
Abstract
[Cu(NH2CH2CH2NEt2){B6O7(OH)6}]·5H2O (1) and [Cu(NH3)2{B6O7(OH)6}]·2H2O (2) have been obtained as crystalline materials from aqueous solutions of Dynamic Combinatorial Libraries (DCLs) originating from B(OH)3 and appropriate Cu(II) amine complexes. These two Cu/O/B clusters are formed through self-assembly processes and contain tridentate hexaborate(2−)-κ3O ligands. Both compounds have been characterized by TGA/DSC, magnetic susceptibility measurements, IR spectroscopy, and single-crystal XRD studies. The intermolecular H-bond interactions between neighbouring hexaborate units are implicated in their templated synthesis. Compound 2 is a coordination polymer and stabilization is also gained through formation of an additional O–Cu coordinate bond. Steric congestion in 1 blocks formation of this bond resulting in insular complexes.
Similar content being viewed by others

Introduction
Boron is always found in nature combined with oxygen and these compounds are generally known as ‘borates’ [1,2,3,4,5,6,7]. There are more than two hundred known borate minerals, more than a hundred of which have been structurally characterized, and additionally there are several hundred known synthetic borate salts [4, 6]. Borate anions are structural diverse with insular anions through to anionic polymeric chains, layers, clusters, or networks, all well-represented and new structural types are always of interest. Two general synthetic approaches have been used to prepare new polyborate materials and these are either self-assembly (with templating cations) from aqueous solution or high temperature solvothermal methods [7]. Polyborate salts obtained from aqueous solution are usually comprised of discrete, insular anions, partnered by the templating cations, whereas solvothermal methods often lead to more condensed polymeric structures. These templated self-assembly processes are possible since it is well known that B(OH)3 exists in aqueous solution as a Dynamic Combinatorial Library (DCL) [8, 9] of rapidly interconverting B(OH)3, [B(OH)4]− and polyborate anions [10, 11]. The templating cation interacts with this DCL of borate anions and crystallization of energetically favourable salts occur. Stoichiometry, pH, and energetics associated with crystal packing, steric congestion, hydrogen bonding, and coordination bond formation all have important roles to play in these self-assembly processes [12, 13]. However, pentaborate(1−) salts are often formed in these systems since this anion is well suited to forming a wide variety of supramolecular crystalline lattices which are held together by strong H-bond interactions [14,15,16,17]. In our search for novel polyborate anions we have adopted a strategy of using transition-metal complexes as templating cations [18,19,20,21,22,23]. Transition-metal complexes have been chosen since they offer unique opportunities to affect the self-assembly processes in polyborate salt formation and in turn, this will lead to a better understanding of crystal-engineering in general. Following this strategy we have recently reported the synthesis of several salts containing isolated polyborate anions partnered with transition-metal complexes and have described the synthesis and structures of two novel isolated polyborate anions containing the heptaborate(3-) [19] and octaborate(2−) [18] anions. Copper(II) borates [20, 22, 24, 25] are an interesting subclass of metal polyborates and ammonium copper(II) borates have been used as fungicides [4, 6] and have other potential commercial applications. Copper(II) complexes are generally labile [26] and in aqueous solution will generate a DCL of cations to interact with the DCL equilibrium mixture of polyborate anions adding a further dimension to self-assembly synthons present in the reactions solutions. We have also recently reported some novel species containing Cu(II) centers [20, 22] and in this manuscript we describe the self-assembly of two new Cu(II) compounds containing hexaborate(2−) anions and their solid-state structures. The hexaborate(2−) borate anion observed in these Cu(II) compounds is drawn schematically in Fig. 1.

Schematic structures of the hexaborate(2−) anion, [B6O7(OH)6]2−, found in 1 and 2
Results and Discussion
Synthesis and General Characterization
The two new Cu(II) hexaborates [Cu(NH2CH2CH2NEt2){B6O7(OH)6}]·5H2O (1) and [Cu(NH3)2{B6O7(OH)6}]·2H2O (2) have been prepared through self-assembly processes from aqueous solutions originally containing appropriate templating Cu(II) amine complexes and B(OH)3 and Ba(OH)2 (Scheme 1). The Ba(OH)2 is added to the reaction mixture to convert the sulphate salt to the hydroxide salt by removal (precipitation) of BaSO4. Products 1 and 2 crystallize from the solution, after several days at room temperature, in moderate yields. Schematic drawings of the coordination compounds in 1 and 2 are shown in Fig. 2.

Synthesis of copper(II) hexaborate(2−) complexes 1 and 2. (Reactions were performed in aqueous solution, deen = N,N-diethylethylenediame)

Schematic structures of coordination compounds present in [Cu(NH2CH2CH2NEt2){B6O7(OH)6}]·5H2O (1) and [Cu(NH3)2{B6O7(OH)6}]·2H2O (2)
Compounds 1 and 2 were characterized by thermal studies (TGA/DSC), magnetic susceptibility measurements, elemental analyses, IR spectroscopy and single-crystal XRD studies. Elemental analyses data were consistent with their single-crystal structures confirming that crystals chosen for XRD studies were representative of the bulk sample.
The thermal TGA/DSC data obtained for 1 and 2 (see supplementary material) were also consistent with the structures determined by single-crystal X-ray diffractions studies (see below) and were interpreted in terms of three-step decomposition processes. For 1 this involved loss of interstitial water, further loss of water with cross-condensation of hexaborate(2−) ligands, and finally oxidation of the organic ligand. For 2 this involved loss of interstitial water, loss of coordinated ammonia, and finally further loss of water with cross-condensation of hexaborate(2−) ligands. The final residues for both compounds had a mass consistent with that expected from the stoichiometry for anhydrous CuB6O10 (=CuO·3B2O3). This behavior has been previously observed for other related Cu(II) complex polyborate salts [20, 22, 27,28,29].
Room temperature magnetic susceptibility measurements were obtained for 1 and 2 and both compounds were paramagnetic with χm values slightly lower than those usually seen for d9 Cu(II) centres and one unpaired electron. IR spectra can be used to help identify polyborate species since B–O stretches are generally characteristically strong and are observed in diagnostic areas [30]. In particular, 1 and 2 both show a bands at ~ 960(m) cm−1 and 808(s) cm−1 which have been tentatively assigned as diagnostic of the hexaborate(2−) anion [20, 30]. Compounds 1 and 2 are insoluble in organic solvents and dissolve, with decomposition, in aqueous solution. NMR was attempted for these solutions but it was not possible to observe 1H and 13C spectra for the deen ligand in 1, possibly due to its continued coordination to the paramagnetic Cu(II) centre. 11B spectra of 1 and 2 both showed just a single signal at + 16.3 and + 14.4 ppm, respectively. This signal for 2 is in accord with that calculated [16] for a boron/charge ratio of three for a hexaborate(2−) system, assuming fast B(OH)3/[B(OH)4]− exchange [11] and associated with the pH of the solution. The signal for 1 is slightly more downfield than expected.
X-Ray Structural Studies
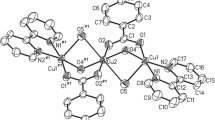
Compound 1 (Fig. 3) is formulated as a neutral molecule containing a tridentate hexaborate(2−)-κ3O ligand coordinated to a Cu(II) centre which is also coordinated by a bidentate H2NCH2CH2NEt2 ligand. Other transition metal complexes containing the hexaborate (2−) ligands include [Me2NHCH2CH2NHMe2][Zn{B6O7(OH)6}2]·2H2O [31], [C4N2H12][Co{B6O7(OH)6}2]·6H2O [32], [Zn(en){B6O7(OH)6}]·2H2O [33], [Zn(pn){B6O7(OH)6}]0.1.5H2O [33] and two hexaborate/Cu(II) complexes: [Cu(NH2CH2CH2NMe2){B6O7(OH)6}]·4H2O [20] and [Cu(Me2NCH2CH2NMe2)·{B6O7(OH)6}]·6H2O [20]. Compound 1 and these reported copper(II) hexaborate(2−) complexes all contain relatively bulky substituted diamine ligands (cf.2 below). The Cu(II) centre in 1 has a distorted square-based pyramidal geometry (τ-index [34] = 0.25) with a hexaborate(2−) O donor (O13) axial. The Cu1–O13 distance (2.1941(10) Å) is significantly longer than the other two Cu–O distances of 1.9968(10) Å (Cu1–O11) and 1.9762(10) Å (Cu1–O12). The N atom bearing the two Et groups (N2) has a bondlength to Cu1 of 2.0663(12) Å whereas the Cu1–N1 bond-length is shorter at 1.9838(12) Å. The bond lengths and bond angles within the hexaborate(2−) unit are similar to those observed in 1 and to those previously reported for structures containing hexaborate(2−) ligands [20, 31,32,33]. Compound 1 also contains five interstitial H2O molecules and these molecules H-bond to one another, the hexaborate(2−) units and an amino H-atom. The hexaborate(2−) anion has six potential H-bond donor sites which are associated with either ‘coordinated’ or ‘peripheral’ hydroxyl groups. Two of the three coordinated hydroxyl group hydrogen atoms of the hexaborate(2−) dianion, (H11 and H12) and one of the amino hydrogen (H1A) atoms H-bonds to water molecules. The third coordinated hydroxyl hydrogen atom (H13) group is involved in an unusual reciprocal R 22 (12) interaction (Etter [35] nomenclature) with a neighbouring hexaborate(2−) unit involving O10* as an acceptor. Two of the peripheral hydroxyl groups hydrogens (H8 and H9) are involved in R 22 (8) interactions with neighboring hexaborate(2−) units whereas as the third hydroxyl hydrogen (H10) is a donor to a H2O molecule (O21). The other amino hydrogen atom (H1B) is also H-bonded to an adjacent hexaborate(2−) unit. Multiple intermolecular R 22 (8) H-bond interactions have been shown to be influential in stabilizing solid-state structures of self-assembled pentaborate(1−) salts [14,15,16,17] and these and the more unusual H-bond interaction displayed in 1 clearly stabilize its formation from the DCL moieties present in the reaction solution. Details of these H-bond interactions are available in the supplementary information.

Crystal structure of [Cu(NH2CH2CH2NEt2){B6O7(OH)6}]·5H2O (1) showing heavy atom numbering scheme (ellipsoids at 50%)
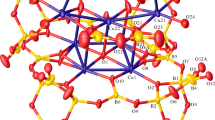
Compound 2 (Fig. 4) is formulated as a 1-D coordination polymer with the repeating formula [Cu(NH3)2{B6O7(OH)6}]·2H2O. Compound 2 contain a tridentate hexaborate(2−)-κ3O ligand coordinated to the Cu(II) centre, as also observed for 1. The Cu(II) centre also has a pair of cis NH3 ligands and completes its coordination sphere by bonding with O9* of ‘adjacent’ heptaborate(2−) ligand by formation of a 1-D coordination polymer (Fig. 5). O9* is trans to O13 and the overall coordination geometry about the Cu(II) centre is an elongated tetragonal distorted octahedron (T6 = 0.78 [36]). The Cu1–O13 and Cu1–O9* distances of 2.5557(10) Å and 2.5469(10) Å are significantly longer than the Cu1–O11 and Cu1O12 distances of 1.9896(10) Å and 1.9630(10) Å. The Cu–N distances average 2.0056(10) Å. The hexaborate(2−) unit has bond lengths and bond angles within the expected ranges associated with other hexaborate(2−) compounds [20, 31,32,33]. Compound 2 also contains two interstitial H2O molecules and one of which is disordered over two sites (Fig. 4). There are many potential H-bond donor sites in 2 and these are involved in multiple intrachain or interchain interactions.

Crystal structure of the monomeric unit of [Cu(NH3)2{B6O7(OH)6}]·2H2O (2) showing heavy atom numbering scheme (ellipsoids at 50%). The water molecules associated with O23 and O22 are disordered with S.O.F of 0.53(3) and 0.47(2), respectively

A 1-D polyborate/Cu(II) chain runs through the structure of 2 with the [B6O7(OH)6]2− ligand bridging two Cu(II) centres (κ3O and κ1O coordination modes)
All six hydroxyl hydrogen atoms of the hexaborate(2−) ligand are involved with H-bond donor interactions. The three coordinated hydroxyl hydrogen atoms link with two adjacent hexaborate(2−) units (H11 and H12) and H13 with a H2O molecule (O21). The three peripheral hydroxyl hydrogen atoms all link with neighbouring hexaborate(2−) units with hydrogens H9, H10 and H12 involved in familiar R 22 (8) interactions. O11H11 and O13H13 are linked in a unique R 33 (10) ring involving a neigbouring hexaborate(2−) unit (O10* acceptor) and H2O (O21 acceptor). O8H8 H-bonds to O13* on an adjacent hexaborate(2−) unit as part of a unique R 44 (10) ring that includes two hexaborate(2−) ligands and two H2O molecules. As noted above, multiple intermolecular R 22 (8) H-bond interactions have been shown to be influential in stabilizing solid-state structures of self-assembled polyborate salts from the DCL moieties present in the reaction solution, and the unique H-bond interactions present in 2 are likely to be strong and will contribute further to the stabilization of the structure. The structure of 2 is further stabilized by formation of an additional Cu–O coordinate bond as a 1-D coordination polymer chain. This is facilitated by sterically less demanding NH3 ligands around the Cu(II) centre in 2 and the involvement of the six amino hydrogens of the coordinated NH3 ligands with H-bond donation to two H2O (O22/23) or four hexaborate(2−) units. Details of these H-bond interactions are available in the supplementary information.
Conclusion
Two new Cu(II)/hexaborate(2−) clusters have been obtained as crystallisation materials from aqueous solutions of DCLs originating from B(OH)3 and appropriate Cu(II) amine complexes. The two compounds are formed through self-assembly processes and are structurally quite similar and are neutral species that contain hexaborate(2−) anions coordinated (tridentate) to Cu(II) centres. There are numerous energetically favourable H-bond interactions in both 1 and 2 with neighbouring moieties and this myriad of H-bond interactions are implicated in the their templated synthesis. Compound 2 is a 1-D coordination polymer and there is additional stabilization in this compound gained from the formation of an O–Cu coordinate bond. This is unavailable to 1 since there is more steric congestion around the Cu(II) centre with the diethylamino group on the diamine ligand effectively blocking the formation of this bond.
Experimental
General
All chemicals were commercially obtained. TGA and DSC analysis was performed between 10 and 800 °C (in air) on an SDT Q600 V4.1 Build 59 instrument using Al2O3 crucibles, with a ramp temperature rate of 10 °C min−1. Magnetic susceptibility measurements were performed on a Johnson–Matthey magnetic susceptibility balance at room temperature (18 °C). Fourier transform Infrared spectra (FTIR) were obtained as KBr pellets on a Perkin–Elmer 100 FTIR spectrometer over 450–4000 cm−1. NMR spectra were obtained on a Bruker Avance 400 spectrometer and reported in ppm with positive chemical shifts (d) to high frequency (downfield) of TMS (1H, 13C) and BF3.OEt2 (11B). Single-crystal X-ray crystallography was carried out at the EPSRC National Crystallography service at the University of Southampton. CHN analysis data were obtained from OEA laboratories Ltd in Callington, Cornwall.
Synthesis, Spectroscopic and Analytical Data for 1
N,N-Diethylethylenediamine (2.8 mL, 20 mmol) was added to an aqueous solution of copper(II) sulphate pentahydrate (2.5 g, 10 mmol) in distilled water (10 mL). The reaction mixture was stirred at room temperature for 60 min, and then a solution of barium hydroxide octahydrate (3.16 g, 10 mmol) in water (20 mL) was added. This mixture was then stirred for a further 30 min and then filtered. A solution of boric acid (6.2 g, 100 mmol) in water (30 mL) was then added to the filtrate. The resulting solution was stirred at room temperature for 3 h before its volume was reduced to 20 mL using a rotary evaporator. This final solution was distributed over a few small vials and left for 14 days to yield blue crystals of [Cu(NH2CH2CH2NEt2){B6O7(OH)6}] 5H2O (1) (1.9 g, 35%). M.p. > 300 °C (dec.). χm = 6.8 × 10−4 cm3 mol−1. C6H32B6CuN2O18. Anal. Calc.: C = 13.1%, H = 5.9%, N = 5.1%. Found: C = 13.2%, H = 5.8%, N = 5.0%. 11B/ppm: 16.3. IR (KBr/cm−1): 3432(s), 1655(m), 1435(s), 1347(s), 1167(m), 1133(s), 1080(s), 951(m), 895(m), 808(s). TGA: 100–190 °C, loss of five interstitial H2O 17.9% (16.4% calc.); 190–280 °C, condensation of hexaborate which loss of three further H2O 29.2% (26.3% calc.); 250–650 °C, oxidation of organic content 50.1% (47.5% calc.); residue CuB6O10 49.9% (52.5% calc.).
Synthesis, Spectroscopic and Analytical Data for 2
A solution of ammonium hydroxide (2.2 mL, 20 mmol, 35%) was added to an aqueous solution of copper(II) sulphate pentahydrate (1.25 g, 5 mmol) in distilled water (5 mL). The reaction mixture was stirred at room temperature for 60 min, and then a solution of barium hydroxide octahydrate (1.58 g, 5 mmol) in water (10 mL) was added. The mixture was then stirred for a further 30 min and filtered. A solution of boric acid (3.09 g, 50 mmol) in water (10 mL) was added to the filtrate and the solution was stirred at room temperature for 3 h before its volume was reduced to 15 mL by using a rotary evaporator. The final solution was distributed over a few small vials and left for 14 days to yield dark blue crystals of [Cu(NH3)2{B6O7(OH)6}]·2H2O (2) (0.94 g, 45%). M.p. 297 °C (dec.). χm = 5.8 × 10−4 cm3 mol−1. B6CuH16N2O15. Anal. Calc.: H = 3.9%, N = 6.8%. Found: H = 4.2%, N = 6.8%. 11B/ppm: 14.4. IR (KBr/cm−1): 3364(s), 1369(s), 1277(m), 1239(m), 1128(s), 1088(s), 966(m), 809(s). TGA: 100–190 °C, loss of two interstitial H2O 8.9% (7.8% calc.); 190–280 °C, loss of two NH3 molecules 18% (16% calc.); 250–650 °C, condensation of hexaborates which loss of three further H2O 30.1% (28.3% calc.); residue CuB6O10 69.9% (71.7% calc.).
X-Ray Crystallography
Suitable crystals of 1 and 2 were selected and mounted on a MITIGEN holder in perfluoroether oil on a Rigaku FRE + equipped with VHF Varimax confocal mirrors an AFC12 goniometer and HG Saturn 724 + detector diffractometer. Crystals were kept at T = 100(2) K during data collection. Cell determination and data collection was carried out using CrystalClear [37] for 1 or CrysAlisPro [38] for 2, with data reduction, cell refinement and absorption correction carried out using CryAlisPro [38]. Using Olex2 [39], the structures were solved with the ShelXT [40] structure solution program, using the Intrinsic Phasing solution method. The models were refined with version 2014/7 of ShelXL [41] using Least Squares minimisation.
Crystal data. 1
C6H32B6CuN2O18, Mr = 548.73, monoclinic, P21/n (No. 14), a = 9.5275(2) Å, b = 15.1317(3) Å, c = 15.7826(3) Å, β = 96.294(2)°, α = γ = 90°, V = 2261.62(8) Å3, T = 100(2) K, Z = 4, Z’ = 1, μ(MoKα) = 1.049 mm−1, of 33,041 reflections measured, 5181 were unique (Rint = 0.0251) used in all calculations. The final wR2 was 0.0704 (all data) and R1 was 0.0250 (I > 2(I)). 2: B6CuH16N2O15, Mr = 412.55, triclinic, P-1 (No. 2), a = 7.4470(2) Å, b = 7.9290(2) Å, c = 13.2276(3) Å, α = 95.649(2)°, β = 93.320(2)°, γ = 113.485(2)°, V = 708.83(3) Å3, T = 100(2) K, Z = 2, Z’ = 1, μ(MoKα) = 1.624, of 15,983 reflections measured, 3245 were unique (Rint = 0.0188) and used in all calculations. The final wR2 was 0.0568 (all data) and R1 was 0.0203 (I > 2(I)).
Associated content
CCDC1890710 (1) and CCDC1890711 (2) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Crystallographic data for 1 and 2 are also available as supplementary material together with 11B NMR, IR, TGA data.
References
C. L. Christ and J. R. Clark (1977). Phys. Chem. Miner. 2, 59.
J. B. Farmer (1982). Adv. Inorg. Chem. Radiochem. 25, 187.
G. Heller (1986). Top. Curr. Chem. 131, 39.
D. M. Schubert (2003). Struct. Bond. 105, 1.
E. L. Belokonova (2005). Crystallography Rev. 11, 151.
D. M. Schubert Kirk-Othmer Encyclopedia of Chemical Technology, 5th ed (Wiley, New York, 2011), p. 1.
M. A. Beckett (2016). Coord. Chem. Rev. 323, 2.
P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders, and O. Otto (2006). Chem. Rev. 106, 3652.
J. Sola, M. Lafuente, J. Atcher, and I. Alfonso (2014). Chem. Commun. 50, 4564.
J. L. Anderson, E. M. Eyring, and M. P. Whittaker (1964). J. Phys. Chem. 68, 1128.
C. G. Salentine (1983). Inorg. Chem. 22, 3920.
G. R. Desiraju (1995). Angew. Chem. Int. Ed. Engl. 34, 2311.
J. D. Dunitz and A. Gavezzotti (2012). Cryst. Growth Des. 12, 5873.
M. Wiebcke, C. C. Freyhardt, J. Felsche, and G. Engelhardt (1993). Z. Naturforsch. 48b, 978.
M. Z. Visi, C. B. Knobler, J. J. Owen, M. I. Khan, and D. M. Schubert (2006). Cryst. Growth Des. 6, 538.
M. A. Beckett, S. J. Coles, R. A. Davies, P. N. Horton, and C. L. Jones (2015). Dalton Trans. 44, 7032.
M. A. Beckett, C. C. Bland, P. N. Horton, M. B. Hursthouse, and K. S. Varma (2007). J. Organomet. Chem. 692, 2832.
M. A. Altahan, M. A. Beckett, S. J. Coles, and P. N. Horton (2015). Inorg. Chem. 54, 412.
M. A. Altahan, M. A. Beckett, S. J. Coles, and P. N. Horton (2015). Inorg. Chem. Commun. 59, 95.
M. A. Altahan, M. A. Beckett, S. J. Coles, and P. N. Horton (2017). Polyhedron 135, 247.
M. A. Altahan, M. A. Beckett, S. J. Coles, and P. N. Horton (2018). Pure Appl. Chem. 90, 625.
M. A. Altahan, M. A. Beckett, S. J. Coles, and P. N. Horton (2018). J. Clust. Sci. 29, 1337.
M. A. Altahan, M. A. Beckett, S. J. Coles, and P. N. Horton (2016). Phosphorus, Sulfur Silicon Relat. Elem. 191, 572.
H. Behm (1983). Acta Cryst. C39, 20.
H. H.-Y. Sung, M.-M. Wu, and I. D. Williams (2000). Inorg. Chem. Commun. 3, 401.
H. Taube (1952). Chem. Rev. 50, 69.
L. Zheng, J. Zhang, and Z. Liu (2009). Chin. J. Chem. 27, 494.
Z.-H. Liu, J.-J. Zhang, and W.-J. Zhang (2006). Inorg Chim. Acta 51A, 519.
Y. Yang, Y. Wang, J. Zhu, R.-B. Liu, J. Xu, and C.-G. Meng (2011). Inorg. Chim. Acta 376, 401.
J. Li, X. S. Xia, and S. Gao (1995). Spectrochim. Acta 51A, 519.
S. Natarajan, W. Klein, M. Panthoefer, L. V. Wuellen, and M. Jansen (2003). Z. Anorg. Allg. Chem. 629, 959.
N. Jemai, S. Rzaigui, and S. Akriche (2014). Acta Cryst. E70, m167.
M. A. Altahan, M. A. Beckett, S. J. Coles, and P. N. Horton (2019). Crystals 8, 470.
A. W. Addison, T. N. Rao, J. Reedik, J. van Rin, and G. C. Verschoor (1984). J. Chem. Soc Dalton Trans. 7, 1349.
M. C. Etter (1990). Acc. Chem. Res. 23, 120.
B. J. Hathaway and P. G. Hodgson (1973). J. Inorg. Nucl. Radiochem. 35, 4071.
CrystalClear (Rigaku Corporation, The Woodlands, 2008-2014).
CrystAlisPro (Software System, Rigaku Oxford Diffraction 2018).
O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, and H. Puschmann (2009). J. Appl. Cryst. C27, 3.
G. M. Sheldrick (2015). Acta Crystallogr. A 71, 3.
G. M. Sheldrick (2015). Acta Crystallogr. C 27, 3.
Acknowledgements
We thank the EPSRC for use of the NCS X-ray crystallographic service (Southampton).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Altahan, M.A., Beckett, M.A., Coles, S.J. et al. Copper(2+) Complexes of Hydroxyoxidoborates. Synthesis and Characterization of Two Clusters Containing the Hexaborate(2−) Ligand: [Cu(NH2CH2CH2NEt2){B6O7(OH)6}]·5H2O and [Cu(NH3)2{B6O7(OH)6}]·2H2O. J Clust Sci 30, 599–605 (2019). https://doi.org/10.1007/s10876-019-01519-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-019-01519-6