m, 9H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 55.39, 112.77, 114.02, 128.17, 128.70, 129.35, 129.61, 131.96, 138.16, 155.46, 156.38, 156.58, 160.33; MS m/z 313.77 [M + 2]+; Anal. Calcd. for C17H14ClN3O: C, 65.49; H, 4.53; N, 13.48; Found: C, 65.51; H, 4.50; N, 13.50.
4-(4-chlorophenyl)-6-(2,4-dimethoxyphenyl)pyrimidin-2-amine ( 5g )
Cream solid; Yield 86%; m.p.: 188–190 °C; IR: (KBr vmax in cm−1): 3455 (NH2), 3020 (CH of Aromatic ring), 2415 (C=N), 1230 (C–OCH3 of aromatic rings), 760 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 3.35 (s, 3H, OCH3), 3.85 (s, 3H, OCH3), 6.99 (s, 2H, NH2), 7.00–8.10 (m, 8H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 55.39, 56.09, 98.71, 108.77, 118.03, 128.70, 129.35, 130.84, 131.96, 138.16, 155.72, 159.83, 160.45, 164.35; MS m/z 343.79 [M + 2]+; Anal. Calcd. for C18H16ClN3O2: C, 63.25; H, 4.72; N, 12.29; Found: C, 63.27; H, 4.70; N, 12.31.
4-(4-chlorophenyl)-6-(3,4,5-trimethoxyphenyl)pyrimidin-2-amine ( 5h )
Cream solid; Yield 84%; m.p.: 200–202 °C; IR: (KBr vmax in cm−1): 3450 (NH2), 3020 (CH of Aromatic ring), 2415 (C=N), 1230 (C–OCH3 of aromatic rings), 765 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 3.35 (s, 6H, OCH3), 3.85 (s, 3H, OCH3), 6.80 (s, 2H), 6.99 (s, 2H, NH2), 7.00–8.10 (m, 5H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 55.39, 56.09, 111.31, 113.35, 128.70, 129.35, 130.28, 131.96, 138.16, 142.36, 153.68, 156.47, 156.65, 160.44; MS m/z 373.10 [M + 2]+; Anal. Calcd. for C19H18ClN3O3: C, 61.38; H, 4.88; N, 11.30; Found: C, 61.39; H, 4.86; N, 11.33.
4-(4-chlorophenyl)-6-(pyridin-2-yl)pyrimidin-2-amine ( 5i )
Brown solid; Yield 86%; m.p.: 154–156 °C; IR: (KBr vmax in cm−1): 3450 (NH2), 3020 (CH of Aromatic ring), 2415 (C=N), 765 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 6.99 (s, 2H, NH2), 7.36–8.77 (m, 9H, Aromatic); 13C NMR (100 MHz, CDCl3, δC ppm): 116.17, 123.89, 125.44, 128.70, 129.35, 131.96, 138.16, 141.68, 146.15, 153.72, 155.10, 155.97, 159.21; MS m/z 284.70 [M + 2]+; Anal. Calcd. for C15H11ClN4: C, 63.72; H, 3.92; N, 19.82; Found: C, 63.74; H, 3.90; N, 19.85.
4-(4-chlorophenyl)-6-(furan-2-yl)pyrimidin-2-amine ( 5j )
Brown solid; Yield 86%; m.p.: 98–100 °C; IR: (KBr vmax in cm−1): 3455 (NH2), 3025 (CH of Aromatic ring), 2420 (C=N), 760 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 6.68 (dd, 1H, furan ring), 6.97 (s, 2H, NH2), 7.30 (dd, 1H, furan ring), 7.69–7.89 (m, 3H, Aromatic), 8.07 (dd, 1H, furan ring); 13C NMR (100 MHz, CDCl3, δC ppm): 102.34, 111.63, 112.63, 128.70, 129.35, 131.96, 138.16, 146.64, 148.14, 150.56, 157.14, 161.26; MS m/z 273.70 [M + 2]+; Anal. Calcd. for C14H10ClN3O: C, 61.89; H, 3.71; N, 15.47; Found: C, 61.92; H, 3.70; N, 15.49.
4-(4-chlorophenyl)-6-(thiophen-2-yl)pyrimidin-2-amine ( 5k )
Brown solid; Yield 88%; m.p.: 118–120 °C; IR: (KBr vmax in cm−1): 3455 (NH2), 3025 (CH of Aromatic ring), 2420 (C=N), 760 (C–Cl); 1H NMR (400 MHz, CDCl3, δH ppm): 6.99 (s, 2H, NH2), 7.17–7.53 (m, 2H, thiophene ring), 7.63–7.89 (m, 3H, Aromatic), 8.09 (dd, 1H, thiophene ring); 13C NMR (100 MHz, CDCl3, δC ppm): 105.42, 111.77, 128.70, 129.29, 129.38, 129.46, 131.96, 138.16, 138.74, 155.05, 156.50, 161.74; MS m/z 289.77 [M + 2]+; Anal. Calcd. for C14H10ClN3S: C, 58.43; H, 3.50; N, 14.60; Found: C, 58.45; H, 3.49; N, 14.63.
General procedure for the synthesis of 3-(4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-ylimino)indolin-2-one (7a–7k)
Classical synthesis of 3-(4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-ylimino)indolin-2-one (7a–7k)
A mixture of 4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-amine derivatives (5a–5k) (0.5 mmol) and isatin (6) (0.5 mmol), in absolute ethanol as solvent, was refluxed for 6–10 h in the presence of glacial acetic acid as a catalyst. Then, the reaction mixture was allowed to cool, poured into ice-cold water and filtered under suction. The precipitate thus obtained was washed with water and recrystallized from ethanol.
Ultrasonic mediated synthesis of 3-(4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-ylimino)indolin-2-one (7a–7k)
In a 50 mL borosil beaker a mixture of 4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-amine derivatives (5a–5k) (0.5 mmol) and isatin (6) (0.5 mmol), in absolute ethanol, was sonicated at a temperature of 50 °C and frequency 20 kHz for a specified time given in Table 3. Then, the reaction mixture was allowed to cool, poured into ice-cold water and filtered under suction. The precipitate thus obtained was washed with water and recrystallized from ethanol. The authenticity of the synthesized compounds was established by IR, NMR, mass spectral analysis and elemental analysis.
(Z)-3-(4-(4-chlorophenyl)-6-phenylpyrimidin-2-ylimino)indolin-2-one ( 7a )
Light orange solid; Yield 90%; m.p.: 156–158 °C; IR (KBr vmax in cm−1): 3280 (NH), 2945 (C–H of Aromatic), 2440 (C=N), 1710 (C=O), 760 (C–Cl); 1H NMR (400 MHz, DMSO, δH ppm): 7.27–7.63 (m, 13 H), 8.47 (s, 1H, pyrimidine ring), 10.47 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 113.26, 117.86, 122.15, 125.15, 125.56, 127.36, 128.70, 128.73, 129.25, 129.35, 130.04, 131.96, 138.16, 138.34, 138.77, 142.09, 157.53, 157.63, 161.47, 167.95; MS m/z 412.10 [M + 2]+; Anal. Calcd. for C24H15ClN4O: C, 70.16; H, 3.68; N,13.64; Found: C, 70.18; H, 3.66; N, 13.68.
(Z)-3-(4,6-bis(4-chlorophenyl)pyrimidin-2-ylimino)indolin-2-one ( 7b )
Light orange solid; Yield 89%; m.p.: 160–162 °C; IR (KBr vmax in cm−1): 3285 (NH), 2945 (C–H of Aromatic), 2440 (C=N), 1720 (C=O), 760 (C–Cl); 1H NMR (400 MHz, DMSO, δH ppm): 7.27–7.63 (m, 12 H), 8.46 (s, 1H, pyrimidine ring), 10.46 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 113.26, 117.86, 122.15, 125.15, 125.56, 128.70, 129.35, 130.35, 131.96, 138.16, 138.77, 142.09, 157.53, 157.63, 161.47, 167.95; MS m/z 448.05 [M + 4]+; Anal. Calcd. for C24H14Cl2N4O: C, 64.73; H, 3.17; N, 12.58; Found: C, 64.75; H, 3.15; N, 12.59.
(Z)-3-(4-(4-chlorophenyl)-6-(4-fluorophenyl)pyrimidin-2-ylimino)indolin-2-one ( 7c )
Orange solid; Yield 90%; m.p.: 158–160 °C; IR (KBr vmax in cm−1): 3280 (NH), 2945 (C–H of Aromatic), 2440 (C=N), 1710 (C=O), 1350 (C-F), 760 (C–Cl); 1H NMR (400 MHz, DMSO, δH ppm): 7.07–7.63 (m, 12 H), 8.45 (s, 1H, pyrimidine ring), 10.46 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 113.26, 116.01, 117.86, 122.15, 125.56, 128.70, 129.16, 129.25, 129.35, 130.29, 130.32, 131.96, 138.16, 138.77, 142.09, 157.53, 157.63, 161.47, 163.22, 167.95; MS m/z 430.08 [M + 2]+; Anal. Calcd. for C24H14ClFN4O: C, 67.22; H, 3.29; N, 13.06; Found: C, 67.24; H, 3.26; N, 13.09.
(Z)-3-(4-(4-bromophenyl)-6-(4-chlorophenyl)pyrimidin-2-ylimino)indolin-2-one ( 7d )
Orange solid; Yield 90%; m.p.: 166–168 °C; IR (KBr vmax in cm−1): 3285 (NH), 2945 (C–H of Aromatic), 2440 (C=N), 1720 (C=O), 760 (C–Cl), 545 (C–Br); 1H NMR (400 MHz, DMSO, δH ppm): 6.99–7.77 (m, 12 H), 8.49 (s, 1H, pyrimidine ring), 10.43 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 113.33, 117.80, 121.18, 123.18, 125.50, 128.75, 129.15, 129.44, 130.01, 132.24, 132.68, 138.88, 139.09, 142.14, 157.33, 157.56, 161.47, 167.95; MS m/z 493.08 [M + 4]+; Anal. Calcd. for C24H14BrClN4O: C, 58.86; H, 2.88; N, 11.44; Found: C, 58.89; H, 2.86; N, 11.46.
(Z)-3-(4-(4-chlorophenyl)-6-(4-hydroxyphenyl)pyrimidin-2-ylimino)indolin-2-one ( 7e )
Orange solid; Yield 91%; m.p.: 150–152 °C; IR (KBr vmax in cm−1): 3550 (OH of Aromatic), 3285 (NH), 2945 (C–H of Aromatic), 2440 (C=N), 1725 (C=O), 755 (C–Cl); 1H NMR (400 MHz, DMSO, δH ppm): 5.80 (s, 1H, OH), 6.79–7.63 (m, 12 H), 8.49 (s, 1H, pyrimidine ring), 10.46 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 113.26, 115.71, 117.86, 122.15, 125.56, 128.70, 129.25, 129.35, 129.47, 131.96, 138.16, 138.77, 142.09, 157.53, 157.63, 159.01, 161.47, 167.95; MS m/z 428.85 [M + 2]+; Anal. Calcd. for C24H15ClN4O2: C, 67.53; H, 3.54; N, 13.13; Found: C, 67.57; H, 3.47; N, 13.19.
(Z)-3-(4-(4-chlorophenyl)-6-(4-methoxyphenyl)pyrimidin-2-ylimino)indolin-2-one ( 7f )
Orange solid; Yield 89%; m.p.: 150–152 °C; IR (KBr vmax in cm−1): 3280 (NH), 2945 (C–H of Aromatic), 2445 (C=N), 1720 (C=O), 1230 (C–OCH3 of aromatic rings), 1020 (–O–), 765 (C–Cl); 1H NMR (400 MHz, DMSO, δH ppm): 3.80 (s, 3H, OH), 6.97–7.62 (m, 12 H), 8.45 (s, 1H, pyrimidine ring), 10.46 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 55.39, 113.69, 114.02, 117.86, 122.15, 125.56, 128.17, 128.70, 129.25, 129.35, 129.61, 131.96, 138.16, 138.77, 142.09, 155.46, 157.53, 157.63, 161.47, 167.95; MS m/z 442.15 [M + 2]+; Anal. Calcd. for C25H17ClN4O2: C, 68.11; H, 3.89; N, 12.71; Found: C, 68.15; H, 3.86; N, 12.74.
(Z)-3-(4-(4-chlorophenyl)-6-(2,4-dimethoxyphenyl)pyrimidin-2-ylimino)indolin-2-one ( 7g )
Orange solid; Yield 94%; m.p.: 146–148 °C; IR (KBr vmax in cm−1): 3280 (NH), 2940 (C–H of Aromatic), 2450 (C=N), 1725 (C=O), 1230 (C–OCH3 of aromatic rings), 1020 (–O–), 765 (C–Cl); 1HNMR (400 MHz, DMSO, δH ppm): 3.38 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 6.65–8.00 (m, 11H), 8.25 (s, 1H, pyrimidine ring), 10.43 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 55.35, 55.40, 101.71, 108.99, 113.41, 115.49, 117.72,119.42, 122.81, 125.46, 128.91, 129.56, 129.67, 130.21, 131.32, 138.99, 139.31, 150.11, 142.77, 156.36, 160.11, 161.32, 163.83, 164.88, 167.77; MS m/z 472.15 [M + 2]+; Anal. Calcd. for C26H19ClN4O3: C, 66.31; H, 4.07; N, 11.90; Found: C, 66.33; H, 4.04; N, 11.93.
(Z)-3-(4-(4-chlorophenyl)-6-(3,4,5-trimethoxyphenyl)pyrimidin-2-ylimino)indolin-2-one ( 7h )
Orange solid; Yield 88%; m.p.: 140–142 °C; IR (KBr vmax in cm−1): 3280 (NH), 2945 (C–H of Aromatic), 2450 (C=N), 1720 (C=O), 1230 (C–OCH3 of aromatic rings), 760 (C–Cl); 1H NMR (400 MHz, DMSO, δH ppm): 3.81 (s, 6H, OCH3), 3.92 (s, 3H, OCH3), 6.77–7.89 (m, 10H), 8.49 (s, 1H, pyrimidine ring), 10.47 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 56.47, 58.76, 113.31, 114.15, 117.86, 122.14, 125.66, 128.78, 129.45, 130.28, 131.96, 138.16, 138.77, 142.19, 143.77, 153.68, 157.55, 157.88, 161.53, 167.96; MS m/z 502.13 [M + 2]+; Anal. Calcd. for C27H21ClN4O4: C, 64.74; H, 4.23; N, 11.18; Found: C, 64.76; H, 4.20; N, 11.19.
(Z)-3-(4-(4-chlorophenyl)-6-(pyridin-2-yl)pyrimidin-2-ylimino)indolin-2-one ( 7i )
Dark Orange solid; Yield 90%; m.p.: 162–164 °C; IR (KBr vmax in cm−1): 3280 (NH), 2940 (C–H of Aromatic), 2450 (C=N), 1725 (C=O), 765 (C–Cl); 1H NMR (400 MHz, DMSO, δH ppm): 7.26–7.26 (m, 9H,), 8.02–8.85 (m, 4H, pyridine ring), 8.87 (s, 1H, pyrimidine ring), 10.95 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 113.26, 115.03, 117.86, 122.15, 123.89, 125.44, 125.56, 128.70, 129.25, 129.35, 131.96, 138.16, 138.77, 141.68, 142.09, 146.15, 153.72, 156.43, 157.49, 159.19, 165.72, 167.95; MS m/z 412.84 [M + 2]+; Anal. Calcd. for C23H14ClN5O: C, 67.08; H, 3.43; N, 17.00; Found: C, 67.10; H, 3.40; N, 17.03.
(Z)-3-(4-(4-chlorophenyl)-6-(furan-2-yl)pyrimidin-2-ylimino)indolin-2-one ( 7j )
Dark Orange solid; Yield 85%; m.p.: 156–158 °C; IR (KBr vmax in cm−1): 3280 (NH), 2950 (C–H of Aromatic), 2440 (C=N), 1720 (C=O), 765 (C–Cl); 1H NMR (400 MHz, DMSO, δH ppm): 6.93–8.07 (m, 11H), 8.28 (s, 1H, pyrimidine ring), 10.95 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 107.42, 111.63, 113.26, 117.86, 122.15, 125.56, 128.70, 129.25, 129.35, 131.96, 138.16, 138.77, 142.09, 146.04, 148.15, 153.13, 158.29, 163.03, 166.13, 167.95; MS m/z 402.04 [M + 2]+; Anal. Calcd. for C22H13ClN4O2: C, 65.92; H, 3.27; N, 13.98; Found: C, 65.94; H, 3.25; N, 13.99.
(Z)-3-(4-(4-chlorophenyl)-6-(thiophen-2-yl)pyrimidin-2-ylimino)indolin-2-one ( 7k )
Dark Orange solid; Yield 87%; m.p.: 148–150 °C; IR (KBr vmax in cm−1): 3285 (NH), 2950 (C–H of Aromatic), 2445 (C=N), 1725 (C=O), 765 (C–Cl); 1H NMR (400 MHz, DMSO, δH ppm): 6.88–7.22 (m, 3H, thiophene ring), 7.30–7.63 (m, 8H), 8.36 (s, 1H, pyrimidine ring), 10.99 (s, 1H, NH); 13C NMR (100 MHz, DMSO, δC ppm): 111.06, 111.77, 113.26, 117.86, 122.15, 125.56, 128.70, 129.25, 129.29, 129.35, 129.45, 131.96, 138.16, 138.74, 138.77, 142.09, 157.27, 157.52, 164.50, 167.95; MS m/z 418.88 [M + 2]+; Anal. Calcd. for C22H13ClN4OS: C, 63.38; H, 3.14; N, 13.44; Found: C, 63.39; H, 3.13; N, 13.46.
Biological evaluation
In vitro anticancer screening
The newly synthesized derivatives (7a–7k) were evaluated for their in vitro anticancer activity against selected human cancer cell lines viz. human breast cancer cell line MCF-7, human cervical cancer cell line HeLa, human prostate cancer cell line PC-3, human lung cancer cell line A-549 and normal prostate epithelial cells RWPE-1 by Sulforhodamine B (SRB) assay, using Sunitinib as a standard drug [32].
Computational studies
Molecular docking
In order to explore binding affinity, binding mode and molecular interactions of the synthesized derivatives molecular docking study was carried out. Tubulin α/β hetrodimer serves as an important drug target in breast cancer [33]. Tubulin heterodimers of α- and β-tubulin (50 kDa each in size) are the basic structural components of microtubules, which are hollow tubes approximately 25 nm in diameter. Microtubules are cytoskeletal polymers involved in many cell functions such as mitosis, organization of intracellular structure and intracellular transport, as well as ciliary and flagellar motility. The α/β dimer in relation to the polarity of the microtubule lattice displays a β-tubulin monomer at the plus end and an α-tubulin at the minus end. In humans, there are six isotypes of α-tubulin and seven isotypes of β-tubulin, and the level of expression of each isotype varies in different tissues and cells [34,35,36]. Of course, tubulin-binding drugs have different affinities for different isotypes, which affects the overall efficacy in different cancers. There are many chemically diverse compounds that bind to the tubulin–microtubule system. Tubulin-binding agents are potent mitotic poisons [37, 38].To perform molecular docking three dimensional X-ray crystal structure of tubulin (PDB ID: 1SA0 Resolution 3.58 Å) complex with colchicine and a stathmin-like domain and was used VEGFR2 in complex with a novel 4-amino-furo[2,3-d]pyrimidine (PDB ID: 1YWN Resolution 1.71 Å) was used [39, 40]. Docking study was carried out using Surflex-Dock module of Sybyl 2.1.1 package following the standard procedure.
The molecular docking study was initiated with sketching of 2D form of structure of all the synthesized compounds using sketch modules of SYBYL-X 2.1.1 later on these 2D forms automatically converted into 3D forms and later on this optimized 3D forms were used for molecular docking.
In silico ADMET prediction
In silico ADMET predictor FAFDrugs2 which runs on Linux OS was used. Prepared ligand library for molecular docking and reference co-crystallized ligands and standards used for biological evaluations are subjected for ADMET testing.
In vivo acute oral toxicity study and behavioral study
The in vivo acute oral toxicity study for the newly synthesized compounds 7b and 7c was carried out by the following OECD guideline no. 425 using Swiss albino mice (18–22 g weight) quarantined in animal housing at Y.B. Chavan College of Pharmacy, Aurangabad IAEC approval number CPCSEA/IAEC/P’col-52/2015-16/115. Each group consisted of six mice (overnight fasted) and was kept in a colony cage at 25 ± 2 °C with 55% relative humidity and a 12-h light/dark cycle. A specified dose of 100, 250, 500, 750, 1000, 1500 and 2000 mg/kg body weight of mice was administered orally as a single dose. The acute toxic symptoms and the behavioral changes produced by the test compounds were observed continuously for 4 h periods at the 8th, 12th and 24th h of onset of toxic symptoms and the gross behavioral changes were also recorded. These animals were maintained for a further 10 days with observation made daily. In case the animal appeared moribund (dying), the animal was euthanized in a humane way and was considered to have died because of toxicity.
Result and discussion
Chemistry
According to Claisen-Schmidt condensation method, the 1-(4-chlorophenyl)-3-(substituted phenyl/heteryl)prop-2-en-1-one (3a–3k) were synthesized by condensation of 4-chloroacetophenone (1) (0.5 mmol) with suitable aldehyde (2a–2k) (0.5 mmol) in ethanol (5–8 mL) by adding drop-wise a cold solution of 40% KOH until a turbid solution formed. Then the turbid mixture was treated with sonication until the completion of reaction. In order to justify the use of ultrasound, these reactions were also carried out in the absence of ultrasound at room temperature, and the results are presented in Table 1. The 4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-amine (5a–5k) derivatives were synthesized by reacting 1-(4-chlorophenyl)-3-(substituted phenyl/heteryl)prop-2-en-1-one (3a–3k) (0.5 mmol) with guanidine hydrochloride (4) (0.5 mmol) by using 40% KOH solution as a base in an ethanolic medium under relatively “greener” reaction conditions by using ultrasound irradiation. In order to justify the use of ultrasound, these reactions were also carried out in the absence of ultrasound under reflux conditions, and the results are presented in Table 2. Finally, the designed compounds 3-(4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-ylimino)indolin-2-one (7a–7k) derivatives were made by reacting 4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-amine derivatives (5a–5k) (0.5 mmol) and isatin (6) (0.5 mmol), in absolute ethanol under relatively “greener” reaction conditions by using ultrasound irradiation as shown in scheme 1. In order to justify the use of ultrasound, these reactions were also carried out in the absence of ultrasound under reflux conditions, and the results are presented in Table 3. Further, all the synthesized final compounds (7a–7k) were unambiguously characterized by mass spectra, 1H NMR spectroscopy, 13C NMR spectroscopy and elemental analysis.
Biological activity
In vitro anticancer evaluation
The newly synthesized derivatives (7a–7k) were evaluated for their in vitro anticancer activity against selected human cancer cell lines viz. human breast cancer cell line MCF-7, human cervical cancer cell line HeLa, human prostate cancer cell line PC-3 and human lung cancer cell line A-549 by Sulforhodamine B (SRB) aasay, using Sunitinib as the standard drug [32]. The synthesized compounds 7b and 7c, which were found to be most active on human prostate cancer cell line PC-3, were further tested on normal prostate epithelial cells (RWPE-1) to find their selectivity towards the cancer cells. Concentrations required to cause 50% inhibition of cancer cell growth are expressed as GI50 values. The GI50 (μM) values of the test compounds 7(a–k) and reference standard Sunitinib are summarized in Table 4.
From the close examination of GI50 values, it is observed that 7b, 7c, 7d, and 7g were active on most of the tested cancer cell lines. Specifically, compounds 7b, 7c and 7d displayed potent in vitro anticancer activity against HeLa, PC-3 and A-549 cancer cell lines. Gratifyingly, the compound 7b bearing a chloro group was the most active compound in this series, exhibiting potent in vitro anticancer activity with GI50 values of 15.38, 19.67 and 4.37 μM on HeLa, PC-3 and A-549 cancer cell lines, respectively, whereas, the compound 7h bearing trimethoxy group was found to be most active against MCF-7 cancer cells with GI50 values of 15.10 μM. The compound 7h was found to be 1.6 times better against MCF-7 cancer cells than the standard drug Sunitinib with GI50 values of 24.18 μM. From the close examination of GI50 values (μM), it can be inferred that the compounds 7a, 7i, 7j and 7k were less active against MCF-7 cancer cells.
From the in vitro anticancer result data shown in Table 4, the most active compounds 7b and 7c were tested on normal prostate epithelial cells (RWPE-1) to find the selectivity towards the cancer cells. It is very important for cancer therapy that the chemotherapy agents have the properties of high effectiveness and low toxicity. The GI50 values of the most active synthesized compounds 7b and 7c is 98.5 and 65.7 µM against RWPE-1 normal cell lines, respectively, which indicates the safety, less toxic nature and selectivity towards its cytotoxic activity. The compound 7b showed almost 5.00 times more selectivity for PC-3 cancer cell lines in comparison to the RWPE-1 normal prostate epithelial cells. The compound 7c showed almost 3.00 times more selectivity for PC-3 cancer cell lines in comparison to the RWPE-1 normal prostate epithelial cells.
From the in vitro anticancer result data as shown in Table 4, the most active compound 7h was tested on non-tumorigenic breast epithelial cell lines (MCF-10A) to find the selectivity towards the cancer cells. The compound 7h showed almost 5.00 times more selectivity for MCF-7 cancer cell lines in comparison to the MCF-10A normal breast epithelial cell lines.
Structure activity relationship (SAR) revealed that the scaffolds Indole-2-one, N-4-Chlorobenzyl moiety and pyrimidine pharmacophores are responsible for in vitro anticancer activity. From the structure activity relationships (SARs), it could be inferred in general that the modifications on the phenyl ring or replacement of phenyl ring with other heterocyclic ring significantly influence the in vitro anticancer activity. Structure activity relationship (SAR) studies for these compounds demonstrated that phenyl ring with electron-withdrawing groups such as chloro (7b), fluoro (7c) and bromo (7d) exhibited maximum in vitro anticancer activity against HeLa, PC-3 and A-549 cancer cell lines compared to electron-donating groups. Introduction of electron donating polar groups on phenyl ring such as in 4-OCH3 (7f), 2,4-di OCH3 (7g) and 3,4,5-tri OCH3 (7h) augmented the in vitro anticancer activity against MCF-7 cancer cell lines. In general, the in vitro anticancer activity against MCF-7 cancer cell line decreases with phenyl ring substituents in the order of 3,4,5-tri OCH3 (7h) > 2,4-di OCH3 (7g) > 4-OCH3 (7f) > 4–OH (7e) > 4-Cl (7b) > 4-F (7c) > 4–Br (7d). Replacement of the phenyl ring with heterocyclic rings such as a furan ring in compound 7j and with a thiophene ring as in 7k decreased the in vitro anticancer activity against MCF-7 cancer cell line.
The induction of apoptosis by chemotherapeutic agents has always been a superlative choice in developing a anti-cancer therapeutics. To determine whether the treatment with these compounds could lead to loss of cell viability and induction of apoptosis, MCF-7, HeLa and PC-3 cancer cell lines were treated with GI50 concentration of the synthesized compounds (7a–7k). Cell morphology was observed at the GI50 concentration of the synthesized compounds (7a–7k) and photographs were taken under an Eclipse Ti-S Inverted Research Microscope (Nikon), and the images were processed using NIS-Elements software. It can be inferred from Fig. 2 that at the GI50 concentration of the most active compound 7b there were distinctive morphological changes such as cell detachment, cell wall deformation, cell shrinkage and reduced number of viable cells in HeLa cancer cell lines in comparison to control cells. Similarly there were also distinctive morphological changes observed in PC-3 cancer cell lines at the GI50 concentration of the most active compound 7b as shown in Fig. 3. When the MCF-7 cancer cell lines were treated with the most active compound 7h had shown characteristic apoptotic features like cell shrinkage, cell wall deformation and reduced number of viable cells in comparison to control cells at its GI50 concentration as shown in Fig. 4.

Morphological changes in HeLa cancer cell lines a not treated with compound 7b used as control for 48 h; b treatment with compound 7b (15.59 μM) for 48 h

Morphological changes in PC-3 cancer cell lines a not treated with compound 7b used as control for 48 h; b treatment with compound 7b (19.81 μM) for 48 h

Morphological changes in MCF-7 cancer cell lines a not treated with compound 7h used as control for 48 h; b treatment with compound 7h (15.09 μM) for 48 h
Computational studies
Molecular docking
In order to explore binding affinity, binding mode and molecular interactions of the molecular docking study for the synthesized compounds was carried out on microtubules and vascular endothelial growth factor receptors (VEGFR2). The number of compounds showing potent anticancer activity indicated by GI50 value on the selected human cancer cell lines had very high total docking score, polar score and low crash score indicates non-covalent interactions such as hydrogen bond interaction and π interactions [41]. To represent the details of docking score following terms are used: (a) Total score as total docking score; (b) Crash score as degree of inappropriate penetration by the ligand into the protein and of interpenetration between ligand atoms that are separated by rotatable bonds of compounds; (c) Polar score gives an idea about the contribution of the polar non-hydrogen bonding interactions to the total score are shown in Table 5.
The detail analysis of binding affinity (− log Ki) values and molecular interactions of the synthesized derivatives such as 7b (5.63), 7d (5.61) and 7c (5.50) suggesting that they are the most active amongst all the synthesized derivatives and compared with reference co-crystallised ligand colchicines (CN2) and sunitinib. The most active synthesized derivatives such as 7b (5.63), 7d (5.61) and 7c (5.50) shown efficient binding mode and penetrating active site cavity by forming the hydrogen bond interactions and π interactions with active site residues.
The active site amino acid LEU252 forms hydrogen bond interactions with indol-2-one carbonyl (–C=0) and THR376 forms hydrogen bond interaction with pyrimidine and chlorine atoms of phenyl ring whereas CYS241 forming π-donor hydrogen bond interaction with heterocyclic pyrimidine ring. The active site amino acids, namely VAL318, LEU255, LEU248, ILE378, ALA250, LEU242, ALA354, LYS352, ALA316 and VAL328, form various π interactions with indole, pyrimidine and the chloro-phenyl ring present in compound 7b as shown in Fig. 5a.


a The binding position and the molecular interactions of the compound 7b in the active site of tubulin. b The binding position and the molecular interactions of the compound 7d in the active site of tubulin. c The binding position and the molecular interactions of the compound 7c in the active site of VEGFR2. d The binding position and the molecular interactions of the compound 7d in the active site of VEGFR2
The second most active synthesized compound 7d (5.6155) forms hydrogen bond interactions with amino acids ALA317 and LYS352 with the nitrogen atom of the indole ring. The amino acids ILE378, VAL318, LEU248, LYS254, LEU255, LEU242 and LEU252 form π interactions with indole, pyrimidine, chloro-phenyl ring and bromine atoms of the phenyl ring as shown in Fig. 5b. On the basis of GI50 anticancer activity data and molecular docking analysis, it was found that the synthesized derivatives 7b, 7d and 7c had potential to inhibit enzyme tubulins α/β.
The molecular docking study was also carried out against vascular endothelial growth factor receptors (VEGFR2) in order to understand mechanism of inhibition and the inhibition potential of synthesized lead molecules.
The compounds 7e, 7f, 7g, 7i and 7k had potential anticancer activity indicated by total docking score 3.6–3.9 as shown in Table 6.
The most active synthesized analogue 7c against VEGFR2 has shown π and halogen bond interactions with active site residues PHE843, GLY841, VAL846, LYS866, ALA864, VAL914, VAL912, ILE913 and LEU838. The amino acids VAL912 and ILE913 interact with fluorine atoms of the phenyl ring. The amino acids PHE843, GLY841, VAL846, LYS866, ALA864, VAL914 and LEU838 form various π interactions with aryl rings and heterocyclic rings, such as indole and pyrimidine ring systems, as shown in Fig. 5c. The analogue 7d forms conventional hydrogen bond interactions and numerous π interactions with active site amino acids such as GLU848, PHE916, VAL846, ALA864, VAL914, LYS866, VAL912, GLY839, LEU838, CYS917, as shown in Fig. 5d.
In silico ADMET prediction
In silico ADMET study was performed to evaluate the pharmacokinetic properties and safety potential of the synthesized derivatives (7a–7k) and standard such as 4-amino-furo[2,3-d]pyrimidine (ILF), Colchicines (CN2) and Sunitinib. ADMET properties were predicted using ADMET predictor FAFDrugs2 which runs on Linux OS [42]. In particular, we have calculated the compliance of synthesized compounds to the Lipinski’s rule of five [43]. We have assessed parameters such as percent absorption (% ABS), molecular weight (MW < 500), partition coefficient (log P < 5), polar surface area (PSA), number of rotatable bonds (< 10), hydrogen bond donor (HBD) and hydrogen bond acceptors (HBA). All the mentioned parameters signify oral bioavailability and good intestinal absorption [44]. The values obtained are depicted in Table 7. All the synthesized compounds exhibited a very good % ABS ranging from 76.05 to 85.80%. Moreover, none of the synthesized derivatives (7a–7k) violated Lipinski’s rule of five. Each of the synthesized derivatives (7a–7k) has the potential to be developed as an orally active drug candidate.
In vivo acute oral toxicity study and behavioral study
The toxicity study of the synthesized compounds at the early stage of research simplifies the path to clinical trials and decreases the failure of potential therapeutics at later stages of testing. Therefore, in vivo acute oral toxicity study and behavioral study of the most promising compounds 7b and 7c were evaluated. Animals treated with the newly synthesized compounds 7b and 7c were free of any toxicity as per acceptable range given by the OECD guideline no. 425 and no mortality was found up to 2000 mg/kg, which indicates that the lethal dose of the compounds is above 2000 mg/kg body weight in mice and that the compounds can be considered to be safe and can be developed in future as good anticancer agents. The results of the in vivo acute oral toxicity study and gross behavioral studies of the newly synthesized compounds 7b and 7c is as shown in Table 8.
Conclusion
In conclusion, we have synthesized a suite of 11 novel 3-(4-(4-chlorophenyl)-6-(substituted phenyl/heteryl)pyrimidin-2-ylimino)indolin-2-one derivatives (7a–7k) using ultrasound irradiation as a green protocol. The in vitro anticancer activity results revealed that most of the synthesized hybrid compounds were active on all the tested cancer cell lines. Among the tested compounds, 7b exhibited promising anticancer activity against HeLa, PC-3 and A-549. Gratifyingly, the compound 7h exhibited potent anticancer activity against MCF-7 cancer cell line. The treatment of HeLa, PC-3 and A549 cancer cells with 7b compound and treatment of MCF-7 cancer cells with 7h compound showed apoptosis and morphological changes such as cell shrinkage, cell wall deformation and reduced number of viable cells. The compound 7b showed almost 5.00 times more selectivity for PC-3 cancer cell lines in comparison to the RWPE-1 normal prostate epithelial cells. The compound 7c showed almost 3.00 times more selectivity for PC-3 cancer cell lines in comparison to the RWPE-1 normal prostate epithelial cells. Computational molecular docking study highlight and supports the experimental results for in vitro anticancer activity and demonstrated that 7b, 7c and 7d are the most active compounds of the synthesized derivatives and have the potential anticancer activity towards tested cancer cells. Overall, the green synthetic protocol, significant anticancer activity, good in silico ADMET properties and in vivo non toxic nature makes these compounds a promising starting point for the development of potent cancer chemotherapeutic agents in the drug discovery process.
References
A. Jemal, F. Bray, M.M. Center, J. Ferlay, E. Ward, D. Forman, CA Cancer J. Clin. 61, 69 (2011)
R. Siegel, D. Naishadham, A. Jemal, CA Cancer J. Clin. 62, 10 (2012)
D. Hanahan, J. Folkman, J. Cell. 86, 353 (1996)
R. Kerbel, J. Folkman, J. Nat. Rev. Cancer 2, 727 (2002)
P. Carmeliet, R.K. Jain, Nature 473, 298 (2011)
R.K. Jain, P. Carmeliet, Cell 149, 1408 (2012)
J. Ma, D.J. Waxman, Mol. Cancer Ther. 7, 3670 (2008)
C. Dumontet, M.A. Jordan, Nat. Rev. Drug Discov. 9, 790 (2010)
M. Hall, C. Gourley, I. McNeish, J. Ledermann, M. Gore, G. Jayson, T. Perren, G. Rustin, S. Kaye, Br. J. Cancer 108, 250 (2013)
C. Holohan, S. Van Schaeybroeck, D.B. Longley, P.G. Johnston, Nat. Rev. Cancer 13, 714 (2013)
Glaxo SK (2000) Combination of lapatinib with carboplatin, paclitaxel and trastuzumab in metastatic breast cancer. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US) [cited 2015 Mar 06]. NLM Identifier: NCT00367471
Twenty eight clinical trials were found on clinicaltrials.gov (accessed in March 2015) site in which RTK inhibitors were being used in combination with antitubulins and other chemotherapeutic agents. Of these, 16 trials are currently in progress and the identification numbers in clinical trials.gov are provided below: NCT01855750; NCT01804530; NCT01606878; NCT01746277; NCT02326285; NCT01620190; NCT01974440; NCT01683994; NCT02378389; NCT01939054; NCT01719302; NCT02191059; NCT02191059; NCT01876082; NCT00567554; NCT00367471 (03/06/2015)
U.O. Vermont Docetaxel, Gemcitabine and Pazopanib as Treatment for Soft Tissue Sarcoma. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2015 Mar 06]. NLM Identifier: NCT01719302
A. Gangjee, N. Zaware, S. Raghavan, M. Ihnat, S. Shenoy, R.L. Kisliuk, J. Med. Chem. 53, 1563 (2010)
A. Millemaggi, R.J.K. Taylor, Eur. J. Org. Chem. 24, 4527 (2010)
G.S. Singh, Z.Y. Desta, Chem. Rev. 112, 6104 (2012)
G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar, P.T. Perumal, Eur. J. Med. Chem. 51, 79 (2012)
Y. Kia, H. Osman, R.S. Kumar, V. Murugaiyah, A. Basiri, S. Perumal, H.A. Wahab, C.S. Bing, Biorg. Med. Chem. 21, 1696 (2013)
A. Leoni, A. Locatelli, R. Morigi, M. Rambaldi, Expert Opin. Ther. Pat. 26, 149 (2016)
H. Prenen, J. Cools, N. Mentens, C. Folens, R. Sciot, P. Schoffski, A.V. Oosterom, P. Marynen, M. Debiec-Rychter, Clin. Cancer Res. 12, 2622 (2006)
C. Sessa, L. Vigano, G. Grasselli, J. Trigo, I. Marimon, A. Llado, A. Locatelli, N. Ielmini, S. Marsoni, L. Gianni, Eur. J. Cancer 42, 171 (2006)
C.A. London, P.B. Malpas, S.L. Wood-Follis, J.F. Boucher, A.W. Rusk, M.P. Rosenberg, C.J. Henry, K.L. Mitchener, M.K. Klein, J.G. Hintermeister, Clin. Cancer Res. 15, 3856 (2009)
K. Spiekermann, F. Faber, R. Voswinckel, W. Hiddemann, Exp. Hematol. 30, 767 (2002)
R. Griffith, M. Brown, A. Mc Cluskey, L. Ashman, Mini. Rev. Med. Chem. 6, 1101 (2006)
R. Dudhea, P.K. Sharma, P. Verma, A. Chaudhary, J. Adv. Sci. Res. 2, 10 (2011)
S.K. Prajapti, A. Nagarsenkar, S.D. Guggilapu, K.K. Gupta, L. Allakonda, M.K. Jeengar, V.G. Naidu, B.N. Babu, Bioorg. Med. Chem. Lett. 26, 3024 (2016)
S.V. Tiwari, J.A. Seijas, M.P. Vazquez-Tato, A.P. Sarkate, D.K. Lokwani, A.P.G. Nikalje, Molecules 21, 894 (2016)
A.P.G. Nikalje, S.V. Tiwari, J.G. Tupe, V.K. Vyas, Q.B. Gulamnizami, Lett. Drug Des. Discov. 14, 1195 (2017)
B. Meunier, Acc. Chem. Res. 41, 69 (2008)
X. Zhang, S. Raghavan, M. Ihnat, E. Hamel, C. Zammiello, A. Bastian, S.L. Mooberry, A. Gangjee, Bioorg. Med. Chem. 23, 2408 (2015)
B.G. Raimond, B. Gigant, P.A. Curmi, I. Jourdain, S. Lachkar, A. Sobel, M. Knossow, Nature 248, 198 (2004)
V. Vichai, K. Kirtikara, Nat. Protoc. 1, 1112 (2006)
C.M. Lin, H.H. Ho, G.R. Pettit, E. Hamel, Biochemistry 28, 6984 (1989)
S.A. Lewis, M.E. Gilmartin, J.L. Hall, N.J. Cowan, J. Mol. Biol. 182, 11 (1985)
M.I. Nicoletti, G. Valoti, P. Giannakakou, Z. Zhan, J.H. Kim, V. Lucchini, F. Landoni, J.G. Mayo, R. Giavazzi, T. Fojo, Clin. Cancer Res. 7, 2912 (2001)
P.G. McKean, S. Vaughan, K. Gull, J. Cell Sci. 114, 2723 (2001)
E.A. Perez, Mol. Cancer Ther. 8, 2086 (2009)
M. Kavallaris, Nat. Rev. Cancer 10, 1 (2010)
A. Chaudhary, P.P. Sharma, G. Bhardwaj, V. Jain, P.V. Bharatam, B. Shrivastav, R.K. Roy, Med. Chem. Res. 12, 5654 (2013)
E.S. Tamany, F.A. Shahed, B.H. Mohamed, J. Serb. Chem. Soc. 64, 9 (1999)
Y. Huang, R.P. Hickey, J.L. Yeh, D. Liu, A. Dadak, L.H. Young, R.S. Johnson, F.J. Giordano, FASEB J. 18, 1138 (2004)
D. Lagorce, H. Sperandio, M. Miteva, B.O. Villoutreix, Bioinformatics 9, 396 (2008)
C. Lipinski, F. Lombardo, B. Dominy, Adv. Drug Deliv. Rev. 46, 3 (2001)
Y. Zhao, M.H. Abraham, J. Lee, A. Hersey, N.C. Luscombe, G. Beek, B. Sherborne, I. Cooper, Pharm. Res. 19, 1446 (2002)
Acknowledgements
The authors are thankful to Mrs. Fatima Zakaria, Chairman Maulana Azad Educational Trust and Dr. Zahid Zaheer, Principal, Y.B. Chavan College of Pharmacy, Dr. Rafiq Zakaria Campus, Aurangabad 431 001 (M.S.), India for providing the laboratory facility.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors confirm that this article content has no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nikalje, A.P.G., Tiwari, S.V., Sangshetti, J.N. et al. Ultrasound-mediated synthesis, biological evaluation, docking and in vivo acute oral toxicity study of novel indolin-2-one coupled pyrimidine derivatives. Res Chem Intermed 44, 3031–3059 (2018). https://doi.org/10.1007/s11164-018-3292-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-018-3292-5