
Abstract
The system Rb3PO4–Ba3(PO4)2 was investigated by thermoanalytical methods, X-ray powder diffraction, ICP, and FT-IR. On the basis of the obtained results its phase diagram was proposed. For this system with one intermediate compound, BaRbPO4, we found that this compound melts congruently at 1700 °C, exhibits a polymorphic transition at 1195 °C and is high-temperature unstable. Also, the intermediate compound was subject to gradual decomposes to Ba3(PO4)2 (the solid phase) and vaporization (with conversion of phosphorus and rubidium oxides into vapor phase). We also found that Rb3PO4 melts congruently at 1450 °C and shows a polymorphic transition at 1040 °C. Regarding Ba3(PO4)2, we have confirmed that it melts congruently at 1605 °C and exhibits a polymorphic transition at 1360 °C.
Similar content being viewed by others

Introduction
In the present article, the results of investigations pertaining to the equilibria which occur in the system Rb3PO4–Ba3(PO4)2 are described. Its phase diagram was not published previously. There is literature, however, concerning the initial orthophosphates Rb3PO4, Ba3(PO4)2 as well as the barium–rubidium orthophosphate BaRbPO4. The literature reports mainly concern on the methods of obtaining, crystal structure, polymorphism, and the possibility of application. Elammari, Elouadi, and Müller-Vogt have noted [1] that some of the mixed orthophosphates of the formula AIBIIPO4 (where AI is a monovalent cation and BII is a divalent cation) show ferroelectric properties; it can be mentioned that the BaRbPO4 compound belongs to this group.
Orthophosphate Rb3PO4 has been found to be dimorphic. The low-temperature form is orthorhombic (s.g. Pnma; the lattice parameters are: a = 1.17362(2), b = 0.81046(1), c = 0.615167(9) nm) [2]. The high-temperature form crystallizes in the cubic system (s.g. Fm 3; a = 8.44 Å) [3]. We found no reported data on the melting temperature of Rb3PO4. The orthophosphate Ba3(PO4)2 melts congruently at 1605 ± 2 °C [4] and >1620 °C [5]; it shows a polymorphic transition at 1360 ± 2 °C [4] and 1390 ± 15 °C [5]. The crystal structure of the low-temperature form of the Ba3(PO4)2 has been reported in [6] (s.g.\( {\text{R}}\mathop 3\limits^{ - } {\text{m}} \); a = 5.6038(7), c = 21.000(5) Å). According to [7], diffraction pattern of Ba3(PO4)2 corresponds to rhombohedral translation lattice (the unit cell dimensions are: a = 7.696 ± 0.002 Å, α = 42°35′ ± 2′). The orthophosphate BaRbPO4 exhibits a reversible phase transition at 1060 °C [1]. According to [1, 8] this compound crystallizes in the orthorhombic system with Pnma space group (a = 7.812(2), b = 5.740(1), c = 10.056(2) Å). We found no reported data on its melting point, too.
Studies of phase equilibria which occur in different systems, particularly for a wide temperature range, are of great importance in science and technology. Such new data can afford possibilities for identification of unknown phases with their physicochemical properties, and for offering different methods of obtaining. On the one hand they contribute to better knowledge and, on the other, help to search for new, suitable and low-cost materials with properties required in different technologies. Hence, the results of phase-equilibria studies can be applied to further specialized or interdisciplinary research work.
Experimental
In the present investigations of the system Rb3PO4–Ba3(PO4)2 the following commercial reagents (all analytical pure) were used: BaHPO4, BaCO3, Rb2CO3, (NH4)2HPO4, NH4H2PO4, Ba(NO3)2, citric acid monohydrate and ethylene glycol.
Ba3(PO4)2, BaRbPO4, and Rb3PO4 compounds were prepared in our laboratory. Barium diphosphate Ba2P2O7 was obtained from BaHPO4 by heating at 900 °C for 1 h. The barium orthophosphate Ba3(PO4)2 was obtained from stoichiometric amounts of Ba2P2O7 and BaCO3. The substrates, thoroughly mixed and rubbed, were sintered in a platinum crucible at 1300 °C for 6 h. Rubidium orthophosphate Rb3PO4 was obtained from a stoichiometric mixture of dry Rb2CO3 (dried at 250 °C) and (NH4)2HPO4. After mixing and rubbing, it was heated at 200 °C for 4 h, then at 500 °C for 4 h, thereafter rubbed and heated at 900 °C for 10 h. After a new rubbing it was still heated at 1000 °C for 20 h.
The phase equilibria in the system Rb3PO4–Ba3(PO4)2 were investigated using thermoanalytical methods, X-ray powder diffraction, ICP, and FT-IR technique. Samples for investigation were prepared from the parent substances mixed in the fixed amounts. In order to ensure their homogeneity, the mixtures were shaken in a weighing bottle and ground in an agate mortar, pelletized, placed in platinum crucibles, and then sintered. The conditions under which the samples were prepared (i.e., the sintering temperature and time) had been determined experimentally. The obtained sinters were crushed and finely ground.
The thermoanalytical investigations in the solid phase were carried out using a SETSYSTM (SETARAM) differential thermal analyzer (calorimeter) with a balance. The device enables one to perform simultaneous TGA-DTA or TGA-DSC measurements in the temperature range 20–1300 °C. Samples from 1 to 20 mg were placed in platinum crucibles and heated at a rate of 10 °C min−1 in an atmosphere of argon. DTA, TG, analyses in the solid phase were also carried out in the air, up to 1400 °C, by using a derivatograph type 3427 (MOM, Hungary) with a heating rate of 5 °C min−1, platinum crucibles, sample weight 0.3–0.6 g. Temperature was measured by means of a Pt/PtRh10 thermocouple which was calibrated against the melting points of NaCl, K2SO4, Ca2P2O7, and the transition point of K2SO4. In order to determine the temperature for the thermal effect, the peak parameter on the DTA-heating curve was taken into account. The high-temperature thermal investigations (above 1400 °C) were carried out under argon in a horizontal, resistance furnace with a molybdenum winding. Presynthesized samples were pressed into pellets of 1–2 g weight and loaded in boats of a PtRh30 alloy. The temperature point at which a sample was diffusing and got blurred in the field of observation to finally disappear was read by means of an optical pyrometer calibrated against the melting points of Na3PO4 and Ca3(PO4)2. For the samples that were melted in a temperature range, the points determined were approximate. After melting, the samples were cooled down to room temperature. The quenching-in-ice technique (both for the sinters and for the melted samples) was also employed; a high-temperature vertical-tubular furnace (20–1750 °C; Nabertherm RHTV 120-300/18) was used.
The phase purity of the commercial and the self-prepared parent phosphates, and the phase composition of both the sintered and the melted samples of the investigated system, were controlled by X-ray powder diffraction at room temperature. A SIEMENS D 5000 diffractometer with CuKα radiation and a Ni filter was used. The quantitative analysis was performed using an emission spectrometer with argon plasma excitation (ICP Model ARL 3410). The vaporization of the orthophosphates Rb3PO4 and BaRbPO4 has been studied by using the thermal analysis (Netzsch 409C) combined with mass spectrometry (Balzers Instruments ThermoStar). FT-IR spectra were measured over the range 1400–400 cm−1 (with KBr as diluent; Perkin-Elmer System 2000 FT-IR).
Results and discussion
Finding the phase equilibria existing in the Rb3PO4–Ba3(PO4)2 system was preceded by investigation of thermal stability, polymorphism and other properties of the orthophosphates Ba3(PO4)2, Rb3PO4, and BaRbPO4. This was aimed at confirmation or verification or complementing the literature data. It is well known that the character of phase equilibria which arise in binary systems essentially depends on the properties of parent components.
Regarding Rb3PO4, we have found that the compound melts congruently at about 1450 °C and shows two polymorphic modifications with the transformation point at 1040 °C. The orthophosphate is strongly hygroscopic; for a presynthesized sample of Rb3PO4, TG/DTA-heating curves indicated water losing in a few stages in the temperature range 100–430 °C. Also, a slow gradual loss of mass was observed on TG/DTA-heating curves (as shown in Fig. 1) during continued heating, from a temperature of ~1150 °C. The loss noted was ~5.5 wt% as a result of melting up the Rb3PO4 sample. In order to explain the deficit, applied was thermal analysis combined with mass-spectrometric analysis. Presynthesized Rb3PO4 was heated from 20 to 1400 °C at a rate of 10 °C min−1. The mass-spectrometric analysis of the gaseous phase revealed the presence of the ions PO+, PO2 +, P4O10 +, RbO+, RbO2 +, Rb2O2 +. The FT-IR spectra both for the presynthesized Rb3PO4 and for the melted sample, were taken, too. It appeared that their infrared spectra were the same (see Fig. 2). Those results indicated that the stoichiometric composition of the condensed phase did not change.

TG and DTA curves of presynthesized Rb3PO4, in air atmosphere

FT-IR spectra of Rb3PO4; a melted, b presynthesized
It was confirmed, in accord with the results of [4], that the orthophosphate Ba3(PO4)2 melts congruently at 1605 °C and occurred in two polymorphic modifications; the transition point being 1360 °C. Our thermal investigations confirmed the stability of Ba3(PO4)2 compound. No weight loss in TG curves was noted in the temperature range 20–1400 °C during the DTA-heating of the orthophosphate samples of both the presynthesized and the melted ones. No difference was found in powder X-ray diffractograms for Ba3(PO4)2 between the presynthesized samples and the melted ones via slow crystallization (at a rate of 3 °C min−1). It was noted that the melted Ba3(PO4)2 had a tendency to appear partially in the glassy form. An amorphous form appeared for a sample of Ba3(PO4)2, which was crystallized fast (~15 °C min−1) after melting. This was observed using reflected light microscopy of polished sections.
BaRbPO4 orthophosphate was obtained by the standard method of solid-phase reaction according to the following reaction networks.
The conditions under which the above reaction was made were those given in paper [1]. It appeared, however, that additional roasting at 900 °C for 10 h was necessary to attain the BaRbPO4 of phase purity.
In the method (2) the parent orthophosphates were thoroughly mixed (by shaking in a weighing bottle), rubbed in an agate mortar, pressed into pellets, and heated at 1000 °C for 2.5 h. The conditions (i.e., temperature and time) of both reactions were experimentally found.
X-ray powder diffraction of the sinters obtained by reactions (1) and (2) showed the phase-pure BaRbPO4 structure in agreement with [1, 8]. We found that the orthophosphate melts congruently at about 1700 °C. Next, its thermal stability and polymorphism were investigated. During the DTA/DSC-heating of the presynthesized BaRbPO4 a slight, gradually proceeding loss of mass (noticeable in the TG curve) was observed from ~1230 °C. Figure 3 shows the TG/DTA-heating curves for the presynthesized BaRbPO4. It was also noted that the BaRbPO4 sample lost ~10 wt% of its original mass as a result of melting. Its phase composition was verified by X-ray powder analysis. The diffractogram revealed reflections typical of Ba3(PO4)2, apart from BaRbPO4 diffraction lines. This evidenced that the orthophosphate BaRbPO4 is high-temperature unstable and subjected to a gradual decomposition and vaporization. To verify the conclusion:

TG and DTA curves of presynthesized BaRbPO4, in air atmosphere
-
A quantitative determination of rubidium, barium and phosphorus content was made for the samples of both the presynthesized compound and the melted one. The quantitative analysis was performed using an emission spectrometer with argon plasma excitation. It appeared that the presynthesized and the melted samples differed in rubidium, barium and phosphorus content. Rubidium and phosphorus content decreased by ~5 and 0.6 wt%, respectively, for the melted sample and that of barium increased by ~4.7 wt% (with respect to the stoichiometric composition of the compound).
-
The thermal analysis combined with mass spectrometry was performed. A sample of presynthesized BaRbPO4 was heated from 20 to 1600 °C at a rate of 10 °C min−1. The mass spectrometry of the gaseous phase showed the presence of the ions: PO+, PO2 +, P4O10 +, RbO+, RbO2 +, Rb2O2 +.
In view of all the results of this study the decomposition and vaporization of orthophosphate BaRbPO4 occur according to the reaction:
The appearance of the BaRbPO4 in two modifications was confirmed in our thermoanalytical investigations. However, a disagreement was concerning the temperature of the transition. The differential thermal analysis of heating was performed for both presynthesized and melted BaRbPO4. The DTA/DSC-heating curve of sinter in the temperature range 20–1350 °C revealed an endothermic effect to which a temperature of 1195 °C corresponded (see Fig. 3). Accordingly, a melted sample of BaRbPO4 was investigated by using DTA/DSC-heating (being aware that stoichiometry of the melted sample and BaRbPO4 are different). This time the DTA/DSC-heating curves showed a single endothermic effect, too, but at a corresponding temperature of ~1090 °C (see Fig. 4). As was subsequently found, the effect was present on the DTA/DSC-heating curves for all the presynthesized samples with composition ranging between 65 and 99 wt% Ba3(PO4)2. In view of all results of the present study we ascribe a temperature point of 1195 °C to the polymorphic transformation of BaRbPO4.

TG and DTA curves of the melted BaRbPO4, in air atmosphere
The previously unknown phase equilibria in the Rb3PO4–Ba3(PO4)2 system were investigated in the entire composition range up to a temperature of 1800 °C. BaRbPO4 appeared as an intermediate compound in the system. Due to the synthesis conditions of the orthophosphate, the thermal instability of the phosphates Rb3PO4 and BaRbPO4 at high temperatures, the hygroscopicity of Rb3PO4, and also to meet the requirement of equilibrium composition, the following two series of samples were used.
-
Heteromolar mixtures of (NH4)2HPO4, dry Rb2CO3, and BaRbPO4—to determine the phase equilibria within the composition range 0–60 wt% Ba3(PO4)2. Those mixtures, after mixing and grinding, were heated at 200 °C for 4 h, at 500 °C for 4 h, at 900 °C for 10 h and at 1000 °C for 20 h with intermediate grindings to ensure a total reaction.
-
Heteromolar mixtures of BaRbPO4 and Ba3(PO4)2 orthophosphates—to determine the phase equilibria within the composition range 65–100 wt% Ba3(PO4)2. Those mixtures were presynthesized via the solid-phase reaction by heating at 950 °C for 2 h.
The sinters obtained by the above methods were cooled down to room temperature and homogenized by rubbing, then were subjected to thermal analyzing. The phase composition of the sinters was identified by X-ray powder diffraction at room-temperature. Testing procedure showed that all samples of the system of interest melted in excess of 1400 °C. Accordingly, to draw the liquidus curves the samples after pressing into pellets were placed in PtRh30 boats and heated under argon in the horizontal furnace. The temperature of the pellets’ diffusion was read by means of optical pyrometer. The outline of both the liquidus and the solidus curves, therefore, is approximate as shown by dashed lines.
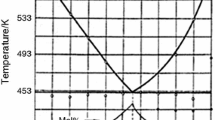
The liquidus curve of the system Rb3PO4–Ba3(PO4)2, at ~30 wt%, had a maximum at ~1560 °C (Fig. 5, point B of the phase diagram). This indicated, for such a composition, the possibility of formation of another intermediate compound which would congruently melt. The compound’s formation seemed likely since either the molar ratio Rb3PO4:Ba3(PO4)2 equal to 4:1 or the molar ratio Rb3PO4:BaRbPO4 = 1:1 (i.e., theoretical formula of the compound BaRb4(PO4)2) correspond to the percentage composition in question (30 wt% Ba3(PO4)2 and 70 wt% Rb3PO4). Compounds of the type M I4 MII(PO4)2 (where MI = Na, K; MII = Mg, Ca) are known in the literature [9–12].

Phase diagram of the system Rb3PO4–Ba3(PO4)2
In order to verify the hypothesis the following was done.
-
An heteromolar mixture of Rb3PO4 and BaRbPO4 (after a thorough homogenization) was sintered at 1150 °C for 2 h and quenched.
-
A mixture of 4 mol Rb3PO4 and 1 mol Ba3(PO4)2 (after a thorough homogenization) was sintered at 1100 °C for 5 h and for 1 h at 1150 °C, then quenched.
-
A modified Pechini method was employed; stoichiometric amounts of Ba(NO3)2, Rb2CO3, NH4H2PO4 were dissolved in a small portion of distilled water, then citric acid and ethylene glycol were added as the complexing agent. The mixture was dried at 120 °C for 24 h, then heated at 450 °C for 24 h, rubbed, heated at 1150 °C for 2 h and quenched.
Phase composition of reaction products from the above processes was verified by X-ray powder diffraction. Diffractograms revealed only reflections from Rb3PO4 and BaRbPO4 phosphates. Consequently, the maximum present on the liquidus curve (at ~30 wt% Ba3(PO4)2) can account for division of the liquid phase into the two liquid solutions L1 and L2. Hence, the ABC area (in the composition range 17–36 wt% Ba3(PO4)2) represents a mixture of liquid solutions L1 + L2. A transformation at a constant temperature of ~1510 °C can be ascribed to point C, according to the reaction network: L2C → L1A + BaRbPO4 (where L2C denotes the liquid L2 with a composition of point C, and L1A – liquid L1 with a composition of point A). An eutectic occurs in the Rb3PO4-rich part of the system of interest at ~1.0 wt% Ba3(PO4)2, which melts at ~1440 °C. In the Rb3PO4–Ba3(PO4)2 system, within the composition range 63.14–100 wt% Ba3(PO4)2; high-temperature, continuous solid solutions α exhibit their maximum melting point at ~1720 °C.
In order to find the phase equilibria for the system Rb3PO4–Ba3(PO4)2, the DTA/DSC-heating of solid-phase presynthesized samples was performed in the subsolidus area. Thermal instability of Rb3PO4 and BaRbPO4 phosphates hindered dealing with melted samples because of changes in original composition. The elaborated phase diagram of the Rb3PO4–Ba3(PO4)2 system is shown in Fig. 5.
References
Elammari L, Elouadi B, Müller-Vogt G. Study of phase transitions in the system AIBIIPO4 with AI = Li, Rb and BII = Mg, Ca, Sr, Ba, Zn, Cd, Pb. Phase Transit. 1988;13:29–32.
Voronin VI, Berger IF, Proskurnina NV, Sheptyakov DV, Goshchitskii BN, Burmakin EI, Stroev SS, Shekhtman GSh. Crystal structure of the low-temperature forms of cesium and rubidium orthophosphates. Inorg Mater. 2008;44(6):646–52.
Hoope R, Seyfert HM. Zur Kenntnis wasserfreier Orthophosphate der höheren Alkalimetalle: K3PO4, Rb3PO4, Cs3PO4. Z Naturforsch. 1973;28b:507–8.
McCauley RA, Hummel FA. Phase relationships in a portion of the system BaO–P2O5. Trans Brit Ceram Soc. 1968;67:619–28.
Kreidler ER. Phase equilibria and tin-activated luminescence in the system Ca3(PO4)2–Ba3(PO4)2. J Electrochem Soc. 1971;118:923–9.
Sugiyama K, Tokonami M. The crystal structure refinements of the strontium and barium orthophosphates. Mineral J. 1990;15:141–6.
Zachariasen WH. The crystal structure of the normal orthophosphates of barium and strontium. Acta Crystallogr. 1948;1:263–5.
Elammari L, Elouadi B. Crystal structure of the orthophosphate RbBaPO4. J Alloys Comp. 1992;188:99–101.
Berak J, Znamierowska T. Phase equilibria in the system CaO–Na2O–P2O5. Part I. The system Ca(PO3)2–Na2O. Roczniki Chemii (Ann Soc Chim Polonorum). 1967;41:2065–9.
Znamierowska T. Phase equilibria in the system CaO–K2O–P2O5. Part IV. Partial system CaKPO4–CaK4(PO4)2–K4P2O7–CaK2P2O7. Polish J Chem. 1979;53:1415–23.
Berak J, Podhajska-Kaźmierczak T. Phase equilibria in the ternary system MgO–K2O–P2O5. Part I. The partial system MgO–Mg3(PO4)2–K3PO4. Polish J Chem. 1991;65:1137–49.
Podhajska-Kaźmierczak T, Znamierowska T. Phase equilibria in the system MgO–Na2O–P2O5: the binary system Mg3(PO4)2–Na3PO4. Polish J Chem. 1999;73:279–86.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Radomińska, E., Znamierowska, T. & Szuszkiewicz, W. Phase equilibria in the system Rb3PO4–Ba3(PO4)2 . J Therm Anal Calorim 103, 761–766 (2011). https://doi.org/10.1007/s10973-010-0962-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-010-0962-y