
Abstract
The study describes the thermal properties of functional microspheres composed of glycidyl methacrylate (GMA) and crosslinking agent ethylene glycol dimethacrylate (EGDMA). Copolymeric poly(GMA-co-EGDMA) microspheres were prepared via suspension–emulsion polymerization in the presence of toluene and decan-1-ol as porogens. In order to introduce functional groups, the porous methacrylate network was modified by epoxy ring opening with the use of sodium cyclopentadienide and then the Diels–Alder addition with maleic anhydride. The thermal properties of poly(GMA-co-EGDMA) materials were evaluated by thermogravimetry and differential scanning calorimetry. By TG/FTIR, it was observed that new functional materials exhibited multi-staged decomposition patterns, different from parent poly(GMA-co-EGDMA) microspheres. The synthesized poly(GMA-co-EGDMA) microspheres exhibited rather high thermal stability in inert atmosphere. Their initial decomposition temperature determined at the temperature of 2% of mass loss was about 210 °C; however, after the chemical modification it was slightly lower. The thermal degradation of parent poly(GMA-co-EGDMA) copolymer runs mainly according to the depolymerization mechanism, while functionalized by cyclopentadienyl group and maleic anhydride microspheres decompose through the chain scission mechanism.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polymers having functional groups in their structural units and capable of further transformations are termed as reactive polymers. In some cases, the presence of the reactive glycidyl methacrylate in these copolymers made them quite convenient for further changes. Glycidyl methacrylate (GMA) is an attractive vinyl monomer because of its low toxicity, lower cost compared with other acrylic monomers, versatile properties and especially due to the presence of dual functionality, containing both methacrylic and epoxide groups [1, 2]. The methacrylic group containing the reactive C=C bond readily reacts with a wide range of monomers including multifunctional ones which can create the crosslinked network. On the other hand, the glycidyl fragment is a convenient group which in a mild condition is able to react with a numerous of strong and weak nucleophiles to provide the user with maximum freedom and flexibility in designing the polymer structure. However, the mechanical and thermal stability of polymers synthesized with GMA monomer is not quite up to the mark.
Since the pioneering work of Švec et al. [3] on macroporous copolymer based on GMA and ethylene glycol dimethacrylate, a large number of papers have been focused on more detailed studies of the synthesis, properties, chemical modification and applications of copolymers based on GMA. For decades, copolymers of glycidyl methacrylate crosslinked with ethylene glycol dimethacrylate have been widely used as sorbents for chromatography [4, 5], solid-supported catalysis [6], enzyme immobilization [7, 8], ion–resin exchange [9], solid-phase peptide synthesis (SPPS) and solid-phase organic synthesis (SPOS) [10]. The thermal resistance of materials such as poly(glycidyl methacrylate) [11, 12], its copolymers with styrene [13], methyl methacrylate [12, 14], vinyl acetate [15], acrylonitrile [16] and α-methyl styrene [17] were reported. However, the use of polymers based on glycidyl methacrylate as adsorbents is limited by their thermal stability. To increase the thermal resistance of porous adsorbents, Maciejewska proposed the synthesis of porous microspheres of GMA crosslinked with trimethylolpropane trimethacrylate and functionalized with pyrrolidone [18] and diethylenetriamine [19]. The interesting porous copolymer of GMA crosslinked with bis[4(2-hydroxy-3-methacryloyloxypropoxy)phenyl]sulphide and modified by diethylenetriamine and triethylenetetramine [20] possesses high thermal stability and the chemical structure suitable for use as an adsorbent.
In our study, we intended to obtain porous microspheres of glycidyl methacrylate crosslinked with ethylene glycol dimethacrylate, which were able to further chemical modification. The final microspheres bear anhydride groups that can be transferred in a simple way into carboxyl one. Copolymeric porous beads with carboxyl groups are attractive materials used as column packings for liquid chromatography. On the other hand, the synthesis of crosslinked microspheres via suspension or emulsion polymerization from monomers possessing carboxyl group, e.g. methacrylic or acrylic acid, is ineffective. In this study, we propose the synthesis of functional crosslinked microspheres possessing anhydride moieties which can be used as stationary phases in liquid chromatography and solid-phase extraction. Poly(GMA-co-EGDMA) copolymeric microspheres were prepared via suspension–emulsion polymerization. Their structure was modified firstly by cyclopentadienyl group and finally with the use of Diels–Alder cycloaddition reaction with maleic anhydride. The aim of this work is to present the thermal properties of a series of poly(GMA-co-EGDMA) functional polymeric microspheres. Furthermore, the project shows the influence of chemical structure on the thermal stability of copolymeric microspheres. To the best of our knowledge, we demonstrate for the first time the thermal characterization of copolymers prepared in this way. The TG/FTIR-coupled method and DSC method were employed to study the thermal behaviour of the materials discussed in inert atmosphere.
Experimental
Materials
Glycidyl methacrylate (GMA, 97%), ethylene glycol dimethacrylate (EGDMA, 98%), azobisisobutyronitrile (AIBN, 98%), maleic anhydride (99%), sodium cyclopentadienide (NaCp, 2 M in THF), decan-1-ol (99%) and sodium bis(2-ethylhexyl) sulfosuccinate (97%) were obtained from Sigma-Aldrich. Toluene, ethanol, tetrahydrofuran, acetone, hexane and hydrochloric acid were of analytical reagent grade and were from POCh (Gliwice, Poland). Monomers: GMA and EGDMA, were purified of inhibitor by vacuum distillation and stored in a refrigerator until use. Other reagents were used as received without further purification.
Preparation of poly(GMA-co-EGDMA) polymeric microspheres
Polymeric microspheres were synthesized by the suspension–emulsion polymerization in accordance with the procedure described earlier [21, 22]. Polymerization reactions were carried out in an aqueous solution of sodium bis(2-ethylhexyl) sulfosuccinate. Toluene and decan-1-ol were used as pore-forming agents, AIBN acted as an initiator of polymerization. The monomers: GMA and EGDMA, were used at different molar ratios in the range of (4, 1.5, 1):1, respectively. The polymerization process was carried out at 80 °C and stirring at approx. 300 rpm for 10 h. After the reaction was completed, the obtained spheres were extracted with hot THF for 4 h. Based on the molar ratio of GMA to EGDMA, the obtained materials poly(GMA-co-EGDMA) were denoted as: poly(GMA-co-EGDMA)8/2; poly(GMA-co-EGDMA)6/4; and poly(GMA-co-EGDMA)5/5.
Preparation of functional polymeric microspheres
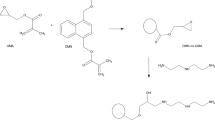
The chemical modification of primary poly(GMA-co-EGDMA)8/2 microspheres was performed in several steps, according to scheme shown in Fig. 1. Firstly, the reaction with sodium cyclopentadienide (NaCp) was carried out [23, 24]. Generally, the reaction was performed at about − 7 °C, in dry THF, using the twofold molar excess of NaCp relative to the number of epoxide groups in the polymer matrix. The epoxide number was calculated according to the method described in our previous papers [22, 23]. Subsequently, microspheres denoted as poly(GMA-co-EGDMA)-Cp were filtered off, washed with acetone, the solution of EtOH/water (50:50), 3% HCl in water, water, tetrahydrofuran and hexane and then dried at 50 °C overnight.

Scheme of the synthesis of primary microspheres and their modification
In the next step, the poly(GMA-co-EGDMA)-Cp material was reacted with maleic anhydride in the Diels–Alder reaction. The process was run in acetone at 50 °C for 2.5 h [23]. Afterwards, microspheres were filtered off, extracted with acetone and dried at 50 °C overnight. As a result, poly(GMA-co-EGDMA)-MA microspheres were produced.
Methods of analysis
ATR-FTIR spectra were recorded using the Tensor 27 spectrometer (Bruker, Germany), equipped with a diamond crystal. The spectra were made in the spectral range of 600–4000 cm−1 with a resolution of 4 cm−1 and 16 scans per spectrum.
Parameters characterizing the porosity of parent copolymers were determined by nitrogen adsorption at − 196 °C using a Micromeritics ASAP 2420 analyser. Before measurements, samples were degassed at 60 °C under vacuum.
Differential scanning calorimetry measurements were performed using the DSC 204 calorimeter (Netzsch, Germany) operating in a dynamic mode. The dynamic scans were performed at a heating rate of 10 K min−1 from 20 to 550 °C under argon atmosphere (20 mL min−1). The empty aluminium crucible was applied as the reference.
Thermogravimetric analysis of materials was carried out on a STA 449 F1 Jupiter (Netzsch, Germany) at the heating rate of 10 K min−1, in the temperature range of 20–850 °C, with the sample mass of ≈ 10 mg in inert (helium) atmosphere. The gas flow was 20 mL min−1. As the reference, the empty Al2O3 crucible was used.
Results and discussion
Copolymeric microspheres poly(GMA-co-EGDMA) were obtained using suspension–emulsion polymerization. Our aim was to prepare polymeric beads with epoxide ring in their structure, so that glycidyl methacrylate was used as a functional monomer and ethylene glycol dimethylacrylate was chosen as the crosslink agent. Firstly, primary poly(GMA-co-EGDMA) microspheres with a different degree of crosslinking were synthesized. Their porosity parameters: specific surface area (S BET), pore volume (V p) and average pore diameters (D p), were determined by means of the nitrogen adsorption–desorption method. The most developed porous structure with S BET equal 242 m2 g−1 and pore volume of 0.34 cm3 g−1 is observed in the case of the copolymer poly(GMA-co-EGDMA)5/5 which is the most crosslinked one. Along with the decreasing amount of EGDMA in the structure of copolymers, their porosity also decreases. Thus, for poly(GMA-co-EGDMA)6/4 S BET is 152 m2 g−1 and V p is 0.30 cm3 g−1 and for poly(GMA-co-EGDMA)8/2 S BET is 36 m2 g−1 and V p is 0.12 cm3 g−1. The copolymers poly(GMA-co-EGDMA)5/5 and poly(GMA-co-EGDMA)6/4 are mesoporous, whereas poly(GMA-co-EGDMA)8/2 possesses a macroporous structure. Due to the largest diameters of pores, the copolymer poly(GMA-co-EGDMA)8/2 was chosen for further modification. As it was described earlier [23, 24], the epoxide group is able to react with sodium cyclopentadienide, and after that cyclopentadienyl moiety is attached to the surface of microspheres named as poly(GMA-co-EGDMA)-Cp. Afterwards, the Diels–Alder reaction of Cp-functionalized microspheres with maleic anhydride was performed and poly(GMA-co-EGDMA)-MA material was obtained. In Fig. 2, the structure of synthesized polymers is presented.

The chemical structure of primary poly(GMA-co-EGDMA), poly(GMA-co-EGDMA)-Cp and poly(GMA-co-EGDMA)-MA materials
The chemical structure of prepared primary and modified microspheres was assessed by ATR-FTIR analysis, and in Fig. 3 selected spectra are presented. In the spectrum of primary poly(GMA-co-EGDMA)8/2 copolymer, the absorption bands at 907 and 845 cm−1 attributed to the epoxy ring vibrations confirm the presence of epoxide group in the copolymer network. The decreased intensity of these absorption bands is observed in the spectrum of poly(GMA-co-EGDMA)-Cp, obtained after the reaction of epoxide ring opening. However, the presence of mentioned signals in the spectrum suggests that only some part of the epoxides reacted with NaCp, most probably the surface groups. The spectrum of poly(GMA-co-EGDMA)-MA shows the intense characteristic absorption bands at 1777 cm−1 and a weak absorption bands at 1855 cm−1 [25]. They are due to symmetric (strong) and asymmetric (weak) carbonyl C=O stretching vibration of the anhydride rings. Additional characteristic for C–O–C stretching vibrations in cyclic anhydrides strong absorption bands appears at 909 cm−1 and weak at 1082 cm−1 [25]. It confirms that the reaction of cyclopentadienyl group with maleic anhydride was performed successfully.

ATR-FTIR spectra of primary poly(GMA-co-EGDMA), poly(GMA-co-EGDMA)-Cp and poly(GMA-co-EGDMA)-MA materials
Thermal properties of the obtained copolymers were determined using differential scanning calorimetry and thermogravimetric methods. The TG, DTG and DSC curves for copolymeric poly(GMA-co-EGDMA) microspheres are present in Fig. 4 whereas the temperature of 2% of mass loss (T 2%) and the residual mass as well as the temperature of the maximum rate of mass loss (T max) obtained from the DTG curves and the mass losses corresponding to them (W loss) are collected in Table 1. As follows from the DTG and DSC curves, the copolymers exhibit at least a three-step degradation process with endothermic and exothermic effects. The main mass loss of copolymers is found in the temperature range of about 245–290 °C with the value of W loss1 of 78.8% for the least crosslinked poly(GMA-co-EGDMA)8/2 material and 69.4% for the largest crosslinked poly(GMA-co-EGDMA)5/5. The first DTG peak with T max at about 290 °C (Fig. 4) is evidently composed of two poorly resolved peaks. This suggests that in the temperature range of 245–290 °C at least two different processes proceed. It follows from the DSC curves that at about 250 °C the endothermic process takes place whereas at about 308 °C the maximum of exothermic effect followed by the fast endothermic one is observed. It was earlier reported that the thermal degradation of homopolymer poly(GMA) starts with the endothermic depolymerization followed by the exothermic isomerization of the epoxy group to the carbonyl one [15]. It is assumed that in the case of the copolymers poly(GMA-co-EGDMA) degradation also starts with the depolymerization and is accompanied by the isomerization reaction in the gaseous phase. To confirm these expectations, the simultaneous analysis of the evolved volatile products of degradation was done. Taking into account the 3D FTIR diagrams (Fig. 5) presenting spectra of gaseous products evolved during copolymers degradation, it can be seen that in the temperature range about 250–300 °C two poorly resolved maxima of emission are present. Additionally, in Fig. 6 the FTIR spectra extracted at the maxima of decomposition for the exemplary poly(GMA-co-EGDMA)8/2 are shown. The FTIR spectra collected at 250 and 290 °C are almost the same. The absorption bands: at 1741 cm−1 attributed to the vibrations of C=O group in esters, at 1638 cm−1 to the vibrations of C=C bond in the methacrylate group, at 1156 cm−1 to the vibrations of C–O–C group in esters, at 940 cm−1 to the vibrations of =CH2 and at 846 cm−1 to the vibrations of epoxide ring, confirm that depolymerization to monomers (mainly GMA) occurs at the beginning of decomposition. It is assumed that the degradation of ester bonds in the crosslinked part of copolymer takes part at about 290 °C according to the random chain scission mechanism. Furthermore, at 290 °C new absorption bands at 3464, 2815, 2781 and 1883 cm−1 are visible. Their appearance is due to the emission of acrolein, which is a product of breaking of ester bonds and further isomerization of the gaseous degradation products [15, 26]. Additionally, the increasing absorption band at 3581 cm−1 indicates the emission of allyl alcohol or glycidol as products of ester and epoxide decomposition [26].

TG, DTG and DSC curves of poly(GMA-co-EGDMA) copolymers obtained under helium atmosphere

3D diagrams of FTIR absorption of gases evolved from the poly(GMA-co-EGDMA) copolymers under helium atmosphere

FTIR spectra of gases evolved from poly(GMA-co-EGDMA)8/2 at the maxima of decomposition
The second mass loss of copolymers is observed in the temperature range of 360–470 °C. The calculated mass losses are dependent on the amount of crosslinker (EGDMA) used for the synthesis of copolymers and are in the range of 19.5–28.9%. The FTIR spectrum corresponding to the T max2 (410 °C) shows an emission of the organic compounds derived from the decomposition of residual crosslinked part (including a small amount of ethylene glycol dimethacrylate) and also release of significant amount of carbon dioxide (absorption bands at 2357 and 670 cm−1) as a product of degradation of ester bonds present in the EGDMA units.
The TG and DTG curves obtained in helium atmosphere of the unmodified poly(GMA-co-EGDMA)8/2 copolymer and modified materials are shown in Fig. 7. The thermal degradation profile of poly(GMA-co-EGDMA)-Cp material obtained after the reaction of primary microspheres with sodium cyclopentadienide is completely different from the profile of starting material. The main stage of decomposition of poly(GMA-co-EGDMA)8/2 occurs at about 200–350 °C whereas poly(GMA-co-EGDMA)-Cp displays a broad degradation stage between 260 and 480 °C with the maximum at 412 °C and 94.1% mass loss. On the other hand, considering the Gram–Schmidt diagram for poly(GMA-co-EGDMA)-Cp, three maxima of emission are visible. This suggests that its degradation proceeds in at least three overlapping stages with the maxima at 322 °C, 412 °C (corresponding to the maximum on the DTG curve) and 531 °C. The FTIR spectra extracted at these temperatures are present in Fig. 8. Considering the spectrum extracted at the first maximum of emission at 322 °C, the presence of the absorption bands at 2812, 2781, 2747, 1731, 1707, 958 and 924 cm−1 confirms the evolution of acrolein which is a product of isomerization of the epoxide group to the carbonyl group [15, 26]. This phenomenon suggests that the poly(GMA-co-EGDMA)-Cp units derived from glycidyl methacrylate are present. It also confirms that the reaction of epoxide groups with NaCp was not completed. It is interesting that the evolution of the monomer GMA is not observed, as it is in the case of primary microspheres, so it can be concluded that depolymerization does not take place for the material modified with cyclopentadienyl group but the degradation proceeds according to the chain scission starting with the ether bond cleavage at glycidyl moiety [27]. At 412 °C, the emission of acrolein is still observed; however, new absorption bands in the spectrum region of methylene and methyl groups vibrations (3015–2870 cm−1), and at 3099 cm−1 (characteristic of –CH=CH2 vibration) appear. Moreover, in the region of C=O group vibrations a new band at 1764 cm−1 appears. The latter absorption bands justify the emission of species containing cyclopentadienyl moiety, the most probably it is cyclopentadienyl propanone [26]. It is noteworthy that the emission of carbon dioxide (2359, 2310 cm−1) and carbon monoxide (2179, 2120 cm−1) is significantly larger in comparison with the unmodified material. Simultaneously with the increasing CO2 emission, the decrease in absorption bands intensity at 1148 cm−1 (C–O group stretching vibrations) and 1743 cm−1 (C=O group stretching vibrations) is observed, testifying that the degradation of modified material runs according to the random chain scission. Moreover, it is clearly visible that in the spectrum extracted at 442 °C the bands derived from released acrolein disappear whereas new absorption bands at 948 and 1641 cm−1 occur probably from the emission of alkene gaseous compounds. The spectrum taken out at 531 °C, corresponding to the last emission maximum on the Gram–Schmidt plot, shows the absorption bands derived from the evolution of carbon dioxide, and also strong bands at 3580 and 1017 cm−1 which most probably are connected with the emission of glycol or alcohol compounds, the bands at 1764 and 1127 cm−1 are derived from the carbonyl compounds formed during degradation of the crosslinked part of material [28].

TG, DTG and Gram–Schmidt curves of primary poly(GMA-co-EGDMA)8/2 and modified copolymers obtained under helium atmosphere

FTIR spectra of gases evolved from poly(GMA-co-EGDMA)-Cp at the maxima of emission
The TG and DTG profiles of poly(GMA-co-EGDMA)-MA material obtained after the Diels–Alder reaction of poly(GMA-co-EGDMA)-Cp with maleic anhydride are changed after the modification reaction. Compared with the DTG of poly(GMA-co-EGDMA)-Cp material in which a broad overlapping peak is present, on the DTG curve for poly(GMA-co-EGDMA)-MA a not well-separated peak with the maximum at 346 °C and the mass loss of 36.9% is observed (Fig. 7; Table 1). It is assumed to be connected: firstly, with the degradation of residual epoxide groups (as in the case of poly(GMA-co-EGDMA)-Cp material) and secondly, with a loss of anhydride groups, which is in line with the previous reports [29, 30]. The Gram–Schmidt plots of these two materials are of similar character, and also 3D FTIR diagrams (Fig. 9) of volatile products of their degradation show the same absorption bands. However, taking into consideration the absorption bands at 1148 and at 1743 cm−1 at 452 °C (Fig. 10), differences in their intensities are clearly visible. It is probable that at temperature above 250 °C anhydride groups can undergo esterification reaction with the hydroxyl groups present on the surface of microspheres (Fig. 2) [29,30,31]. To confirm this expectation, sample of the poly(GMA-co-EGDMA)-MA copolymer was heated to 300 °C under helium atmosphere and then the ATR-FTIR spectrum of preheated sample was taken out. The spectra present in Fig. 11 show the differences in the structure of heated and unheated sample. The absorption bands at 1777 and 1858 cm−1 (stretching vibrations of C=O in anhydrides) and at 1082 and 909 cm−1 (stretching vibrations of C–O–C in anhydrides) disappear. Moreover, in the spectrum of evolved volatile products any gasses connected with degradation of anhydride spices cannot be found; thus, it can be concluded that esterification runs probably out at higher temperature on the surface of microspheres, prior to their total thermal degradation.

3D diagrams of FTIR absorption of gases evolved from the poly(GMA-co-EGDMA)-Cp and poly(GMA-co-EGDMA)-MA materials under helium atmosphere

FTIR spectra of gases evolved from poly(GMA-co-EGDMA)-Cp (blue curve) and poly(GMA-co-EGDMA)-MA (red curve) at 451 °C

FTIR spectra of poly(GMA-co-EGDMA)-MA (red curve) and poly(GMA-co-EGDMA)-MA preheated to 300 °C (blue curve)
Conclusions
The DSC- and TG/FTIR-coupled methods were used to monitor the thermal behaviour of porous primary and modified copolymeric microspheres of GMA crosslinked with EGDMA. The poly(GMA-co-EGDMA) copolymers are thermally stable up to 210 °C in inert atmosphere. Their thermal resistance is dependent on the amount of the crosslinked monomer. The degradation of parent microspheres starts with the depolymerization process initiated at the glycidyl fragments in the copolymer network and is followed by the isomerization process at gaseous phase. The residual crosslinked part of copolymers decomposes above 360 °C. The modification steps of poly(GMA-co-EGDMA) microspheres including the reaction of epoxide group with sodium cyclopentadienide followed by the Diels–Alder reaction with maleic anhydride changed the thermal properties of starting material. The value of T 2% is slightly lower for modified microspheres; however, the main degradation process is shifted to higher temperature region. The poly(GMA-co-EGDMA)-Cp as well as the poly(GMA-co-EGDMA)-MA materials reveal complex degradation processes which run according to the random chain scission mechanism. Although some part of epoxide groups remained in the interior of microspheres after the modification, the products of the depolymerization are not observed during the thermal degradation. Moreover, the thermal analysis of the copolymer with grafted maleic anhydride shows that the esterification takes place probably at higher temperature on the surface of microspheres, prior to their total thermal degradation. In this study, we present the model reaction of functionalized microspheres with maleic anhydride; however, the presence of cyclopentadienyl group in the structure of poly(GMA-co-EGDMA)-Cp copolymer allows to the addition of any dienophile with the use of Diels–Alder reaction. Moreover, the anhydride group in the structure of poly(GMA-co-EGDMA)-MA microspheres can be easily transferred into carboxyl group. The final microspheres possessing anhydride groups or such with carboxyl one can be used as column packings in liquid chromatography and solid-phase extraction.
References
Rahman A, Iqbal M, Rahman F, Fu D, Yaseen M, Lv Y, Omer M, Garver M, Yang L, Tan T. Synthesis and characterization of reactive macroporous poly(glycidyl methacrylate-triallyl isocyanurate-ethylene glycol dimethacrylate) microspheres by suspension polymerization: effect of synthesis variables on surface area and porosity. J Appl Polym Sci. 2012;124:915–26.
Jin JM, Lee JM, Ha MH, Lee K, Choe S. Highly crosslinked poly(glycidyl methacrylate-co-divinyl benzene) particles by precipitation polymerization. Int J Polym Mater. 2007;11:3107–15.
Švec F, Hradil J, Čoupek J, Kálal J. Reactive polymers I. Macroporous methacrylate copolymers containing epoxy groups. Macromol Mater Eng. 1975;48:135–43.
Ferreira A, Bigan M, Blondeau D. Optimization of a polymeric HPLC phase: poly(glycidyl methacrylate–co-ethylene dimethacrylate): influence of the polymerization conditions on the pore structure of macroporous beads. React Funct Polym. 2003;56:123–6.
Švec F, Fréchet JMJ. Modified poly(glycidyl methacrylate-co-ethylene dimethacrylate) continuous rod columns for preparative-scale ion-exchange chromatography of proteins. J Chromatogr A. 1995;702:89–95.
Saluzzo C, Lamouille T, Hérault D, Lemaire M. Polymer-supported catalysts: enantioselective hydrogenation and hydrogen transfer reduction. Bioorg Med Chem Lett. 2002;12:1841–4.
Miletić N, Vuković Z, Nastasović A, Loos K. Macroporous poly(glycidyl methacrylate-co-ethylene glycol dimethacrylate) resins-versatile immobilization supports for biocatalysts. J Mol Catal B Enzym. 2009;56:196–201.
Petro M, Švec F, Fréchet JMJ. Immobilization of trypsin onto “molded” macroporous poly(glycidyl methacrylate-co-ethylene dimethacrylate) rods and use of the conjugates as bioreactors and for affinity chromatography. Biotechnol Bioeng. 1996;49:355–63.
Hainey P, Sherrington DC. Oligoamine-functionalized poly(glycidyl methacrylate-ethyleneglycol dimethacrylate) resins as moderate base extractants for gold from cyanide solutions. React Funct Polym. 2000;43:195–210.
Kita R, Švec F, Fréchet JMJ. Hydrophilic polymer supports for solid phase synthesis: the preparation of poly(ethylene glycol) methacrylate polymer beads using “classical” suspension polymerization in aqueous medium and their application in the solid phase synthesis of hydantoins. J Comb Chem. 2001;3:564–71.
Zulfiqar S, Zulfiqar M, Nawaz M, McNeill IC, Gorman JG. Thermal degradation of poly(glycidyl methacrylate). Polym Degrad Stab. 1990;30:195–203.
Ananthalakshmi NR, Wadgaonkar PP, Sivaram S, Varma IK. Thermal behaviour of glycidyl methacrylate homopolymers and copolymers. J Therm Anal Calorim. 1999;58:533–9.
Zulfiqar M, Paracha A, Zulfiqar S. Thermal degradation of glycidyl methacrylate-styrene copolymers. Polym Degrad Stab. 1994;43:403–8.
Piracha A, Zulfiqar S. The thermal degradation of glycidyl methacrylate-methyl methacrylate copolymers. Polym Degrad Stab. 1996;51:27–34.
Piracha A, Zulfiqar S, McNeill IC. The thermal degradation of copolymers of glycidyl methacrylate and vinylacetate. Polym Degrad Stab. 1996;51:319–26.
Iqbal MS, Jamil Y, Kausar T, Akhtar M. Thermal degradation study of glycidyl methacrylate acrylonitrile copolymers. J Therm Anal Calorim. 2009;96:225–33.
Ahmad S, Zulfiqar S. Synthesis, characterization and thermal degradation of glycidyl methacrylate-α-methyl styrene copolymers. Polym Degrad Stab. 2002;76:173–7.
Maciejewska M. Characterization of thermal properties of porous microspheres bearing pyrrolidone units. J Therm Anal Calorim. 2015;119:1147–55.
Maciejewska M. Thermal properties of TRIM–GMA copolymers with pendant amine groups. J Therm Anal Calorim. 2016;126:1777–85.
Podkościelna B. Synthesis, modification, and porous properties of new glycidyl methacrylate copolymers. J Appl Polym Sci. 2011;120:3020–6.
Gawdzik B. Comparison of the selectivity of di(methacryloyloxymethyl)-naphthalene-divinylbenzene copolymers in reversed-phase high-performance liquid chromatography. J Chromatogr. 1991;549:77–88.
Grochowicz M, Bartnicki A, Gawdzik B. Preparation and characterization of porous polymeric microspheres obtained from multifunctional methacrylate monomers. J Polym Sci, Part A: Polym Chem. 2008;46:6165–74.
Grochowicz M, Pączkowski P, Gawdzik B. Diels-Alder reaction as a tool to modify the surface of polymeric microspheres. Adsorpt Sci Technol. 2015;33:677–84.
Kaupp M, Vogt AP, Natterodt JC, Trouillet V, Gruendling T, Hofe T, Barner L, Barner-Kowollik C. Modular design of glyco-microspheres via mild pericyclic reactions and their quantitative analysis. Polym Chem. 2012;3:2605–14.
Socrates G. Infrared and Raman characteristic group frequencies: tables and charts. 3rd ed. Chichester: Wiley; 2001.
NIST chemistry webbook standard reference database number 69 (2011). http://webbook.nist.gov/chemistry. Accessed 14 July 2017.
Chiniwalla P, Bai Y, Elce E, Shick R, McDougall WC, Bidstrupallen SA, Kohl PA. Crosslinking and decomposition reactions of epoxide functionalized polynorbornene. Part I. FTIR and thermogravimetric analysis. J Appl Polym Sci. 2003;89:568–77.
Grochowicz M. Investigation of the thermal behavior of 4-vinylpyridine–trimethylolpropane trimethacrylate copolymeric microspheres. J Therm Anal Calorim. 2014;118:1603–11.
Huang J-W, Lu W-C, Yeh M-Y, Lin C-H, Tsai I-S. Unusual thermal degradation of maleic anhydride grafted polyethylene polymer. Polym Eng Sci. 2008;48:1550–4.
Borah JS, Chaki TK. Thermogravimetric and dynamic mechanical analysis of LLDPE/EMA blends. J Therm Anal Calorim. 2011;105:365–73.
Roumeli E, Terzopoulou Z, Pavlidou E, Chrissafis K, Papadopoulou E, Athanasiadou E, Triantafyllidis K, Bikiaris DN. Effect of maleic anhydride on the mechanical and thermal properties of hemp/high-density polyethylene green composites. J Therm Anal Calorim. 2015;121:93–105.
Acknowledgements
The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/under REA Grant agreement No. PIRSES-GA-2013-612484. The research was carried out with the equipment purchased thanks to the financial support of the Operational Programme Development of Eastern Poland 2007–2013 (contract No. POPW.01.03.00-06-017/09-00).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Grochowicz, M., Pączkowski, P. & Gawdzik, B. Investigation of the thermal properties of glycidyl methacrylate–ethylene glycol dimethacrylate copolymeric microspheres modified by Diels–Alder reaction. J Therm Anal Calorim 133, 499–508 (2018). https://doi.org/10.1007/s10973-017-6785-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6785-3