
Abstract
The steady-state absorption and fluorescence, as well as the time-resolved fluorescence properties of bisdemethoxycurcumin dissolved in several solvents differing in polarity and H-bonding capability were measured. The photodegradation quantum yield of the compound in acetonitrile and methanol was determined. The bisdemethoxycurcumin decay mechanisms from the S 1 state were discussed and compared with those of curcumin. The differences in S 1 dynamics observed between bisdemethoxy-curcumin and curcumin could be ascribed to a difference in H-bond acceptor/donor properties of the phenolic OH and a difference in strength of the intramolecular H-bond in the keto-enol moiety within the two molecules.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
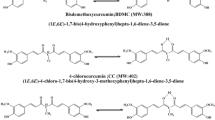
The yellow orange pigment derived from the rhizome of the plant Curcuma longa L., (turmeric) consists of three diarylheptanoids: curcumin (CURC), demethoxy-curcumin (DMC), and bisdemethoxy-curcumin (bisDMC) (Fig. 1). The demethoxy- and bidemethoxy compounds can amount to nearly 40% of what is known as commercially available curcumin [1]. In some turmeric extracts bisdemethoxycurcumin is even shown to be the major constituent [2]. Curcumin is by far the most investigated curcuminoid, although some comparative studies on the naturally occurring curcuminoids have been performed. A constantly increasing number of publications have shown that curcumin displays notable effects not only as an anti-inflammatory compound [3–5] and a potent antioxidant [4, 5], but also as a chemopreventive [6, 7] and chemotherapeutic [8, 9] agent. Moreover, it seems to have a potential in the treatment of Alzheimer disease [10] and cystic fibrosis [11], as well as being considered a model substance for the treatment of HIV-infections [12–14] and as an immune-stimulating agent [12]. CURC was shown to be significantly more effective than DMC [15, 16], which in turn is more effective than bisDMC, as an antioxidant. The molecular mechanisms underlying the antioxidant effects are still not fully understood, but it is apparent that, even if the major antioxidant activity has been associated to electron withdrawal from the keto-enol group to the phenolic hydroxyl moieties, the phenolic methoxy substituents also play a significant role [15–18]. Moreover, CURC is both a powerful metal chelating agent and an efficient radical scavenger. Metal chelation occurs at the central keto-enol group, and is strongly affected by the enol proton mobility and acidity [19]. Scavenging effects are connected with the formation of phenoxyl radicals [19] that are formed by deprotonation of the phenolic hydroxyl groups [20]. In spite of the fact that the methoxy phenolic substituents are not directly involved in either metal chelation or radical scavenging, DMC has been proven to be less effective than CURC, and bisDMC to be almost inactive, with respect to both of these biologically relevant activities [21]. Finally, the three compounds vary dramatically in their ability to suppress the nuclear cell factor κB activation in vitro [22]: CURC is by far the most efficient suppressor while bisDMC is almost inert. This is relevant with respect to their chemopreventive and chemotherapeutic potentials.

Naturally occurring curcuminoids
Combination with light induces additional biological activities in both CURC and its analogues [1, 23–25]. Upon excitation to the S 1-state CURC becomes phototoxic to bacteria [24–28] and to mammalian cells, both cancerous [23] and healthy [1], via mechanisms that are still to be elucidated [23, 29, 30]. Determination of the S 1 dynamics and identification of the deactivation pathways of biologically active curcumin analogues, including bisDMC, may be relevant in assessing the molecular mechanism that a tentative photosensitizing drug should be prone to undergo. Of course, any mechanism leading to photochemical degradation, which has been reported to be significant for CURC in certain environments [31, 32], would be particularly undesirable.
The present work describes the ground- and excited-singlet state characteristics of bisDMC compared to CURC. Absorption, steady-state fluorescence and fluorescence decay measurements were performed on pure, synthetic bisDMC dissolved in several solvents differing in polarity and H-bonding capability. Photodegradation quantum yield of bisDMC in acetonitrile and methanol was also determined. Assessment of the S 1-decay mechanism and the dependence on both the molecular substituents and the environmental conditions is a relevant step towards full exploitation of the photosensitizing potential of curcumin analogues and to a rational design of synthetic curcuminoids featuring enhanced biological activity and photostability. In this work the most relevant decay mechanisms of bisDMC are identified by taking advantage of previous studies on both CURC [33] and dicinnamoylmethane (DCMeth) [30].
Materials and Methods
Chemicals and Sample Preparation
CURC and bisDMC were synthesized as previously described [33, 34]. All the solvents were ≥99.5% pure and were used as received, except ethyl acetate, which was dried over sodium sulfate. Samples in organic solvents were prepared the same day they were used for measurements.
Absorption and Fluorescence Spectra, Fluorescence Quantum Yield
The UV-VIS absorption spectra were measured by an UV-2401 PC UV-VIS recording spectrophotometer (Shimadzu, Tokyo, Japan).
Steady-state fluorescence measurements were carried out with the PTI modular Fluorescence System (PTI, London, Ontario, Canada) described in [33]. The samples thermostated at 25.0 ± 0.1 °C were excited at 420 nm, which is the wavelength of the laser used as the excitation source in the time-resolved fluorescence measurements. The system was equipped with a software (Felix™ for Windows) performing automatic correction of the acquired spectra with regard to the spectral response of both the excitation lamp and the detector.
Fluorescence quantum yields were determined from the spectrum integrated fluorescence by using, as a reference value, that of quinine sulfate in 0.05 M H2SO4 excited at its 344 nm absorption peak: ΦRef = 0.51 [35]. The calculated quantum yields were corrected for differences in peak absorbance and in refractive index of the solvents (obtained from the product specification). The reported values are calculated as the average of three parallels, with errors given by the maximum spread between the experimental data. Determination of the fluorescence quantum yields has been pursued such as in [35].
Fluorescence-decay Measurements
The fluorescence decays were detected by Time-Correlated Single-Photon Counting (TCSPC). The used TCSPC setup has ~30 ps time resolution (full width at half maximum of the detected excitation pulse) and is fully described elsewhere [30, 33, 36]. The fluorescence of the solutions, which were contained in a 1 × 1 cm2 fluorimeter quartz cuvette, was excited at 420 nm by the second harmonic (SH) output of a mode locked Ti:sapphire laser (Tiger-ps SHG, Time Bandwidth Products, Zurich, CH). The fluorescence at λ > 500 nm was collected at 90° to the excitation beam through a cut-off filter (LL-500, Corion, Holliston, MA) by a 20X microscope objective and focused onto the sensitive area of a PDM50 single-photon avalanche diode (Micro-photon-devices, Bolzano, IT). All fluorescence decays were collected up to 10,000 peak counts in strict single photon regime by suitably attenuating the excitation beam with neutral-density filters. The maximum absorbance of the solutions at the excitation wavelength was 0.05.
The fluorescence decay data were fitted, without deconvolving the system pulse response, to either single, double, or triple exponentials above a constant background, by minimizing the chi-square value through a Levenberg-Marquardt algorithm. For each decay, the number of exponential components was established by adding, one by one, exponential components to the fitting function until the fitting routine converged to yield two components of equal time constant. In Fig. 2 one of the decays obtained for bisDMC in DMSO is plotted, together with the corresponding fitting curve and residuals. The decay of bisDMC in DMSO was chosen as a paradigm of our ability of resolving decay components of negligibly low amplitude, such as the long-lived component with relative amplitude <1%. In Fig. 2 the best fitting curve obtained with a single-exponential decay model is also plotted. It is apparent that the quality of the two-exponential decay fit is superior.

Upper panel: fluorescence decay (dots) of bisDMC in DMSO, single exponential fit (dark-gray solid line) and double exponential fit (light-gray solid line); lower panel: residuals corresponding to the single exponential (dark full dots) and double exponential (light empty circles) fits of above
Six decay curves were acquired for each sample: the means of the values obtained from the fits, with errors given by the standard deviations, were assumed as the time constant, τ i , and initial amplitude, A i , of the i-th decay component, being the A i values calculated at the peak channel of the experimental data.
Photodegradation Quantum Yields
The photodegradation quantum yield of bisDMC in selected solvents was measured using the potassium ferrioxalate chemical actinometer [37]. The samples were irradiated by using a monochromator (Applied Photophysics Ltd., f 3.4, 900 W xenon arc lamp) operated with a bandwidth of 20 nm at the selected wavelength. The number of sample molecules reacted per unit time and per unit volume as a function of exposure time was quantified by means of reversed phase HPLC. The separation was performed on a 150 × 3.9 mm Nova Pak® C18 column (Waters, Milford, USA). The mobile phase was a mixture of acetonitrile and 0.5% citric acid buffer, adjusted to pH 3 with KOH. The samples were detected at 350 nm. This detection wavelength was selected in order to reveal tentative degradation products. The chromatic system consisted of a LC-9A pump, a SP D-10A UV-VIS detector, a SIL-10 DV auto sampler and a C-R3A integrator (Shimadzu, Japan).
Results
The solvents used in the present study were divided into the following categories: non-polar (cyclohexane), polar weakly H-bonding (chloroform, ethyl acetate, acetone, acetonitrile), strong H-bond acceptors (dimethylformamide, DMFA, and dimethylsulfoxide, DMSO), and alcohols (isopropanol, ethanol, methanol, and ethylene glycol). The dielectric constant ε was adopted as the indicator of the solvent polarity. The acidity parameter α and the basicity parameter β, were used as the indicators of the solvent H-bond donating and accepting properties, respectively [38]. The above-mentioned solvent properties are summarized in Table 1. Note that the alcohols display both H-bond donating and accepting properties.
Steady-state Absorption and Emission
The absorption maxima, λ Abs , of bisDMC in the different solvents are shown in Table 2. In all solvents except cyclohexane the absorption spectra were broad and essentially structureless and the absorption maximum was blue-shifted with respect to that of CURC [33]. Some representative spectra are reported in Fig. 3a. In cyclohexane, bisDMC was rather insoluble, and only a noisy spectrum could be recorded even for a saturated solution. The main absorption band was observed in the UV, with peaks at 348 nm and 362 nm, respectively. The spectrum also displayed a shoulder around 380 nm. A second absorption band above 400 nm was barely detectable. The situation was opposite for CURC in cyclohexane [33] where the main absorption band was indentified above 400 nm and the near UV band was minor. For both compounds a systematic red shift was observed when changing from cyclohexane to solvents with higher dielectric constants and from weaker to stronger H-bonding solvents of comparable polarity. However, there was no linear correlation between either ε, α or β and λ Abs . Conversely, λ Abs remained constant within one solvent category (see Table 2 for bisDMC and [33] for CURC), but was markedly different from one category to another.

a) Absorption and b) fluorescence emission spectra of bisDMC in cyclohexane (solid black line), acetone (solid grey line), DMSO (full dots) and methanol (empty circles). In panel b) inset the emission spectrum of bisDMC in cyclohexane obtained upon excitation at 420 nm (full line) is compared with that obtained upon excitation at 350 nm (triangles)
The fluorescence spectra of bisDMC are broad and essentially structureless in all solvents except cyclohexane, in which three emission maxima were identified. Some representative spectra are displayed in Fig. 3b. Further, in cyclohexane bisDMC fluorescence was excited efficiently at 420 nm, and the spectral lineshape was similar to that obtained exciting the sample at the UV absorption peak (see inset of Fig. 3b). The emission maxima, λ Fl , of bisDMC in the selected solvents are listed in Table 2. The bisDMC fluorescence spectra were blue-shifted compared to the CURC spectra [33], so that the Stokes shifts were generally smaller for bisDMC than for CURC in all solvents.
The fluorescence quantum yield, Φ Fl , of bisDMC was generally low (Table 2). The lowest value was obtained in DMSO. This is different from CURC where the lowest value was obtained in cyclohexane [33]. Moreover, CURC had higher Φ Fl values in the polar, weakly H-bonding solvents, ranging from Φ Fl = 0.094 in chloroform to Φ Fl = 0.174 in acetone, than in the strongly H-bonding solvents (from Φ Fl = 0.022 in ethylene glycol to Φ Fl = 0.041 in DMFA). The only exception was isopropanol (Φ Fl = 0.114) which seemed to have scarce affinity to form inter-molecular H-bonds with CURC [33]. On the contrary, bisDMC exhibits the maximum Φ Fl in ethanol (Φ Fl = 0.143, see Table 2). The excited state of bisDMC seems to be stabilized by the non-bonding electron pair of the phenolic OH oxygen, which is given to the ring acting as charge-transfer donor to the excited state. This transfer is facilitated by interaction of bisDMC with hydrogen bond accepting solvents, as was demonstrated by the relatively higher Φ Fl of bisDMC in DMFA and the alcohols except methanol. The latter is however, a poorer hydrogen bond accepting solvent than, e.g., ethanol (see [38] and Table 1). This might partly explain the difference in quantum yield between the two alcohols. The stabilizing effect of H-bond accepting solvents is further emphasized by comparison of strongly H-bond accepting solvents and polar weakly H-bonding solvents of similar polarity; e.g. isopropanol (ε ≈ 20, Φ Fl = 0.109 ± 0.006) and acetone (ε ≈ 21, Φ Fl = 0.065 ± 0.004). In the case of the less stabilizing alcohol (i.e. methanol) the fluorescence quantum yield was comparable to the weakly H-bonding solvent of similar polarity acetonitrile (Table 2).
In CURC, the presence of a methoxy group next to the phenolic group seems to make the OH group more susceptible to interactions with H-bond donating solvents. The non-bonding electron pair on the phenolic oxygen atom is possibly engaged in intermolecular H-bonding instead of being given to the ring, thereby leading to less stabilization of the excited state. This is supported by the generally lower fluorescence quantum yields of CURC in alcohols (except isopropanol) with respect to both CURC in weakly H-bonding solvents and bisDMC in alcohols [33]
Photodegradation Quantum Yield
The quantum yield of photodegradation, Φ Degr , of bisDMC was measured in both acetonitrile (Φ Degr = 0.080 ± 0.013) and methanol (Φ Degr = 0.061 ± 0.004). It is comparable to that measured for CURC in the same solvents (acetonitrile: Φ Degr = 0.061 ± 0.011; methanol: 0.021 ± 0.010 ) [33]. \( {\Phi_{{Degr}}} \cong {\Phi_{{Fl}}} \) in both acetonitrile and methanol in the case of bisDMC, while for CURC, \( {\Phi_{{Degr}}} \cong 0.4{\Phi_{{Fl}}} \) in acetonitrile and \( {\Phi_{{Degr}}} \cong 0.75{\Phi_{{Fl}}} \) in methanol. Thus, in neither of these solvents photochemical decomposition is the most relevant deactivation mechanism of S 1 for these compounds.
Excited-state Dynamics
The experimental decay distributions were fitted to either single, double, or triple exponential decay functions as explained in Materials and Methods in order to derive the decay times, τ i , and the relative initial amplitudes, A i . Average fluorescence lifetimes τ av were calculated for each solvent as \( {\tau_{{av}}} = \sum\nolimits_i {{{{{\tau_i}{A_i}}} \left/ {{\sum\nolimits_i {{A_i}} }} \right.}} \) and are reported in Table 2. The radiative (k Fl ) and non-radiative (k NR ) rate constants were calculated from the Φ Fl and τ av values listed in Table 2:
The obtained values are reported in the same Table. The values of k NR were substantially higher than the corresponding k Fl values in all solvents. Thus, it can be concluded that the deactivation of S 1 occurs predominately via non-radiative pathways. This is consistent with previous observations on CURC [33]. Three exponential components were resolved in the fluorescence decay for bisDMC dissolved in cyclohexane, but the compound essentially displayed single-exponential decays in all the other solvents (traces of a second, long-lived component was detectable only in DMSO). This was different from CURC where three decay components were identified in cyclohexane, a single one in polar, weakly H-bonding solvents and two in strongly H-bonding solvents except isopropanol, in which a single exponential decay was observed. The k Fl values for bisDMC were very similar to those calculated for CURC in all the solvents except DMSO and methanol, in which they were considerably lower (\( {k_{{Fl}}} = 0.04 \times {10^9}\,{{\hbox{s}}^{{ - 1}}} \) compared to \( {k_{{Fl}}} = 0.17 \times {10^9}\,{{\hbox{s}}^{{ - 1}}} \) in DMSO and \( {k_{{Fl}}} = 0.09 \times {10^9}\,{{\hbox{s}}^{{ - 1}}} \) compared to \( {k_{{Fl}}} = 0.17 \times {10^9}\,{{\hbox{s}}^{{ - 1}}} \) in MeOH), and cyclohexane, in which the k Fl of bisDMC was higher than that of CURC (\( {k_{{Fl}}} = 0.16 \times {10^9}\,{{\hbox{s}}^{{ - 1}}} \) compared to \( {k_{{Fl}}} = 0.059 \times {10^9}\,{{\hbox{s}}^{{ - 1}}} \)). The k Fl values did not correlate with the solvent properties that were examined, similarly to the CURC data. The k NR values measured for bisDMC spanned from \( {k_{{NR}}} = 1.09 \times {10^9}{ }{{\hbox{s}}^{{ - 1}}} \) in ethylene glycol to \( {k_{{NR}}} = 4.78 \times {10^9}\,{{\hbox{s}}^{{ - 1}}} \) in cyclohexane. They were generally higher in weakly H-bonding solvents than in strongly H-bonding solvents. Moreover, in the polar weakly H-bonding solvents the k NR values calculated for bisDMC were higher than the corresponding values calculated for CURC [33], while the opposite occurred in strongly H-bonding solvents.
Discussion
In reference [33] we proposed a model to explain the data on the CURC decay from the S 1 state. The following radiationless decay mechanisms were considered to concur with fluorescence emission: (a) direct excited-state intra-molecular proton transfer (ESIPT) from the enol to the keto group, which was postulated to be the fastest possible non-radiative decay mechanism [39, 40] and to take place only if the intra-molecular keto-enol H-bond (KEHB) illustrated in Fig. 4a (see closed cis enol conformer) were formed; (b) reketonization [41, 42]; (c) charge/energy transfer to the solvent molecules [43]; (d) slow, solvent-rearrangement moderated ESIPT. The latter occurs in case a trans enol or open cis enol molecule isomerizes to the closed cis enol conformer while in the S 1 state, and then decays to S 0 by means of ESIPT [33]. The different enol conformers are shown in Fig. 4a. On the basis of the photodegradation results, photodegradation was excluded to be the driving force in the S 1 decay. It was postulated that CURC in solution at room temperature is essentially present in its enol conformers, in agreement with previous studies [29, 44–47]. The H-bonded closed cis enol structure is dominant in non-polar environments, while either the open cis enol or the trans enol conformers, which cannot form the KEHB, are dominant in polar weakly-H-bonding and polar strongly-H-bonding solvents, respectively [47]. Tiny amounts of the minimally polar trans (anti) diketo conformer (see Fig. 4b) can be found in non-polar environments [44]. In [30] we have elucidated the very different multi-exponential fluorescence decays of DCMeth, a curcumin analogue lacking both the methoxy and hydroxyl phenolic substituents of CURC, by invoking the concurrence of the same mechanisms outlined above. We only made the further hypothesis that tiny amounts of the very polar cis diketo conformer (see Fig. 4b) of DCMeth exists in polar environments. This conformer has been shown to be unstable for CURC due to either steric interactions [44] or unfavorable dipole-dipole alignment [48].

a) Enol conformers and b) diketo conformers of the investigated curcuminoids. For R-structures see Fig. 1
The results of the above-mentioned works can be used as a guideline in ascribing a decay mechanism to each of the exponential components of the bisDMC fluorescence decays detected in the various solvents. The quantum yields and decay data suggest that bisDMC and CURC decay through the same deactivation pathways in cyclohexane. Moreover, like CURC, bisDMC displayed minimum Stokes shifts in cyclohexane, which is consistent with formation of KEHB which prevents out-of-plane vibrations. Hence, in analogy to CURC, the three decay components observed for bisDMC in cyclohexane are ascribed to: (a) bisDMC molecules initially in the closed cis enol conformer that are excited to S 1 without undergoing cis-trans isomerization and very rapidly decay to S 0 by direct ESIPT according to the scheme in Fig. 5 (\( {\tau_1} = 107\pm 1\,{\hbox{ps}} \), with relative amplitude 0.81); (b) molecules initially in the closed cis enol conformer, that undergo cis-trans isomerization upon excitation to S 1 [41] and de-excitation by reketonization (\( {\tau_1} = 363\pm 6\,{\hbox{ps}} \), with relative amplitude 0.17); (c) molecules in the trans (anti) diketo conformer (\( {\tau_1} = 2702\pm 49\,{\hbox{ps}} \), with relative amplitude 0.02). However, the bisDMC average decay time was much longer than that of CURC and each of the τ i values (i = 1,2,3) was higher for bisDMC than for CURC. This indicates that in the case of bisDMC both direct ESIPT and reketonization occur on slower time scales. The crystal structure of solvated bisDMC shows that (at least in polar environment) the enol proton is tightly bound to the enol oxygen, and remains quite distant from the keto oxygen [49, 50]. This is very different from CURC, in which the enol proton is very mobile and equally distributed between the two oxygens of the keto-enol moiety [51]. If this also pertains to non-polar environments, it indicates that the KEHB must be quite loose, and consequently ESIPT rather slow: this is probably the reason why τ 1 in cyclohexane was approximately twice as long for bisDMC than for CURC. Moreover, slower reketonization rates compared to CURC are expected and observed for bisDMC in cyclohexane.

Scheme of the excited-state intra-molecular proton transfer (ESIPT) undergone by the H-bonded cis enol conformer, see Fig. 4a
Due to its weakness, KEHB is perturbed by both weakly- and strongly H-bonding polar solvents, in which ESIPT is not observed the case of bisDMC, as previously observed in CURC. The single exponential decays obtained for bisDMC in the polar, weakly H-bonding solvents indicate that, similarly to what was observed for CURC, the only relevant non-radiative decay mechanism in such solvents is solvent-rearrangement moderated ESIPT. However, bisDMC has faster decay and lower quantum yield than CURC (see Table 2). Thus, solvent rearrangement seems to be faster in bisDMC, indicating an overall S 1 excited state molecular dipole moment lower than that of CURC. Indeed, the S 1 dipole moment of CURC has been reported to be exceptionally high, and, in particular much higher that that of other curcuminoids, including DCMeth [52].
On the contrary, in the strongly H-bonding solvents bisDMC (Table 2) had slower decay and higher fluorescence quantum yield than CURC [33]. The lack of a decay component with time constant of the same order of magnitude as that of the shortest CURC decay component suggests that deactivation of the bisDMC excited state by inter-molecular H-bond formation does not occur. The single decay time measured for bisDMC in strongly H-bonding solvents (see τ av in Table 2) was in the range of those ascribed to decay by means of solvent rearrangement moderated ESIPT for both CURC and DCMeth [30, 33]. The value was typically longer than that measured for CURC and similar to that measured for DCMeth. The generally slower decay by solvent rearrangement moderated ESIPT of bisDMC compared to CURC, which indicates tighter inter-molecular H-bonding of bisDMC keto-enol moiety, is independent on whether the solvent displays H-bond donating or accepting properties.
This is not contradictory to the intermolecular H-bond mediated charge decay by charge/energy transfer observed for CURC, but not for bisDMC, as it has been shown [48, 52] that both deprotonation and electron transfer occur at CURC phenolic hydroxyl moieties, and not at the enol/keto groups. As we have already discussed above, the removal of methoxy groups in bisDMC affects the interaction of bisDMC phenyl OH with H-bonding solvents. Namely, H-bonding with H-bond donors is weakened and transfer of the lone pair electrons to the phenyl ring is favoured upon interaction with H-bond acceptors in the excited state. In DMSO, which is the most polar solvent, a residual decay component was detected for bisDMC, with τ 2 = 4463 ps and A 2 < 1% (see Fig. 2). This time constant was very similar to the longest ones observed for DCMeth in solvents with dielectric constant >25 [30]. We thus ascribe this component to the presence of tiny amounts of the cis-diketo conformer of bisDMC in DMSO.
Conclusion
The excited-state dynamics of the naturally occurring curcuminoid bisDMC was investigated by means of steady-state absorption and fluorescence, fluorescence decay, and photodegradation quantum yield measurements performed on the compound dissolved in several solvents differing in polarity and H-bonding capability. The bisDMC decay mechanisms from the S 1 state were elucidated and compared with those of CURC. The main differences observed between the S 1 dynamics of the two compounds are:
-
i.
Slower rates of the decays by direct excited-state intra-molecular proton transfer and by reketonization for bisDMC as compared to CURC in non-polar environment;
-
ii.
Faster rate of decay by solvent-rearrangement moderated excited-state intra-molecular proton transfer for bisDMC as compared to CURC in polar, weakly H-bonding environment;
-
iii.
Slower rate of decay by solvent-rearrangement moderated excited-state intra-molecular proton transfer for bisDMC as compared to CURC in strongly H-bonding environment;
-
iv.
Lack of deactivation through inter-molecular charge/energy transfer interactions in strongly H-bonding environment
The above differences between bisDMC and CURC could be ascribed to either the difference in H-bond acceptor/donor properties of the phenolic OH, or the difference in strength of the intramolecular H-bond in the keto-enol moiety. BisDMC undergoes slower or faster deactivation from the singlet state compared to CURC depending on the environment. The photodecomposition seems to be slightly more extensive than what is reported for CURC under the same conditions. Consequently, modification of the CURC molecule by removal of the methoxy substituents does not necessarily improve photostability or photosensitizing potential of the sensitizer. This is consistent with our latest results on the antibacterial phototoxic effect of bisDMC [28].
References
Bruzell E, Morisbak E, Tønnesen HH (2005) Studies on curcumin and curcuminoids. XXIX. Photoinduced cytotoxicity of curcumin in selected aqueous preparations. Photochem Photobiol Sci 4:523–530
Tønnesen HH, Karlsen J, Adhikary SR, Pandey R (1989) Studies on curcumin and curcuminoids. XVII. Variation in the content of curcuminoids in Curcuma Longa L. From Nepal during one season. Z Lebensm-Unters Forsch 189:116–118
Mukhopadhyay A, Basu N, Ghatak N, Gujzal PK (1982) Anti-inflammatory and irritant activities of curcumin analogs in rats. Agents Actions 12:508–515
Srimal RC, Dhawan BN (1973) Pharmacology of di-ferulyl methane (curcumin), a non-steroidal anti-inflammatory agent. J Pharm Pharmacol 25:447–452
Rao T, Basu N, Ghatak N, Gujral PK (1982) Anti-inflammatory activity of curcumin analogs. Indian J Med Res 75:574–578
Khafif A, Schantz SP, Chou TC, Edelstein D, Sacks PG (1998) Quantification of chemopreventive synergism between epigallocatechin-3-gallate and curcumin in normal, premalignant and malignant human oral epithelial cells. Carcinogenesis 19:419–424
Leu TH, Maa MC (2002) The molecular mechanisms for the antitumorigenic effect of curcumin. Curr Med Chem 2:357–370
Woo J, Kim Y, Choi Y, Kim D, Lee K, Bae JH, Chang DS, Jeong YJ, Lee YH, Park J, Kwon TK (2003) Molecular mechanisms of curcumin-induced cyclotoxicity: induction of apoptosis through generation of reactive oxygen species, down-regulation of Bcl-XL and IAP, the release of cytochrome c and inhibition of Akt. Carcinogenesis 24:1199–1208
Moos PJ, Edes K, Mullally J, Fitzpatrick J (2004) Curcumin impairs tumor suppressor p53 function in colon cancer cells. Carcinogenesis 9:1611–1617
Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen P, Kayed R, Glabe CG, Frautschy SA, Cole GM (2004) Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 280:5892–5901
Egan ME, Pearson M, Weiner SA, Rajendarn V, Rubin D, Glochner-Pagel J, Canney S, Du K, Lukacs GL, Caplan MF (2004) Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 304:600–602
Aggrawal BB, Sundaram C, Malani N, Ichikawa H (2007) Curcumin: the Indian solid gold. Adv Exp Med Biol 595:1–75
Mazumder A, Neamati N, Sunder S, Schulz J, Perez H, Aich E, Pommier Y (1997) Curcumin analogs with altered potencies against HIV-1 integrase as probes for biochemical mechanisms of drug action. J Med Chem 40:3057–3063
Sui Z, Salto R, Li J, Craik C, Ortiz de Montellano PR (1993) Inhibition of the HIV-2 proteases by curcumin and curcumin boron complexes. Bioorg Med Chem 1:415–422
Cai YZ, Sun M, Xing J, Luo Q, Corke H (2006) Structure-radical scavenging activity relationships of phenolic compounds from traditional Chinese medical plants. Life Sci 78:2872–2888
Jayaprakasha GK, Rao LJ, Sakariah KK (2006) Antioxidant activities of curcumin, demethoxycurcumin and bisdemethoxycurcumin. Food Chem 98:720–724
Chen WF, Deng SL, Zhou B, Yang L, Liu ZL (2006) Curcumin and its analogues as potent inhibitors of low density lipoprotein oxidation: H-atom abstraction from the phenolic groups and possible involvement of the 4-hydroxy-3-methoxyphenil groups. Free Radic Biol Méd 40:526–535
Somparn P, Phisalaphong C, Nakornchai S, Unchern S, Morales NP (2007) Comparative antioxidant activities of curcumin and its demethoxy and hydrogenated derivatives. Biol Pharm Bull 30:74–78
Schaich KM, Fisher C, King R (1994) In: Ho CT, Osava T, Huang MT, Gosen RT (eds) Phytochemicals for cancer prevention II, ACS Symp. Ser. 547. American Chemical Society, Washington, pp 204–221
Gorman AA, Hamblett I, Srinivasan VS, Wood PD (1994) Curcumin-derived transients: a pulsed laser and pulse radiolysis study. Photochem Photobiol 59:389–398
Dairam A, Limson JL, Watkins GM, Antunes E, Daya S (2007) Curcuminoids, curcumin, and demethoxycurcumin reduce lead-induced memory deficits in male wistar rats. J Agric Food Chem 55:1039–1044
Sandur SK, Pandey MK, Sung B, Ahn KS, Murakami A, Sethi G, Limtrakul P, Badmaev V, Aggarwal BB (2007) Curcumin, demethoxycurcumin, bisdemethoxycurcumin, tetrahydrocurcumin and turmerones differentially regulate anti-inflammatory anti-proliferative responses through a ROS-independent mechanism. Carcinogenesis 28:1765–1773
Dahl TA, Bilski P, Reszka KJ, Chignell CF (1994) Photocytotoxicity of curcumin. Photochem Photobiol 59:290–294
Dahl TA, Mcgowan WM, Shand MA, Srinivasan VS (1989) Photokilling of bacteria by the natural dye curcumin. Arch Microbiol 151:183–185
Tonnesen HH, de Vries H, Karlsen J, van Henegouwen GB (1987) Studies on curcumin and curcuminoids IX: investigation of the photobiological activity of curcumin using bacterial indicator systems. J Pharm Sci 76:371–373
Haukvik T, Bruzell E, Kristensen S, Tønnesen HH (2009) Photokilling of bacteria by curcumin in different aqueous preparations. Studies on curcumin and curcuminoids. XXXVII. Pharmazie 64:666–673
Haukvik T, Bruzell E, Kristensen S, Tønnesen HH (in press) Photokilling of bacteria by curcumin in selected polyethylene glycol 400 (PEG 400) preparations. Studies on curcumin and curcuminoids XLI. Pharmazie
Haukvik T, Bruzell E, Kristensen S, Tønnesen HH (in press) A screening for antibacterial phototoxic effects of curcumin derivatives. Studies on curcumin and curcuminoids. XLIII. Pharmazie
Chignell CF, Bilski P, Reszka KJ, Motton AG, Sik RH, Dahl TA (1994) Spectral and photochemical properties of curcumin. Photochem Photobiol 59:295–302
Nardo L, Andreoni A, Bondani M, Màsson M, Tønnesen HH (2009) Studies on curcumin and curcuminoids XXXIV. Photophysical properties of a symmetrical, non-substituted curcumin analogue. J Photochem Photobiol B: Biol 97:77–86
Tønnesen HH, Karlsen J, van Henegouwen GB (1986) Studies on curcumin and curcuminoids. VIII. Photochemical stability of curcumin. Z Lebensm-Unters Forsch 183:116–122
Sundaryono A, Nourmamode A, Gardrat C, Grelier S, Bravic G, Chasseau D, Castellan A (2003) Studies on the photochemistry of 1, 7-dipehnyl-1, 6-heptadiene-3, 5-dione, a non-phenolic curcuminoid model. Photochem Photobiol Sci 2:914–920
Nardo L, Paderno R, Andreoni A, Haukvik T, Màsson M, Tønnesen HH (2008) Studies on curcumin and curcuminoids XXXII. Role of H-bond formation in the photoreactivity of curcumin. Spectroscopy 22:187–198
Tomren MA, Màsson M, Loftsson T, Tønnesen HH (2007) Studies on curcumin and curcuminoids. XXXI. Symmetric and asymmetric curcuminoids: stability, activity and complexation with cyclodextrin. Int J Pharm 338:27–34
Velapoldi R, Tønnesen HH (2004) Corrected fluorescence spectra and quantum yields for a series of compounds in the visible spectral region. J Fluoresc 14:465–472
Nardo L, Bondani M, Andreoni A (2008) DNA-ligand binding mode discrimination by characterizing fluorescence resonance energy transfer through lifetime measurements with picosecond resolution. Photochem Photobiol 84:101–110
Moore DE (2004) In: Tønnesen HH (ed) Photostability of drugs and drug formulations. CRC Press, Boca Raton, pp 49-53
Kamlet MJ, Abboud JLM, Abraham MH, Taft RW (1983) Linear solvation energy relationships. 23. A comprehensive collection of the solvatochromic parameters, π*, α, and β, and some methods for simplifying the generalized solvatochromic equation. J Org Chem 48:2877–2887
Emsley J (1984) In: Clarke MJ, Goodenough JB, Ibers JA, Jørgensen CK, Mingos DMP, Neilands JB, Reinen D, Sadler PJ, Weiss R, Williams RJP (eds) Structure and bonding. Springer Verlag, Berlin, pp 148–191
Strandjord AJG, Courtney SH, Friedrich DM, Barbara PF (1983) Excited-state dynamics of 3-hydroxyflavone. J Phys Chem 87:1125–1133
Weedon AC (1990) In: Rappoport Z (ed) The chemistry of Enols. John Wiley & Sons, New York, pp 591–638
Nikolov P, Fratev F, Petkov I, Markov P (1981) Dimer fluorescence of some β-dicarbonyl compounds. Chem Phys Lett 83:170–173
Gilli G, Bertolasi V (1990) In: Rappoport Z (ed) The chemistry of Enols. John Wiley & Sons, New York, pp 713–764
Balasubramanian K (2006) Molecular orbital basis for yellow curry spice curcumin’s prevention of Alzheimer’s disease. J Agric Food Chem 54:3512–3520
Pedersen U, Rasmussen PB, Lawesson SO (1985) Synthesis of naturally occurring curcuminoids and related compounds. Liebigs Ann Chem 8:1557–1569
Ortica F, Rodgers MAJ (2001) A laser flash photolysis study of curcumin in dioxane-water mixtures. Photochem Photobiol 74:745–751
Toullec J (1990) In: Rappoport Z (ed) The chemistry of Enols. John Wiley & Sons, New York, pp 324–398
Wright JS (2002) Predicting the antioxidant activity of curcumin and curcuminoids. J Mol Struct, Theochem 591:207–217
Tonnesen HH, Karlsen J, Mostad A, Pedersen U, Rasmussen PB, Lawesson SO (1983) Structural studies of curcuminoids. II. Crystal structure of 1, 7-Bis(4-hydroxyphenyl)-1, 6-heptadiene-3, 5-dione – Methanol complex. Acta Chem Scand B 37:179–185
Tonnesen HH, Karlsen J, Mostad A, Pedersen U, Rasmussen PB, Lawesson SO (1988) Structural studies of curcuminoids. IV. Crystal structure of 1, 7-Bis(4-hydroxyphenyl)-1, 6-heptadiene-3, 5-dione Hydrate. Acta Chem Scand B 42:23–27
Tønnesen HH, Karlsen J, Mostad A (1982) Structural studies of curcuminoids. I. The crystal structure of curcumin. Acta Chem Scand B 36:475–479
Galasso V, Kovac B, Modelli A, Ottaviani MF, Pichierri F (2008) Spectroscopic and theoretical study of the electronic structure of curcumin and related fragment molecules. J Phys Chem A 112:2331–2338
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Nardo, L., Andreoni, A., Masson, M. et al. Studies on Curcumin and Curcuminoids. XXXIX. Photophysical Properties of Bisdemethoxycurcumin. J Fluoresc 21, 627–635 (2011). https://doi.org/10.1007/s10895-010-0750-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-010-0750-x