
Abstract
Highly porous carbon aerogels with carbon structures additives may be attractive materials for energy storage devices. Traditional resorcinol–formaldehyde-based carbon aerogels modified with graphene, graphene oxide and CNT were compared with respect to their morphology and capacitive properties as electric double layer capacitors. Materials were prepared using drying in supercritical acetone instead of commonly used carbon dioxide. Acetone was not only used for solvent exchange but was also expelled medium at supercritical conditions. This allowed to limit materials shrinkage and enhance specific capacity that in case of CNT-modified carbon aerogel was as high as 326 F/g i.e. more than 4 times bigger than for parent carbon aerogel. The specific surface area increased from 123 to 629 m2/g. Enhancement of the specific capacity was also found for gels modified with graphene and graphene oxide. The reason of such a behavior was a difference in structure and pore size distribution of additives.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Carbon aerogels are unique and relatively new class of compounds considered for energy storage application. This is caused by their main properties i.e. high surface area, low weight, tunable properties and easy synthesis. Requirements that energy storage materials should meet are among others high specific capacity and low cost. According to the charge storage mechanism two groups of materials can be distinguished, namely pseudocapacitors and electric double layer capacitors (EDLC). Pseudocapacitors store energy by means of redox reactions between electrode material and electrolyte (Faradaic reactions) while EDLC by a charge accumulated on the solid–liquid interphase (non-Faradaic reactions). Both types have pros and cons but the key role in EDLC plays high surface area. This criteria is met by carbon aerogels that offer developed pores structure, high porosity and very low weight. Changing precursors, process parameters and additives causes tunable properties of the final product.
History of aerogels dates back to 1931 when Steven Kistler from the College of the Pacific in Stockton, California invented the first aerogel based on silica gel with liquid expelled by a gas [1]. First organic aerogel synthesis is attributed to Pekala from the Lawrence Livermore National Laboratory in Berkeley, who made polycondensation of resorcinol with formaldehyde under alkali conditions. As a result clusters of functionalized polymers were obtained. Material was subsequently dried in a supercritical conditions producing low density carbon aerogel [2]. Process is composed of several stages: gelation, aging, solvent exchange, drying and pyrolysis. During gelation reaction resorcinol at first reacts with formaldehyde under alkaline or acid pH through CH2 bridges, this resorcinol derivatives condense into clusters and finally, crosslink through CH2OH groups into gel [3]. Cluster formation is strongly dependent on pH, temperature and reagents concentration. Base catalyst is responsible for deprotonation of hydroxymethylated resorcinol, leading to a very reactive o-quinone methide intermediate formation. A gelation reaction was schematically presented in a Fig. 1.

Scheme of the gelation process [2]
Gelled material, known as hydrogel, is then left for aging, for several days at around 85 °C. This time is required for curing material that is observed by color change from yellowish to deep red. Then solvent extraction with fresh acetone, repeated several times, is required for residual water removal. Acetone is a convenient drying solvent at ambient conditions because of its low surface tension and low boiling point that reduce hydrogel shrinkage [4]. Finally, supercritical drying is used to expel solvent from a material and fill the space with carbon dioxide and retain porous structure. If the solvent exchanged hydrogel is traditionally dried the capillary pressure within material is increased during solvent evaporation that may totally collapse porous gel structure producing another type of gel material called, xerogel [5]. Material pyrolysis may cause some shrinkage and mass loss but its main target is to improve electric conductivity. As obtained carbon aerogels may be applied in hydrogen storage, electrical energy storage, desalination, fuel cells and catalysis [6]. To enhance physical and electrical properties of carbon aerogels and make them useful in a broad range of activities some additives such as graphene or CNT’s may be involved in their structure [7, 8].
2 Experimental details
2.1 GO preparation
Graphite oxide (GO) was synthesized by a modified Staudenmaier’s method [9]. Briefly 20 g of commercial graphite powder (Fisher Scientific) was magnetically mixed with 350 mL 95–97% sulfuric acid (Acros Organics) and 180 mL 100% fuming nitric acid (Sigma-Aldrich) in a 1 L round-bottomed flask equipped with a reflux and thermometer and ice-cooled at the temperature close to 273 K. After an hour 220 g potassium chlorate was slowly added and reaction mixture was mixed for 330 h. The greenish GO slurry was transferred to a 10 L beaker and diluted with deionized water. Obtained GO powder was dispersed in water and ultrasonicated into graphene oxide.
2.2 Graphene
Several methods of graphene synthesis can be distinguished i.e. mechanical cleavage of graphite, chemical reduction of graphene oxide, epitaxial growth and chemical vapor deposition [10]. However, for this purpose supplied graphene nanopowder in form of multilayer flakes (Graphene Supermarket) was used.
2.3 CNT preparation
Vertically-aligned carbon nanotubes were obtained in a chemical vapor deposition process from toluene (hplc grade, Fisher Scientific) as a carbon precursor and ferrocene (99.5%, Alfa Aesar) as a catalyst [11]. The specified amount of toluene was ultrasonicated with 3 wt% ferrocene just before the process. Reaction was carried out in a horizontal furnace with three heating sections under argon atmosphere. CNTs were synthesized in Department of Materials Science and Metallurgy, Cambridge University. Among other methods used in CNT manufacturing arc discharge and laser ablation should be reported but carbon evaporation requires temperature above 3000 °C and these methods are neither economical nor convenient [12]. CVD technique is accepted to be most common due to simplicity and low cost.
2.4 Carbon aerogel preparation
Carbon aerogel was synthesized using 10.64 g resorcinol, 15.5 mL formaldehyde solution and 0.0532 g sodium carbonate as a catalyst. All components were magnetically mixed with 190 mL demineralized water at 500 rpm for 72 h at room temperature. In case of composite materials about 1 wt% of CNT, graphene or graphene oxide was added. Then samples were poured into vials, closed and kept at 80–85 °C for 7 days. Next cooled to room temperature and immersed in acetone for solvent exchange. Solvent was changed several times. During first solvent exchange 5% acetic acid was added to acetone to acidify solution and enhance crosslinking. As obtained gel was then put to autoclave (Lampart, 0.75 L, max. pressure: 450 atm., max. temperature 350 °C) for high temperature supercritical drying (HTSCD). Sample was once again immersed in 250 mL acetone and temperature of the reactor was increased up to 250 °C, the pressure inside reactor was as high as 6 MPa. Exhaust valve was used to keep pressure constant while acetone vapor was collected in a scrubber. After 1 h reactor was cooled in inert gas flow. This method allowed to obtain materials shrinkage less than in traditional supercritical CO2 drying.
Carbon aerogels were then carbonized in a tubular electric furnace at 900 °C for 0.5 h under argon flow. Synthesized carbon aerogel as well as carbon aerogels modified with CNT, graphene and graphene oxide were denoted as CA, CACNT, CAG, CAGO, respectively.
2.5 Characterization
BET Surface area and pore size distribution were analyzed using pore structure analyzer (3Flex™, Micromeritics). IR spectra were measured on a Nicolet 6700 FT-IR spectrophotometer with attenuated total reflectance (ATR method). Electrochemical experiments were carried out using two-electrode system. The working electrode materials composed of 90 wt% of active material and 10 wt% PTFE (Sigma-Aldrich, 35 μm) were pasted on electrochemical nickel current collectors to form films (mass of active material was dependent on volume of material and generally was in the range 0.003–0.009 g per electrode) and separated with polycarbonate membrane (Whatman) soaked with 6 M KOH. The accurate weight of the electrodes was read by a high-precision balance (Metler Toledo AT 261 DeltaRange). Electrodes, current collectors and separator were pressed by four screws in a poly (methyl methacrylate) casing. Cyclic voltammetry (CV) and galvanostatic charge/discharge (GC) characteristics were performed with Autolab PGSTAT 30 workstation.
3 Results
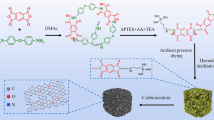
Figure 2 schematically presents steps required to synthesize carbon aerogels according to the proposed method. At first mixing of reagents formed sol either with some additives (graphene, graphene oxide, CNT) or without. Prolonged magnetic mixing was replaced by thermal treatment to start gelation. Shrunk and darker gel was immersed in acetone to exchange solvent. Instead of CO2 the supercritical drying was done in supercritical acetone with subsequent inert gas flow to expel solvent from material pores. The obtained gel was lightweight and dark red in color, however, its carbonization allowed to produce extremely porous carbon aerogel. High pressure and high temperature processes inevitably removed solvent and any functionalities causing huge mass loss within produced material.

Presentation of the unit operations required for carbon aerogel manufacturing
In a parent carbon aerogel mass loss during HTSCD was about 78% (Table 1) while for graphene- and CNT-modified carbon aerogels it varied from 87 to 90%, respectively. The biggest mass loss was observed for GO-modified carbon aerogel. This can be explained on the basis of GO structure possessing oxygen-containing groups and some additional water molecules being intercalated within the layers. These additional water as well as acidic functionalities may be desorbed during drying increasing mass loss of the material. Within carbonization second mass loss was observed that varied from 41 to 45% once again the highest value was recorded for GO-modified aerogel that is caused by removal of residual functionalities.
Figure 3 presents CV curves obtained for tested materials. In all cases shape of the few charge/discharge loops was narrower and asymmetric and changed in time to more box-like. The best supercapacitor characteristics were observed for CNT-modified and graphene oxide-modified carbon aerogel. CAG curves had more rounded corners but still the current intensity for the charging cycle was about 0.054 A in close vicinity to CAGO and CACNT, 0.057 and 0.062 A, respectively. The lowest value was observed for unmodified carbon aerogel, namely 0.037 A.

CV curves for parent carbon aerogel (CA), carbon aerogels modified with graphene oxide (CAGO), graphene (CAG) and CNT (CACNT)
Variation in the specific capacity based on CV measurements for 1, 100, 500 and 1000 cycle is presented in a Fig. 4a. Significant growth of the specific capacity, above 60%, was observed for parent carbon aerogel, similar behavior was found for CNT-modified material while for both GO- and graphene-modified carbon aerogel the capacity was approximately constant during cycling tests.

a Variation of the specific capacity during cyclability tests (1000 cycles); b galvanostatic charge/discharge curves recorded for all materials
This may be interpreted as follows; pores in parent CA are initially only partially used to build up solid electrolyte interface and during next charges more and more space is wetted by an electrolyte that increases the specific capacity.
Although increase in the specific capacity for parent carbon aerogel is relatively big after 1000 cycles (CV) the real specific capacity from galvanostatic charge/discharge measurement (Fig. 4b) is relatively low circa 80 F/g. Both graphene and graphene oxide modified carbon aerogels have specific capacity rather constant through CV tests. In case of CAG about 3% decrease in specific capacity was observed after 1000 cycles while for CAGO less than 2% increase was found. Unexpectedly values recorded by galvanostatic tests were quite similar 227 and 244 F/g for CAGO and CAG, respectively. It was expected that some residual functionalities of graphene oxide may be still present in GO-modified carbon aerogel that increased the specific capacity. However, supercritical drying and carbonization at 900 °C probably converted graphene oxide into reduced graphene oxide, material very similar to graphene. Some discrepancies in the specific capacity between graphene and graphene oxide-enriched materials may arise among others from lower final amount of this additive (graphene and graphene oxide were used in the same initial amount but after thermal treatment most of the oxygen groups desorbed from graphene oxide resulting in diminish of CAGO/CA ratio. About 23% increase in the specific capacity was found for CNT-modified carbon aerogel within 1000 cycles. The real value of specific capacity (based on galvanostatic charge/discharge) was as high as 326 F/g. For CNT- and graphene-modified carbon aerogels GC curves had asymmetric shapes with quite fast charging to 0.8 V, plateau region above it and some iR drop at the beginning of a discharge step. It should be depicted that both materials were able to discharge almost completely. Roughly triangular shape that is characteristic for an ideal supercapacitor was observed for parent carbon aerogels but these electrodes were not able to discharge below 0.4 V that limits its usage. In spite of many trials it was not successful to discharge CAGO below 0.6 V (even at prolonged time). This showed that the calculated specific capacity should be taken with considerable attention.
Difference in the specific capacity can be interpreted based on pore size distribution analysis that showed (Fig. 5) relatively bigger amount of macro- and meso-pores within CNT-modified material. As it is known macropores are responsible for initial wetting of the electrode material and are said to be gates for molecules while mesopores roles are both transportation canals and electrolyte containers within material. In contrary, micropores are very often too small to catch electrolyte molecules during short charge/discharge cycles. Therefore are not fully executed in solid electrolyte layer formation. Electrolyte is adsorbed but not desorbed and net specific capacity is diminished.

Pore size distribution in carbon aerogels modified by various carbon additives
In graphene-modified aerogel amount of these micropores is relatively high (in comparison to CNT aerogel) while macropores content is too small. Graphene oxide-modified material has the highest ca. 80% mesopores but the macropore gates amount is much lower and limits this material. Although parent carbon aerogel and CNT-modified material have quite similar pore size distribution the specific capacity is much different, the reason is the difference in BET surface area for CA and CACNT, being 123 and 574 m2/g, respectively. In case of graphene- and graphene oxide-modified materials BET was as high as 629 and 531 m2/g, respectively.
As all of materials were thermally annealed no significant signals belonging to the oxygen containing groups were identified (Fig. 6). Even signal belonging to water molecules encapsulated within structure and stretching vibration of hydroxyl groups, theoretically located around 3500–3000 cm−1 was almost completely removed. Very small signals located from 2150 to 1900 cm−1, most visible in graphene oxide- and graphene-modified materials can be attributed to the ordered carbon structure (graphene-like layers) while around 1600 cm−1 may be due to C=C stretching vibration present in all samples. Structure of as-prepared modified carbon aerogels is strongly amorphous as a result of harsh conditions during autoclave processing and thermal annealing.

FTiR spectra recorded for carbon aerogel and graphene-, graphene oxide- and CNT-modified counterparts
4 Conclusions
It was demonstrated that incorporation of graphene-like structures in relatively low amount (1 wt%) within typical carbon aerogel strongly enhanced specific capacity and cyclability. Specific capacity recorded for carbon aerogel was 80 F/g while for material modified with CNT it was 326 F/g. This was caused by increase in BET surface area from 123 to 629 m2/g and by improved wettability. An increase was also observed in graphene (227 F/g) and graphene oxide (244 F/g) modified carbon aerogels. For all modified aerogels more box-like shape of CV curves were obtained in comparison to parent carbon aerogel.
This paper also presented acetone supercritical drying method that was used instead of commonly applied carbon dioxide. It allowed to diminish shrinkage of the material during drying and produce high surface area carbon aerogels.
References
S.S. Kistler, Coherent expanded aerogels and jellies. Nature 127, 74 (1931)
R.W. Pekala, J. Mater. Sci. 24, 3221–3227 (1989)
M.A. Aegerter, N. Leventis, M.M. Koebel, Advances in Sol–Gel Derived Materials and Technologies, Aerogels Handbook (Springer, New York, 2011), pp. 216–220. ISBN 978-1-4419-7477-8
S.J. Kim, S.W. Hwang, S.H. Hyun, Preparation of carbon aerogel electrodes for supercapacitor and their electrochemical characteristics. J. Mater. Sci. 40, 725–731 (2005)
D. Wu, R. Fu, S. Zhang, M.S. Dresselhaus, G. Dresselhaus, Preparation of low-density carbon aerogels by ambient pressure drying. Carbon 42, 2033–2039 (2004)
J. Biener, M. Stadermann, M. Suss, M.A. Worsley, M.M. Biener, K.A. Rose, T.F. Baumann, Advanced carbon aerogels for energy applications. Energy Environ. Sci. 4, 656 (2011)
M.A. Worsley, P.J. Pauzauskie, S.O. Kucheyev, J.M. Zaug, A.V. Hamza, J.H. Satcher Jr., T.F. Baumann, Properties of single-walled carbon nanotube-based aerogels as a function of nanotube loading. Acta Mater. 57, 5131–5136 (2009)
X.-L. Wu, A.-W. Xu, Carbonaceous hydrogels and aerogels for supercapacitors. J. Mater. Chem. A 2, 4852–4864 (2014)
L. Staudenmaier, Ber. Dtsch. Chem. Ges. 31, 1481–1487 (1898)
F. Akbar, M. Kolahdouz, Sh Larimian, B. Radfar, H.H. Radamson, Graphene synthesis, characterization and its applications in nanophotonics, nanoelectronics, and nanosensing. J. Mater. Sci. Mater. Electron. 26, 4347–4379 (2015)
S.W. Pattinson, K. Prehn, I.A. Kinloch, D. Eder, K.K.K. Koziol, K. Schulte, A.H. Windle, The life and death of carbon nanotubes. RSC Adv. 2(7), 2909–2913 (2012)
K. Koziol, B.O. Boskovic, N. Yahya, Synthesis of Carbon Nanostructures by CVD Method, Carbon and Oxide Nanostructures of the Series Advanced Structured Materials (Springer, Berlin, 2010), pp. 23–49. ISBN 978-3-642-14672-5
Acknowledgements
Authors would particularly like to thank Prof. Ginter Nawrat from the Department of Inorganic, Analytical Chemistry and Electrochemistry, Silesian University of Technology, Poland for giving opportunity to perform electrochemical tests. Authors are also grateful for opportunity to synthesize CNTs in Dr. Krzysztof Koziol—headed Electric Carbon Nanomaterials Group at Department of Materials Science and Metallurgy, Cambridge University, UK.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ciszewski, M., Szatkowska, E., Koszorek, A. et al. Carbon aerogels modified with graphene oxide, graphene and CNT as symetric supercapacitor electrodes. J Mater Sci: Mater Electron 28, 4897–4903 (2017). https://doi.org/10.1007/s10854-016-6137-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10854-016-6137-2