
Abstract
The aim of this study was to investigate polyurethane foams (PUF) properties obtained from crude glycerol (CG) and polyethylene glycol (PEG) based liquefaction of sawdust. The four types of foam were prepared by producing polyols from different weight loadings of PEG to CG as the liquefaction solvent. The produced polyurethane foams showed densities from 0.042 to 0.08 g/cm3 and compressive strengths from 200 to 311 kPa. The foams obtained from CG/PEG based liquefaction, had more uniform and regular cell structure than foams derived from liquefaction by CG. Also with the increasing percentage of PEG to CG in liquefaction, closed cell content of the synthesized foams increased and the size of cells decreased. The thermal conductivity of the produced foams was between 0.031 and 0.040 W/m K. Foams produced from liquefaction by binary solvent had lower thermal conductivity. However all foams showed approximately similar thermal degradation curves; maximum thermal decomposition temperature was seen for the foam produced from higher weight ratio of PEG to CG in liquefaction. PU foams produced from PEG/CG based liquefaction process had improved properties over from foams derived from sawdust liquefaction by CG.
Similar content being viewed by others

Introduction
Polyurethanes are prepared by the polyaddition processes between poly isocyanates and polyols to form urethane linkages. Rigid polyurethane foam is one of the most important insulating materials used in the construction industry, refrigerators and freezers [1]. Polyurethane foams have unique characteristics the consist of adherence to materials such as steel, wood, thermosetting resins and fibers, varied density ranging 20 to 3000 kg/m3, resistance to petroleum, oils, and other nonpolar solvents [2]. PU foams were considered as structural-thermal material in the appliance and construction industry due to both their thermal insulation and mechanical properties. Polyurethane foam properties are different from foams such as polystyrene, polyolefin and phenolic foams. Today, these foams are produced from polymeric methylene diphenyl diisocyanate (polymeric MDI) and either polyether or polyester polyols that are petro-chemical derived chemicals. Increasing cost of petrochemical feed stocks, concern of future shortage in petroleum supplies and public desire for environmentally friendly green products have encouraged many research projects to explore the polymers from sustainable and renewable resources [3]. Interest in the use of lignocellulosic biomass for preparing PU foams is increasing now. The most popular methods of biomass liquefaction for production of PUF are based on the incorporation of organic solvents with suitable molecular weight and acidic or basic catalysts [4]. Although these methods take advantage of the renewable biomass, the major weakness of these processes is the use of more solvents and chemicals, as well as energy inputs to make the bio polyols [5, 6]. To reduce the production cost of polyols and thus the prevention of future commercialization efforts, the researchers have applied crude glycerol as the liquefaction solvent of lignocellulosic biomass [7, 8]. Vegetable oils or animal fats and methanol reacted with each other via trans-esterification under alkalis such as NaOH and KOH that mainly produced biodiesel as renewable fuel. The byproduct of the biodiesel production process is the unrefined crude glycerol. The produced crude glycerol consists mainly of a mixture of methanol, fatty acids/soap, salts, and alkaline catalyst residues that due to the presence of these impurities have little economic value. Recently researchers applied refined crude glycerol for liquefaction of cellulose that was successfully used to produce polyols and polyurethane foam [9]. In another study, Hu et al. [8] reported the production of bio polyol and PU foams from crude glycerol based liquefaction of soybean straw. In this research maximum liquefaction efficiency was obtained at 360 min, whereas studies on liquefaction by PEG 400/glycerol showed higher liquefaction efficiency for a lower liquefaction time [10]. The relatively lower liquefaction efficiency in the former might be explained by the use of crude glycerol versus PEG 400/glycerol chemical solvent. To combine the advantages of both the solvent in liquefaction process, the present research investigated the PUF properties produced from polyols of binary solvent (CG/PEG) based liquefaction of sawdust. The CG used in this study was based on waste vegetable oils. The recent researches applied the CG derived from a kind of waste vegetable oil for liquefaction and production of polyols. The composition of crude glycerol depends on feedstocks, processes, and post-treatments at biodiesel plants. The varying compositions of crude glycerol let to the change of the produced polyols and PU foams properties. It was hypothesized that adding chemical solvent to crude glycerol as liquefaction solvent could improve the properties of foams in comparison to foams obtained from polyols of CG based liquefaction. The use of waste vegetable oils based CG and its combination with PEG as a liquefaction solvent of biomass has not been reported so far.
Experimental and methods
Materials
The raw materials used in this study include CG based on waste vegetable oils, Poly ethylene glycol (PEG 400), polyols from CG and CG/PEG based liquefaction of sawdust. Crude glycerol was contained different amounts from impurities including free fatty acids (FFAs), methyl esters of fatty acids (FAMEs) glycerides. The Properties of crude glycerol was determined according to the procedure described in study Hu et al. [8] illustrated in Table 1. Chemicals such as polymeric diphenylmathane diisocyanate (PMDI), tin octoate and diamino bicycle octane (DABCO) as Catalyst, HCFC-141b as blowing agent for the preparation of polyurethane foam were purchased from a commercial company.
All experiments were carried out with three replicates then analyzed by SPSS software and ANOVA tests.
Experimental methods
Liquefaction of sawdust
Liquefaction process for bio polyols production was done by different weight loadings of PEG to CG as liquefaction solvent. Before being used as liquefaction solvent, crude glycerol was pretreated to remove salts, water, and methanol. The determined amount of solvent and sulfuric acid were poured in a 500-ml three-neck flask connected to condenser. The pre-weighed sawdust was added into the flask while reaction temperature reached to desired point (120–240 °C). Liquefaction was carried out under atmospheric pressure and certain temperature with constant stirring for 45–360 min. Since temperature reached up to 160 °C, the flask was heated under a constant flow of nitrogen gas. Finally for stopping process, the flask was immediately removed and quenched in tap water, after each liquefaction process, bio polyols properties were measured.
Preparation of polyurethane foams
Foaming process includes the mixture of bio polyol, catalysts, surfactant and blowing agent (water, HCFC-141b) as part A and polymeric MDI as part B. The bio polyols had the hydroxyl numbers from 250 to 300 mg KOH/g, acid numbers below 6 mg KOH/g, and viscosities from 16 to 24 Pa.s. In this study, water was as blowing agent but had limitations for both foam processing and final foam properties. Water blown foams also had a higher thermal conductivity. So, besides water, HCFC-141b was used as blowing agent due to its lower thermal conductivity and good foam surface quality [11]. A determined amount of part A solution was mixed thoroughly in a plastic cup by mechanical stirrer for 3 min under ambient temperature (Table 2). Then a definite amount of polymeric MDI (isocyanate index of 100 wt.%) was added to the components and stirred for another 2 min. After mixing, the resultant mixture was rapidly poured into an open mold and allowed to rise freely at room temperature. Foaming is an exothermic reaction due to polyurethane links formation. The PU foams were kept for 48 h at room temperature before cutting and further testing. Foam 1 as a foam obtained from polyol of sawdust liquefaction by CG and other foams (2, 3, 4) as foams obtained from polyol of liquefaction by various weight ratio of CG to PEG (90/10, 85/15, 80/20 respectively) were defined.
Characterizations
Density and open cell content
The density of PU foams were measured by ASTM D 1622–08 and the open cell contents were determined according to ASTM D6226–05.
Compressive strength of PU foams
Compressive parallel to grain strength was determined using an Instron Universal Testing system in accordance with ASTM D 1621–10 and calculated based on Eq. 1.
Where Pu is final compression stress (N/mm2), Fmax is load (N) and A is area (mm2). The rate of crosshead displacement was 2.5 mm/min with the value at 10% deformation. PU foam samples were cut into square shapes (5 cm × 5 cm × 2.5 cm) and conditioned at 25 °C and 50 ± 10% relatively humidity for at least 1 week before testing.
Scanning Electron microscopy (SEM)
Morphological studies were performed by SEM Cambridge S360. Specimens were cut by using a sharp blade and then were gold-plated with evaporated gold (in 4 nm thickness) prior to scan. The samples were analyzed at an acceleration voltage of 20 kV. The Image J software was used to analyze the obtained figures.
Fourier transform infrared spectroscopy (FTIR)
Infrared spectroscopic analysis was performed with a FT-IR spectrometer Bruker Equinox 55 to indicate any changes in the chemical structure. The foams analyzed were scraped to powder form. The FTIR spectra were carried out within the range of 4000–500 cm−1 at a resolution of 4 cm−1 after 32 scans.
Thermal properties of PU foams
TGA was performed by using a thermogravimetric analyzer Perkin Elmer Pyris 1. Experiments were conducted under nitrogen gas. The samples weighting 10–12 mg were heated from room temperature to 600 °C. The heating rate was adjusted to 10 °C min−1. Thermal conductivity was measured by Taurus TCA 200, according to ASTM C 177–97. The sample was placed between the two plates which were maintained at different temperatures during the test. The size of the samples was 200 × 200 × 20 mm.
Results and discussions
Foaming process kinetics
Table 3 shows foaming process of PU samples obtained from CG and CG/PEG based liquefaction. Foaming characterizations are typically followed by the cream time, rise time, and tack-free time [4]. At the beginning of the foaming process, the color of mixture changes because of gas bubbles formation introduced cream time. The rise time is the time required for foam to reach to its maximum height. At the tack free time the outer surface of the foam loses its stickiness. The polyol viscosity decrease let to the foam mobility and kinetic rate increase. So, lower viscosity polyols had the longest foaming times. Foam 1 (bio polyols of CG based liquefaction) having polyols with the lowest viscosity, showed the shortest cream and tack free time. On the other hand, adding PEG to CG in liquefaction process declined foaming reaction times through the decreasing polyol viscosity caused more foaming efficiency. Therefore for foams derived from liquefaction by binary solvent, the foam prepared from bio polyols of higher ratio of PEG to CG showed lower reaction times. It could be ascribed to higher reactivity of hydroxyl groups of PEG than that of synthesized bio polyols.
Density and compressive strength of PU foams
With similar formulation of foaming, synthesized foams had different densities by various polyols (Fig. 1). It was observed that adding PEG increased liquefaction efficiency subsequently more bio polyol production. With the increasing bio polyol content, the foams density increased. This may be attributed to the liquefied biomass which acts as crosslinking agent having hydroxyl groups rather than as a chain extender in this polymerization [12]. So, the foams produced from bio polyols with higher weight ratio of PEG to CG in liquefaction had higher density than foams prepared from liquefied sawdust by CG. On the other hand, bio polyols of foam 2 because of the lower reactivity in comparison to other synthesized polyols, partly act as adding weight to the foam and less participate in foaming process. A statistical analysis (ANOVA) showed that the effect of the type of polyol on density of synthesized PU foams at confidence level of 95% was significant.

Density of PU foams
The compressive strength of the PU foams was obtained from 200 to 311 kPa. The mechanical properties of PU foams depended on several parameters such as density, cross-linking density, cell geometry and blowing efficiency [13, 14]. The variation in compressive strength with foams density in Fig. 2 was illustrated. The minimum compressive strength was demonstrated at foam 1 with the lowest density. Foams density increase improved compressive strength. So foams obtained from CG/PEG based liquefaction with the higher density had more compressive strength. The similar results reported in previous studies [1, 4]. Besides density, the higher compressive strength of PU foams obtained from polyols of binary solvent based liquefaction could be attributed to higher reactivity of the produced polyols. Hu et al. [8] showed that in addition to density, the compressive strength of PU foams might be affected by factors such as biomass residues or the chemical structures of bio polyols. Having the highest density, foam 2 showed inconsistent behavior. It may be to the less reactive of some of hydroxyl groups of bio polyol of foam 2 that did not participate in forming urethane linkages. So, polyols without the increasing cross-linking density and subsequently improving the foams compressive properties, aggregated as adding weight to the foam. Statistical analysis demonstrated there was significant difference between the compressive strength with the density of foams (at confidence level of 95).

Effect of density on compressive strength of the PU foams
Water absorption of PU foams
Figure 3 shows the effect of density on the water absorption of the samples in volume percentage after soaking in water for 24 h. According to the figure, with the decreasing density the water absorption of foams increased whereas the highest water absorption was obtained for foam 1 with the lowest density. A statistical analysis (ANOVA) suggested that the effect of density on water absorption of the foam was significant in the confidence level of 98%. The cell structure and closed cell content of foams were the other effective factors on water absorption [12]. Foam 2 having higher density and larger cell size had similar water absorption to foam 3 having lower density and smaller cell size. Also Duncan’s grouping put them in one category. This phenomenon was probably stated since, unlike the larger cell size of foam 2, most of the foam cells were closed. Meanwhile, foam 4 having smaller cell size and higher closed-cell percentage, demonstrated lower water absorption than the other foams.

Effect of density on the water absorption of foams
Morphology of PU foams
Figure 4 shows scanning electron micrographs (SEM) images of polyurethane foams prepared from bio polyols of liquefied sawdust. Depending on the type of used polyol in foaming, the cell structure, cell size and closed or open cell content were varied in the synthesized foams. The average cell size was calculated from the SEM photographs (Fig. 5). The cell size of the PU foams decreased from 430 μm of foam 4 to 370 μm of foam 2 by adding PEG to CG in liquefaction process. So depending on foaming process and foaming materials the significant reduction of cell size was observed. Also the increasing ratio of PEG to CG in liquefaction decreased the cell size of foams. The results demonstrated that the higher ratio of PEG to CG as liquefaction contributed the smaller cell size of 170 μm of foam 4.

SEM micrographs of foams with various densities, foam 1 with 0.042 g/cm3 (a), foam 2 with 0.08 g/cm3 (b), foam 3 with 0.062 g/cm3 (c), foam 4 with 0.071 g/cm3 (d)

Average cell size of PU foams produced from polyol of CG and CG/PEG based liquefaction
The foam 1 having lower density showed high percentage of open cells (Fig. 6). The open cell content of PU foam decreased from 89.5% of foam 1 to around 64.3% of foam 2 because of the adding of PEG to CG as liquefaction and the bio polyols formation thereby the varying of foaming material properties. With the increasing ratio of PEG to CG as liquefaction, open cell content of foam 4 decreased more to 10.3%. The addition of chemical solvent as liquefaction process improved foams cell structure and qualify. So foam 4 compared to other foams had more uniform and regular cell structure, higher the number of cells with smaller diameter. In this relation, Xu et al. [15] indicated that with an increasing usage of PEG to polyol produced from sawdust liquefaction by glycerol, the cell structure of the foams became more regular.

Open cell content of PU foams produced from polyol of CG and CG/PEG based liquefaction
FT-IR spectra of PU foams
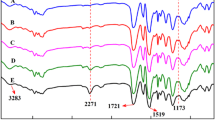
The FTIR spectra of the PU foam samples produced from biopolyols are shown in Fig. 7. Spectra for all the foams formulation did not show significant differences among them. In all samples the formation of urethane linkage was confirmed by FTIR and showed approximately similar signals. The absorption band at 3320 cm−1 is related to N-H groups which are making hydrogen bonds stretching [16]. As can be seen, more band width at foam 2 indicated the presence of the more free OH groups compared to the other foams. With regard to constant ratio of polyol to isocyanate in the foams formulation, this phenomenon can be attributed to less reactivity of polyols and isocyanates during foaming of foam 2, resulting in less activity hydroxyl groups left in the system. The OC=O vibration at 1730 cm−1and the CO-NH vibration at 1600 cm−1 confirms the formation of urethane linkage. Also, the intensities of transmission band at 1530 cm−1 are assigned to the N-H stretching and bending vibrations. Other strong absorption bands at 2932 and 2894 cm−1 are CH2 bridges. The weak peak of unreacted NCO group was at about 2270 cm−1. The band at 1900.0 cm−1 represents the stretching vibration of free carbonyl groups of urethane linkages and the bands at 1510 and 1600 are derived from the aromatic rings of lignin.

FTIR spectra of PU foams
Thermal analysis of PU foams
Figure 8 (A) shows thermal conductivity of PU foams. The thermal conductivity depends on the foam density, cell size, cell structure (percentage of closed and open cell) and the thermal conductivity of the blowing agent [17]. The value of thermal conductivity is observed ranging from 0.031 to 0.040 W/m K at foams with densities of 0.08 to 0.042 g/cm3. Foam 1 with the lowest density and more open cell content had the maximum thermal conductivity due to higher heat transfer in foam cells. However, by increasing the density of the foam the thermal conductivity decreased foam 2 showed different thermal behaviors. This evidence contributed to the bigger pore diameter and thereby higher thermal conductivity. Then having the high density, well-defined cell wall, smaller cell size and the lower open cell content, foam 4 demonstrated the lowest thermal conductivity. Because the considerable amount of air can be trapped that led to an increased passive insulation.

Thermal conductivity (a) and TGA curves (b) of PU foams
Polyurethanes were considered thermally unstable because of the existence of urethane bonds, so TGA measurements were carried out to obtain information on the thermal stability of the foams. The thermal degradation depends on the nature of substituent on the isocyanate and polyol. Figure 8 (B) depicts the TGA of PU foams. In the TGA studies, it was found that, polyurethane degraded in two weight loss stages. The first stage decomposition (5% weight loss of the samples) can be contributed to the decomposition of pyranose rings and isocyanate which usually starts between 150 and 220 °C [18, 19]. One study reported that PU foams were thermally stable at 191.9 °C [20]. The second stage decomposition (50% weight loss) occurred at 400–500 °C as a marker for structural decomposition of foams [21] and also the decomposition of lignin and other difficulty parts. The onset of degradation (5% weight loss) of foams in this study occurred at around 212 °C of foam 1 and 217, 226, 237 °C of foams 2, 3 and 4 respectively. So depending on the type of polyols used in produced foams, the degradation temperature was different. The reason for the increasing of decomposition temperature of the foams produced from CG/PEG based liquefaction may be seen as the result of the existence of polyethylene glycol. Adding PEG to CG lead to an increase in liquefaction efficiency subsequently more bio polyols production. Increasing the percent of bio polyol in the produced foams may cause branching and more cross linking which require more thermal energy to initiate chain movements [22, 23]. Xu et al. [15] indicated that by adding 30% of PEG as high molecular petroleum based polyols to bio-polyols thereby the formation of three dimensional cross-linked polyurethane groups the first stage decomposition temperature of foams increased. The onset of the second stage degradation (about 50% weight loss) occurred at approximately 393 °C for all foams. Previous researches indicated that the second degradation observed at 389 and 400 °C resulted in depolymerization of polyol components such as derivatives of cellulose or lignin [24,25,26].
Conclusion
The biopolyols obtained from liquefied sawdust by CG/PEG had better capability of the PU foams production than CG based liquefaction, most likely due to the presence of chemical solvent resulting in higher liquefaction. Scanning Electron Micrograph showed more uniform and regular cell structure together with higher closed cell area of foam 4, resulting in the lowest water absorption. The mechanical and thermal properties of foams depended on the kind of polyol applied in foaming. Considering the shortage of petroleum supplies and public desire for environmentally friendly green products, the results confirmed that behavior of foams obtained from bio polyols was similar to that of foams based on petrochemical polyols and can probably be used to replace them in similar usages.
References
Cao XD, Habibi Y, Lucia LA (2009) One-pot polymerization, surface grafting, and processing of waterborne polyurethane-cellulose nanocrystal nanocomposites. Mater Chem 19:7137–7145
Ashida K (2007) Polyurethane and related foams: chemistry and technology. CRC press, Boca Raton, FL, 2. Chapter 2:85–88
Fan H, Tekeei A, Suppes GJ, Hsieh FH (2012) Properties of bio based rigid polyurethane foams reinforced with fillers: microspheres and nanoclay. Int J Polym Sci 1:1–8
Yao Y, Yoshioka M, Shiraishi N (1995) Rigid polyurethane foams from liquefaction mixture of wood and starch. Mokuzai Gakkaishi 41:659–668 33
Hu S, Luo X, Li Y (2014) Polyols and polyurethanes from the liquefaction of lignocellulosic biomass. ChemSusChem 7:66–72
Chen F, Lu Z (2009) Liquefaction of wheat straw and preparation of rigid polyurethane foam from the liquefaction products. Appl Polym Sci 111:508–516 16
Hu S, Li Y (2014) Two-step sequential liquefaction of lignocellulosic biomass by crude glycerol for the production of polyols and polyurethane foams. Bioresour Technol 161:410–415
Hu S, Wan C, Li Y (2012) Production and characterization of biopolyols and polyurethane foams from crude glycerol based liquefaction of soybean straw. Bioresour Technol 103:227–233
Kosmela P, Hejna A, Formela K, Haponiuk JK, Piszczyk L (2016) Biopolyols obtained via crude glycerol-based liquefaction of cellulose: their structural, rheological and thermal character. Cellulose 23:2929–2942
Wang H, Chen HZ (2007) A novel method of utilizing the biomass resource. Rapid liquefaction of wheat straw and preparation of biodegradable polyurethane foam (PUF). J Chin Inst Chem Eng 38:95–102
Jung HC, Ryu HC, Kim WN, Lee YB, Choe KH, Kim SB (2001) Properties of rigid polyurethane foams blown by HCFC 141B and distilled water. J. Appl Polym Sci 81:486–493
Thirumal M, Khastgir D, Singha NK, Manjunath BS, Naik YK (2008) Effect of foam density on the properties of water blown rigid polyurethane foam. J Appl Polym Sci 108:1810–1817
Chuayjuljit S, Maungchareon A, Saravari O (2010) Preparation and properties of palm oilbased rigid polyurethane foams. J Reinf Plast Compos 29(2):218–225
Lim L, Kim SK, Kim BK (2008) Effects of silicon surfactant in rigid polyurethane foams. Polym Lett 2:194–200
Xu J, Jiang J, Hse C, Shupe TF (2013) Preparation of polyurethane foams using fractionated products in liquefied wood. Appl Polym Sci:1–7
Goddard RJ, Cooper SL (1995) Polyurethane cationomers with pendant trimethyl ammonium groups. 1. Fourier transform infrared temperature studies. Macromolecules 28:1390–1400
Ahern A, Verbist G, Weaire R, Phelan HJ. (2005) Fleurent, Colloids Surf A: Phys Chem Eng Aspects 6: 263–275
Lee SH, Teramoto Y, Shiraishi N (2002) Biodegradable polyurethane foam from liquefied paper and its thermal stability, biodegradability, and genotoxicity. J Appl Polym Sci 83:1482–1489
Wieweg R, Hchtlen A (1966) Polyurethane in Kunstoff- Handbuch. Carl Hanser Verlag: Munchen, Germany, band VII 17
Hepburn C (1991) Polyurethane elastomers, 2nd ed. London: Elsevier. Appl Sci 16
Gomez EF, Luo X, Li C, Michel FC, Li Y (2014) Biodegradability of crude glycerol-based polyurethane foams during composting, anaerobic digestion and soil incubation. Polym Degrad Stab 102:195–203
Hakim AAA, Nassar M, Emam A, Sultan M (2011) Preparation and characterization of rigid polyurethane foam prepared from sugar-cane bagasse polyol. Mater Chem Phys 129:301–307
Kurimoto Y, Takeda M, Koizumi S, Yamauchi S, Doi S, Tamura T (2000) Mechanical properties of polyurethane films prepared from liquefied wood with polymeric MDI. Bioresour Technol 74:151–157
Zhao Y, Yan N, Feng M (2012) Polyurethane foams derived from liquefied mountain pine beetle-infested barks. J Appl Polym Sci 123:2849–2858
Cinelli P, Anguillesi I, Lazzeri A (2013) Green synthesis of flexible polyurethane foams from liquefied lignin. Euro. Polym J 49:1174–1184
Bernardini J, Cinelli P, Anguillesi I, Coltelli MB, Lazzeri A (2015) Flexible polyurethane foams green production employing lignin or oxypropylated lignin. Eur Polym J 64:147–152
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rastegarfar, N., Behrooz, R. & Barikani, M. Characterization of polyurethane foams prepared from liquefied sawdust by crude glycerol and polyethylene glycol. J Polym Res 25, 154 (2018). https://doi.org/10.1007/s10965-018-1516-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-018-1516-4