
Abstract
To determine the rate and predictors of autism spectrum disorder (ASD) in congenital diaphragmatic hernia (CDH). Between 06/2004 and 09/2015 a total of 110 CDH survivors underwent neurodevelopmental (ND) testing and screening for ASD, followed by a full autism diagnostic evaluation if indicated at our institution. We found a 9 time higher rate of ASD in CDH children compared to the general population (P = 0.0002). Multiple patient-related and clinical variables risk factors of ASD were identified by univariate analysis. However, only short-term and long-term neurodevelopmental delays were strongly associated with ASD in CDH by multivariate comparisons. There is a striking prevalence of ASD in CDH survivors and our findings suggest that all CDH children should be regularly screened for ASD.
Similar content being viewed by others
Introduction
The Center of Disease Control and Prevention estimates 1 in 68 children in the US are affected by autism spectrum disorder (ASD) (Autism and Developmental Disabilities Monitoring Network, 2016). An increased risk for ASD has recently been noted in premature infants (Dodds et al. 2011; Guy et al. 2015; Kuzniewicz et al. 2014; Lampi et al. 2012), as well as in children born with congenital malformations (Danzer et al. 2015; Razzaghi et al. 2015; Wier et al. 2006), and genetic syndromes (Fine et al. 2005) compared to the general population.
Congenital diaphragmatic hernia (CDH) is a birth defect with estimated prevalence of one per 2500 live births. Over the past several decades, innovations in surgical techniques and advances in perinatal care and management have significantly lowered mortality rates for children with CDH (Danzer and Hedrick 2014) With improvements in survival, increased focus has been directed toward neurodevelopmental and neurobehavioral outcomes (Danzer and Hedrick 2011). Among CDH survivors, there is a pattern of developmental, functional, and behavioral impairment characterized by mild to moderate cognitive and language impairment, impaired visual-motor integration, persistent hypotonia, delayed motor coordination, difficulties with emotion and behavior regulation, and delays in the appropriate development of socialization skills (Chen et al. 2007; Danzer et al. 2010, 2013a, b; Friedman et al. 2008; Peetsold et al. 2009; Wynn et al. 2013). Rates of ASD in CDH survivors have not been studied. However, based on anecdotal reports (Danzer et al. 2010, 2013b) and the potential overlap in impairments between ASD and CDH survivors, we hypothesized that children with CDH have an increased risk of ASD.
The objectives of this study were to: (1) determine the rate of ASD in a relatively large cohort of CDH survivors enrolled in a multidisciplinary follow-up program, and (2) identify patient-related and clinical variables risk factors that might explain the association between CDH and increased risk of ASD.
Methods
We reviewed prospectively collected data on demographics, perinatal, perioperative, and postnatal factors, and developmental and diagnostic outcomes in CDH survivors enrolled in our multidisciplinary follow-up program, the Pulmonary Hypoplasia Program, between June 2004 and September 2015. All CDH survivors born during the study period who enrolled in the follow-up program were eligible. Among this cohort, subjects who were at least 2 years of age and who underwent neurodevelopmental evaluations were identified and form the study sample. Both maternal prenatal and neonatal/follow-up medical records were reviewed. The Institutional Review Board, Committee for Protection of Human Subjects of our institution approved this study and all parents or legal guardians gave written informed consent for their children (IRB 2004–003779).
Perinatal and Postnatal Management
As described in previous work from our group, CDH patients at our institution are treated according to specific perinatal and postnatal management guidelines (Danzer et al. 2013a, b; Danzer and Hedrick 2011, 2014). Briefly, all CDH patients referred to our center undergo a comprehensive prenatal imaging evaluation (ultrasonography, echocardiography, MR imaging). After evaluation, all patients undergo nondirective counseling for pregnancy management options. After birth, the postnatal ventilatory management in the neonatal intensive care unit utilizes a lung-preservation strategy similar to that of infants with other causes of pulmonary hypoplasia (e.g., fetal lung lesions, giant omphalocele) (Danzer et al. 2015, 2012a). If medical management fails to prevent ventilator-related lung injury or persistent hypotension/acidosis, extracorporeal membrane oxygenation (ECMO) therapy is initiated. The operating surgeon determines the timing of repair based upon co-morbidities and clinical stability during the neonatal period as well as the need for patch repair based upon the size of the diaphragmatic defect (Tsai et al. 2012).
Follow-up, Neurodevelopmental and ASD Assessment
Standard of care for all neo-natal follow-up patients, including CDH, is regular follow up care at ages 6, 12, and 24 months, and before entering Kindergarten. If a developmental concern arises at the 12 or 24 month visit, then supplemental visits are scheduled for 18 or 36 months, respectively, with annual visits until Kindergarten if indicated. Regular follow-up care includes medical, neuromotor, and psychological assessment with standardized instruments (described below), and ASD screening (with further assessment if indicated; described below).
At each visit, growth parameters including weight, length, and head circumference are measured and compared to standard reference curves. The neuromuscular exam includes assessment of cranial nerves, passive and active muscle tone, strength, protective, postural, deep tendon reflexes and motor skills. Children’s neuromotor functioning is classified as “normal” or “abnormal/suspect”, based on the degree that the tonal abnormalities impact the quality and acquisition of skills.
Developmental assessment is conducted with the Bayley Scales of Infant Development II (prior 2006) or III (after 2006) at the 6, 12, and 24 month visits. Children seen after age 24 months are assessed with either the BSID-II or the Wechsler Preschool and Primary Scale of Intelligence, Third Edition and Fourth Edition (WPPSI-III, WPPSI-IV) depending on their age. These instruments are all widely used and yield individual domain scores, as well as overall composite scores that are norm referenced (see Table 2). For each of the BSID and the WPPSI composite scores the mean standardized score is 100 with a standard deviation (SD) of 15. We categorized scores as average, mildly delayed, and severely delayed based on SD intervals (85–115, 70–84, ≤ 69, respectively) (Danzer et al. 2010, 2013b). Also as previously reported (Danzer et al. 2013a, b), in order to capture the majority of CDH survivors who would be expected to experience at least some degree of impairment, we defined neurodevelopmental delay as a score of ≤ 85 in any of the evaluated composite scores. Severe impairment was defined as a score of ≤ 69 in at least one domain tested.
ASD screening is provided to all children at the 24 month visit with the Modified Checklist for Autism in Toddlers (M-CHAT). An ASD diagnostic evaluation occurs when a child either screens positive on the M-CHAT or when a provider or parent is concerned (regardless of age). ASD evaluations are conducted by two clinical psychologists who have extensive experience in both neonatal follow-up and ASD. The evaluations include the Autism Diagnostic Observation Schedule-2 (ADOS-2; or the original ADOS before the ADOS-2 was available) for children at any age, and the Social Communication Questionnaire (SCQ) for children age 48 months and older. The ADOS-2 (like the ADOS) is a standardized, semi-structured assessment of social interaction, communication, play, and repetitive and restricted behaviors. It is used widely in both clinical and research centers, and is the “gold standard” of observational tools for measuring ASD related characteristics. The SCQ is the most widely used parent report measure of ASD characteristics in ASD research and clinical work.
ASD diagnoses are based on DSM-IV-TR or DSM-5 criteria informed by all available information including the ADOS-2, M-CHAT, SCQ, and developmental testing. Because we obtain developmental testing on all children, we do a careful differentiation between ASD and developmental delay in this complex group of patients. We do not apply an ASD diagnosis unless the child’s social communication and behaviors are impaired relative to his or her current developmental level.
Statistical Analyses
For the purpose of this study, the data from the latest available follow-up visit were used to define outcome.
Continuous data are presented as means ± SD (median, range). Categorical data are presented as proportions. The differences between developmental outcomes were determined using Student’s t, one-way ANOVA, or linear regression, depending on the type of outcome variable. Prediction of outcome variables used logistic regression, or ordinal logistic regression, depending on the type of outcome variable. After univariate analysis, variables with a P value of ≤ 0.05 were included into the final multivariate analysis model. One-sample t-test was used to compare the mean outcome scores to the population mean. P values < 0.05 were considered statistically significant. All analyses were conducted in Stata version 12.0 (College Station, TX, USA).
Results
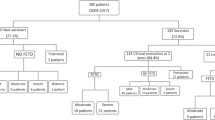
Between June 2004 and September 2015 a total of 183 CDH survivors completed standardized neurodevelopmental assessment as part of their multidisciplinary follow-up in our Pulmonary Hypoplasia Program. Of those, 110 patients (60%) were at least 2 years at their most recent evaluation and represent the study population. A total of 15 (13.6%) CDH patients of the study population (n = 110) were diagnosed with an ASD. All 15 ASD diagnoses were made at our institution. Mean age of ASD diagnosis was 3.8 ± 2.2 years; range 2–6.3 years. Compared to the reported risk of 1 in 68 children born without congenital malformation (Autism and Developmental Disabilities Monitoring Network, 2016), the relative risk of ASD in CDH was 9.3 times the risk of the general population (P = 0.0002). Further, the relative risk for male CDH (18.8%, RR 7.9, P = 0.01) as well as for female CDH (4.9%, RR 9.3, P = 0.02) children was significantly higher than expected risk in normal males (2.4%) and females (0.5%) (Autism and Developmental Disabilities Monitoring Network, 2016). Notably, the relative risk of ASD in CDH patients remained significantly higher than predicted by the incidence of ASD in the general population even when patients with severe neurodevelopmental deficits or associated anomalies were excluded (RR 3.8 and RR 6.8, P = 0.02, P < 0.01, respectively).
Important baseline, clinical, and surgical characteristics of the study participants are shown in Table 1. Three (20%) children with CDH + ASD and 26 (27%) children with CDH alone were diagnosed postnatally and transferred to our institution for postnatal NICU management, surgery, and follow-up. Two CDH + ASD patients (13%) and 3 (3%) CDH only patients were referred to our institution for follow-up evaluations after they received NICU care elsewhere. Children with CDH + ASD were more likely to be males, more likely to be born prematurely, and more likely to have a complicated NICU course compared with children with CDH alone (P < 0.05).
Karyotyping was completed in 11 (73%) of children with CDH + ASD and in 58 (61%) of children with CDH alone. Significantly more children with CDH + ASD had associated major malformations and/or associated genetic syndromes (5 [33%]) compared to the CDH alone (5 [5%], P = 0.004), but the genetic syndromes identified were not among those commonly associated with ASD. Major associated malformations and/or genetic syndromes in the CDH + ASD group included Ehler-Danlos syndrome (n = 1), Goldenhar syndrome (n = 1), IUGR and tethered cord syndrome (n = 1), ventricular septal defect requiring surgical repair (n = 1), and Pentalogy of Cantrell (n = 1). Associated major malformations and/or genetic syndromes in children with CDH alone were ventricular septal defect and coarctation of the aorta requiring surgical repair (n = 1), chromosomal 17p12 deletion (n = 1), Saethre–Chotzen syndrome (n = 1), cleft-lip, cleft palate and micrognathia (n = 1), and Turner syndrome (n = 1).
Contrary to literature on head circumference in ASD (Sacco et al. 2015), microcephaly, defined as head circumference percentile ≤ 5%, was significantly more common in children with CDH + ASD than CDH alone (3 [20%] vs. 2 [2%], P < 0.02). No differences between the groups were found for weight or height. Cranial imaging studies (brain US and/or MRI) were available in 11 (73%) children with CDH + ASD and 90 (95%) of patients without ASD. Small foci of periventricular leukomalacia did not differentiate the two groups (9% in CDH + ASD, 11% in CDH alone, P = 1.0). However, intraventricular hemorrhage was more commonly found in the CDH + ASD group (6 [45%]) compared to CDH alone (14 [16%], P = 0.007).
Neuromuscular hypotonicity was found in 9 (60%) CDH + ASD, and in 31 (33%) without ASD (P = 0.05). Various degrees of delayed motor coordination were documented in seven (47%) of the children with CDH + ASD, while coordination difficulties were noted in 14 (15%) of children without ASD (P = 0.02).
Unadjusted outcomes on the developmental measures for CDH + ASD compared to CDH alone are summarized in Table 2. Mean age at follow-up was similar between the CDH + ASD (3.6 ± 1.4 years [range 2–6.3]) and CDH alone group (3.1 ± 1.4 [range 2–6.2]) (P = 0.27). Children with CDH + ASD had significantly lower mean cognitive, language, and motor BSID-III composite scores compared to CDH alone (P = 0.0001). Mean Full-IQ (P = 0.01) and performance IQ (P = 0.0009) WPPSI scores were also found to be significantly lower in the CDH + ASD group compared to CDH alone. Mean verbal IQ WPPSI scores were similar between groups (P = 0.11). Three children (3%) underwent their latest neurodevelopmental assessment before 2006 and were evaluated using the BSID-II; thus they were excluded from group comparisons.
The prevalence and severity of neurodevelopmental impairment based on proportions of patients that scored 1 SD (mildly delayed) or 2 SD (severely delayed) below the mean were also compared between CDH + ASD and CDH alone (Table 3). Significantly more in the CDH + ASD group scored within the severely delayed range (n = 10, 67%) for neurodevelopmental scores at follow-up in at least one domain compared to CDH children without ASD (n = 6, 6%, P < 0.001) (Fig. 1). Eleven (73%) patients of the CDH + ASD group and 83 (87%) of the CDH alone group underwent serial assessment of neurocognitive outcome. Early developmental findings, defined as those obtained at the first follow-up visit at a median of 11 months (range 5–36 months), indicated that mild and severe neurocognitive delays were already significantly more often found in CDH + ASD group (82% vs. 33% P = 0.006).

Bar graph showing the distribution in percent of CDH survivors with ASD scoring within the average, mildly delayed, and severely delayed range in at least one composite domain tested compared to CDH children without autism
Patient-related and clinical variables associated with ASD in CDH, as well as their univariate statistical significance, are shown in Table 4. Surrogate markers of CDH severity such as prolonged ventilatory support, need for O2 supplementation beyond 30 days of life, inadequate oral intake supplemented by enteral feedings at follow-up, and prolonged NICU stay were associated with an increased risk of ASD. Patient-related factors including male gender, right-sided diaphragmatic defect, preterm delivery, presence of associated malformations and/or genetic syndromes, and history of intraventricular hemorrhage were also associated with an ASD diagnosis. In addition, developmental-related factors including neuromuscular hypotonicity, abnormal brainstem auditory evoked responses, ongoing neurological impairment, and adverse neurodevelopmental outcome identified during infancy were also associated with ASD. Multivariate analysis of these identified risk factors showed that ongoing neurocognitive delay had the strongest association with ASD (OR 25.14; 95% CI 0.95–664.81; P = 0.05). Of note, ND delay identified during earlier evaluation tended to be associated (OR 3.7; 95% CI 0.93–10.19; P = 0.06) with subsequent diagnosis of ASD.
Other risk factors analyzed such as ethnicity, lung-to-head ratio, liver position, type of ventilator support, failure to thrive, parental social-economic status, and others were not significantly associated with increased risk of ASD in this sample (data available upon request).
Discussion
This is the first study to evaluate the association of CDH and ASD. Consistent with our hypothesis, we found that CDH survivors are at increased risk of developing ASD. We have identified ASD in nearly 14% of CDH survivors which is higher than the reported prevalence of 1.5% in the general population, and also approaches the reported prevalence of ASD of 8–22% in known high risk populations, such as children born with severe prematurity, other congenital malformations, and/or genetic syndromes (Danzer et al. 2015; Fine et al. 2005; Johnson et al. 2010; Lindquist et al. 2006; Razzaghi et al. 2015; Wier et al. 2006).
The striking ninefold increased risk of ASD in the current study population is of concern and has implications throughout life for these patients. The risk of ASD remained higher than expected even after we excluded CDH patients with severe neurodevelopmental impairments or with associated congenital malformations and/or genetic abnormalities. The identified additional malformations (i.e. Ehler-Danlos syndrome, Goldenhar syndrome, and cardiac anomalies) have been linked to an increased risk of autism independent of the presence of CDH (Cederlöf et al. 2016; Johansson et al. 2007; Bean Jaworski et al. 2017). Our data suggests, however, that ASD is also more common in CDH patients with no other developmental disabilities than in the general population. Knowledge of ASD risk in CDH survivors is pivotal for perinatal counseling of families and etiologic investigation, as well as tailoring developmental follow up protocols and implementing early intervention to optimize long-term outcomes (Dawson et al. 2010).
It remains unclear whether ASD is emerging at high rates because it is associated with CDH and more infants with CDH are surviving, or whether ASD is emerging because we are specifically screening for it in every single patient. Our analysis of patient-related and clinical variables risk factors suggests that ASD in CDH is heterogeneous in cause. Univariate analysis suggest a possible association between preterm delivery and low birth weight, male gender, prolonged ventilation and intensive care hospitalization during the neonatal period, need for enteral feeding access, and presence of multiple malformations or genetic syndromes and ASD diagnosis in the current study. These patient-related variables have been shown to be important risk factors for ASD in general and may not be specific to patients with CDH. ASD risk is also increased in children born with other congenital malformations that require prolonged NICU care (Danzer et al. 2015; Neufeld et al. 2008; Razzaghi et al. 2015; Timonen-Soivio et al. 2015; Wier et al. 2006) or co-occurring medical conditions (Alexeeff et al. 2017). Of note, several investigators suggest a common etiological pathway (e.g. shared gene and/or environmental insult during development) between congenital malformations and ASD (Hultman et al. 2002; Wier et al. 2006). The association between CDH and ASD may involve a shared rather than causal etiology and warrants further investigation.
Perhaps not surprisingly, ASD in CDH survivors appears to co-occur with intellectual disabilities at a higher rate than seen in the larger ASD population. In the current study, 67% of CDH children with ASD scored in the severely delayed range for the neurodevelopmental domains tested, compared to 32% of children with ASD in population-based studies (Christensen et al. 2016). Furthermore, CDH + ASD had a higher rate of intellectual disability than CDH alone in our sample (67% compared to 6%). Further, 82% of CDH children with ASD were found to have already significant deficits during early life. We previously observed that severe neurological delay during infancy is associated with persistent significant neurodevelopmental and functional impairments during preschool age (Danzer et al. 2013a). Although these findings require corroboration in larger prospective studies, we suggest that careful surveillance and screening for ASD in CDH survivors should start during early toddlerhood, particularly for those patients who are identified with significant developmental delay during early life. Formal screening before 2 years of age for all children has been recommended by the American Academy of Pediatrics (AAP) (Johnson et al. 2007). Earlier identification will lead to greater understanding and insights into the nature of neurodevelopmental and functional deficits that result in subsequent ASD. Based on the results of the current study, our center has changed practice patterns and is screening for early clinical signs of ASD (e.g. poor eye contact, poor attention to the speaker, lack of emerging gestures, or poor joint attention) in CDH patients as early as 16 months of age and is recommending aggressive use of early intervention services. Interestingly, McDonald and associates (McDonald et al. 2017) recently showed that patients with tuberous sclerosis complex that were diagnosed with ASD by age 3 already show significant deficits in social communication behavior as early as 9 months of age compared to patients without ASD diagnosis, further supporting the importance of early ASD screening in high-risk populations.
Growing evidence suggests that aberrant central nervous system development in high-risk infants, which is often associated with neurodevelopmental delays, is also found in children with ASD (Guy et al. 2015; Movsas et al. 2013; Padilla et al. 2015). Congenital and acquired central nervous system malformations are seen in CDH. We previously demonstrated that brain development in CDH patients is structurally delayed and in turn this maturation delay is associated with neurocognitive and language delays (Danzer et al. 2012b). In the current study only history of intraventricular hemorrhage and presence of microcephaly appeared to be associated with an increased risk of ASD. Our patients, however, underwent cranial imaging studies based on clinical indications rather than specific protocols which precludes a more definite conclusion about the impact of even subtle structural brain abnormalities on risk of ASD and/or neurodevelopmental delays. How alteration of brain development relates to changes in cognitive functioning and risk of ASD in CDH children merits exploration in future well designed image-based studies.
The strengths of this study include the large sample size of a particular high-risk population, as well as the use of an extensive, prospectively collected dataset of prenatal, perinatal, socioeconomic, and developmental variables sufficient enough to identify multiple risk factors of ASD by univariate and multivariate analysis. Further, assessment of neurodevelopmental outcome and ASD was performed by only two psychologists (CH, MG) which minimizes observer bias. Their inter-rater administration and scoring is assessed on a yearly basis and has been 100% in agreement. A final strength is our multidisciplinary team of experienced and consistent health care providers who are involved in the care of these children and families from the time of prenatal diagnosis to perinatal management to follow-up.
Despite these strengths, the current study must be viewed in light of its limitations. Although this is the largest study to date reporting on ASD risk in CDH survivors, our results should be interpreted with care. The data of the current report were not population-based and a selection bias may have been introduced due to the specific nature of our tertiary referral center. Data replication and analysis of larger CDH populations is necessary to further define the natural history, as it is currently unknown whether ASD in CDH survivors is a sequelae of congenital abnormalities in general, or CDH in particular. Also, a longer follow-up into adolescence and early adulthood is needed to evaluate the long-term ASD, neurodevelopmental, medical, and psychiatric characteristics in CDH survivors and impact on quality of life. Our study relied on testing data from CDH survivors who were evaluated as part of their clinical follow-up and families’ willingness to return to these appointments. Consequently, we were not able to assess ASD and neurodevelopmental outcomes during precise developmental windows for better between-subject comparisons. To ameliorate this potential bias, comparisons between ASD and neurodevelopmental outcomes were therefore made based on overall developmental categories derived from standard deviation intervals, rather than on individual scores analyzed as continuous variables (Aylward and Aylward 2011). While it may be a disadvantage that our study included children with a range of ages, the advantage is that as in the general population, children in this sample met criteria for diagnosis at a variety of ages, some in early toddler hood, and others not until closer to kindergarten age. By including children’s data from the oldest assessment available, we were able to include some of the later diagnoses. This also means that our estimates of ASD in CDH survivors could be low, as they will likely increase as all of the children in this cohort reach school age. A final limitation of the current study is the inability to directly compare the increased risk of ASD in CDH survivors to the outcomes of patients with a complicated and prolonged NICU course due to other congenital malformations or premature delivery. Without these comparisons we are unable to draw firm conclusions about the significance of the overall clinical course on the outcome in CDH patients. Further research in the CDH population as well as in other patient groups born with congenital abnormalities is necessary to fully delineate whether the underlying malformation, it’s perinatal management, or a combination of both impact on the risk of autism spectrum disorder and adverse neurological outcome in affected children.
In conclusion, ASD was highly prevalent in our sample of extensively followed CDH survivors. Short-term and long-term neurodevelopmental delays were strongly associated with ASD in CDH children. Disease severity, feeding difficulties, prematurity, neurological abnormalities, and the presence of major associated malformations/syndromes were important risk factors. However, children with CDH but without associated syndromes and severe neurodevelopmental delays were also more at risk for ASD than children in the general population. Our data suggest that a heightened surveillance, screening, and monitoring for neurodevelopmental disorders and ASD in all CDH survivors is warranted. Although the presence of CDH might indicate an increased risk for ASD, given the identified risk factors, the relationship between CDH and ASD may not be causal. In other words, a child born with any congenital malformation associated with increased risk of prematurity, neurological injury, prolonged and complicated neonatal course, and presence of additional anatomical or genetic abnormalities will have accumulated multiple risk factors for developmental disorders that might be more important than the underlying congenital defect itself.
Thus, health care providers should be aware that all CDH survivors might need specialized care beyond the neonatal period and deserve close monitoring and follow-up well beyond infancy. They should also be aware that children with multiple risk factors, including a complicated neonatal course, might be at greater risk for ASD. Resources and funding need to be made available for comprehensive evaluations by professionals trained to recognize ASD symptoms and differentiate these from more general developmental delays, as well as for family counseling, support, and intervention programs. Further research using larger CDH cohorts are needed to assess possible behavioral phenotyping for this population, influence standard of care for screening and surveillance as well as looking at additional environmental and genetic factors leading to CDH and ASD.
References
Alexeeff, S.E., et al. (2017). Medical conditions in the first years of life associated with future diagnosis of ASD in children. Journal of Autism and Developmental Disorders. https://doi.org/10.1007/s10803-017-3130-4.
Aylward, G.P., & Aylward, B.S. (2011). The changing yardstick in measurement of cognitive abilities in infancy. Journal of Developmental and Behavioral Pediatrics, 32, 465–468. https://doi.org/10.1097/DBP.0b013e3182202eb3.
Bean Jaworski, J.L., et al. (2017). Rates of autism and potential risk factors in children with congenital heart defects. Congenital Heart Disease, 12, 421–429. https://doi.org/10.1111/chd.12461.
Cederlöf, M., et al. (2016). Nationwide population-based cohort study of psychiatric disorders in individuals with Ehlers–Danlos syndrome or hypermobility syndrome and their siblings. BMC Psychiatry, 16, 207. https://doi.org/10.1186/s12888-016-0922-6.
Chen, C., et al. (2007). Approaches to neurodevelopmental assessment in congenital diaphragmatic hernia survivors. Journal of Pediatric Surgery, 42, 1052–1056, discussion 1056. https://doi.org/10.1016/j.jpedsurg.2007.01.042.
Christensen, D.L., et al. (2016). Prevalence and characteristics of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2012. Morbidity and Mortality Weekly Report: Surveillance Summaries, 65, 1–23. https://doi.org/10.15585/mmwr.ss6503a1.
Danzer, E., et al. (2010). Neurodevelopmental outcome of infants with congenital diaphragmatic hernia prospectively enrolled in an interdisciplinary follow-up program. Journal of Pediatric Surgery, 45, 1759–1766. https://doi.org/10.1016/j.jpedsurg.2010.03.011.
Danzer, E., et al. (2012a). Early neurodevelopmental outcome of infants with high-risk fetal lung lesions. Fetal Diagnosis and Therapy, 31, 210–215. https://doi.org/10.1159/000336228.
Danzer, E., et al. (2012b). Abnormal brain development and maturation on magnetic resonance imaging in survivors of severe congenital diaphragmatic hernia. Journal of Pediatric Surgery, 47, 453–461 https://doi.org/10.1016/j.jpedsurg.2011.10.002.
Danzer, E., et al. (2013a). Longitudinal neurodevelopmental and neuromotor outcome in congenital diaphragmatic hernia patients in the first 3 years of life. Journal of Perinatologyapy, 33, 893–898. https://doi.org/10.1038/jp.2013.47.
Danzer, E., et al. (2013b). Preschool neurological assessment in congenital diaphragmatic hernia survivors: Outcome and perinatal factors associated with neurodevelopmental impairment. Early Human Development, 89, 393–400. https://doi.org/10.1016/j.earlhumdev.2012.12.009.
Danzer, E., et al. (2015). Patient characteristics are important determinants of neurodevelopmental outcome during infancy in giant omphalocele. Early Human Development, 91, 187–193. https://doi.org/10.1016/j.earlhumdev.2014.12.009.
Danzer, E., & Hedrick, H.L. (2011). Neurodevelopmental and neurofunctional outcomes in children with congenital diaphragmatic hernia. Early Human Development, 87, 625–632. https://doi.org/10.1016/j.earlhumdev.2011.05.005.
Danzer, E., & Hedrick, H.L. (2014). Controversies in the management of severe congenital diaphragmatic hernia. Seminars in Fetal and Neonatal Medicine, 19, 376–384. https://doi.org/10.1016/j.siny.2014.10.001.
Dawson, G., et al. (2010). Randomized, controlled trial of an intervention for toddlers with autism: The Early Start Denver Model. Pediatrics, 125, e17–e23. https://doi.org/10.1542/peds.2009-0958.
Developmental Disabilities Monitoring Network Surveillance Year Principal, I., C. Centers for Disease, Prevention (2014). Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2010. Morbidity and Mortality Weekly Report: Surveillance Summaries, 63, 1–21.
Dodds, L., Fell, D.B., Shea, S., Armson, B.A., Allen, A.C., & Bryson, S. (2011). The role of prenatal, obstetric and neonatal factors in the development of autism. Journal of Autism and Developmental Disorders, 41, 891–902. https://doi.org/10.1007/s10803-010-1114-8.
Fine, S.E., et al. (2005). Autism spectrum disorders and symptoms in children with molecularly confirmed 22q11.2 deletion syndrome. Journal of Autism and Developmental Disorders, 35, 461–470. https://doi.org/10.1007/s10803-005-5036-9.
Friedman, S., et al. (2008). Neurodevelopmental outcomes of congenital diaphragmatic hernia survivors followed in a multidisciplinary clinic at ages 1 and 3. Journal of Pediatric Surgery, 43, 1035–1043. https://doi.org/10.1016/j.jpedsurg.2008.02.029.
Guy, A., et al. (2015). Infants born late/moderately preterm are at increased risk for a positive autism screen at 2 years of age. The Journal of Pediatrics, 166, 269–275. https://doi.org/10.1016/j.jpeds.2014.10.053.
Hultman, C.M., Sparen, P., & Cnattingius, S. (2002). Perinatal risk factors for infantile autism. Epidemiology 13, 417–423.
Johansson, M., et al. (2007). Autism spectrum disorder and underlying brain mechanism in the oculoauiculovertebral spectrum. Developmental Medicine and Child Neurology, 49, 280–288. https://doi.org/10.1111/j.1469-8749.2007.00280.x.
Johnson, C.P., Myers, S.M., & American Academy of Pediatrics Council on Children With (2007). Identification and evaluation of children with autism spectrum disorders. Pediatrics, 120, 1183–1215. https://doi.org/10.1542/peds.2007-2361.
Johnson, S., Hollis, C., Kochhar, P., Hennessy, E., Wolke, D., & Marlow, N. (2010). Autism spectrum disorders in extremely preterm children. The Journal of Pediatrics, 156, 525–531. https://doi.org/10.1016/j.jpeds.2009.10.041.
Kuzniewicz, M.W., Wi, S., Qian, Y., Walsh, E.M., Armstrong, M.A., & Croen, L.A. (2014). Prevalence and neonatal factors associated with autism spectrum disorders in preterm infants. The Journal of Pediatrics, 164, 20–25. https://doi.org/10.1016/j.jpeds.2013.09.021.
Lampi, K.M., et al. (2012). Risk of autism spectrum disorders in low birth weight and small for gestational age infants. The Journal of Pediatrics, 161, 830–836. https://doi.org/10.1016/j.jpeds.2012.04.058.
Lindquist, B., Carlsson, G., Persson, E.K., & Uvebrant, P. (2006). Behavioural problems and autism in children with hydrocephalus: A population-based study. European Child and Adolescent Psychiatry, 15, 214–219. https://doi.org/10.1007/s00787-006-0525-8.
McDonald, N.M., et al. (2017). Early autism symptoms in infants with tuberous sclerosis complex. Autism Research, https://doi.org/10.1002/aur.1846.
Movsas, T.Z., et al. (2013). Autism spectrum disorder is associated with ventricular enlargement in a low birth weight population. The Journal of Pediatrics, 163, 73–78 https://doi.org/10.1016/j.jpeds.2012.12.084.
Neufeld, R.E., et al. (2008). Five-year neurocognitive and health outcomes after the neonatal arterial switch operation. Journal of Thoracic and Cardiovascular Surgery, 136, 1413–1421. https://doi.org/10.1016/j.jtcvs.2008.05.011.
Padilla, N., Eklof, E., Martensson, G.E., Bolte, S., Lagercrantz, H., & Aden, U. (2015). Poor brain growth in extremely preterm neonates long before the onset of autism spectrum disorder symptoms. Cerebral Cortex. https://doi.org/10.1093/cercor/bhv300.
Peetsold, M.G., Huisman, J., Hofman, V.E., Heij, H.A., Raat, H., & Gemke, R.J. (2009). Psychological outcome and quality of life in children born with congenital diaphragmatic hernia. Archives of Disease in Childhood, 94, 834–840. https://doi.org/10.1136/adc.2008.156158.
Razzaghi, H., Oster, M., & Reefhuis, J. (2015). Long-term outcomes in children with congenital heart disease: National health interview survey. The Journal of Pediatrics, 166, 119–124. https://doi.org/10.1016/j.jpeds.2014.09.006.
Sacco, R., Gabriele, S., & Persico, A.M. (2015). Head circumference and brain size in autism spectrum disorder: A systematic review and meta-analysis. Psychiatry Research, 234, 239–251. https://doi.org/10.1016/j.pscychresns.2015.08.016.
Timonen-Soivio, L., et al. (2015). The association between congenital anomalies and autism spectrum disorders in a Finnish national birth cohort. Developmental Medicine and Child Neurology, 57, 75–80. https://doi.org/10.1111/dmcn.12581.
Tsai, J., Sulkowski, J., Adzick, N.S., Hedrick, H.L., & Flake, A.W. (2012). Patch repair for congenital diaphragmatic hernia: Is it really a problem?. Journal of Pediatric Surgery, 47, 637–641. https://doi.org/10.1016/j.jpedsurg.2011.11.054.
Wier, M.L., Yoshida, C.K., Odouli, R., Grether, J.K., & Croen, L.A. (2006). Congenital anomalies associated with autism spectrum disorders. Developmental Medicine and Child Neurology, 48, 500–507. https://doi.org/10.1017/S001216220600106X.
Wynn, J., et al. (2013). Developmental outcomes of children with congenital diaphragmatic hernia: A multicenter prospective study. Journal of Pediatric Surgery, 48, 1995–2004. https://doi.org/10.1016/j.jpedsurg.2013.02.041.
Acknowledgments
We greatly appreciate the statistical advice and analysis provided by Mark S. Cary, PhD, Sr. Staff Biostatistician, Biostatistics Analysis Center, Center for Clinical Epidemiology and Biostatistics, The Perelman School of Medicine at the University of Pennsylvania. We also thank Norma Rendon for superb administrative assistance. This study has been in part be presented as poster at the 2016 Annual Meeting of the American Pediatric Surgery Association in San Diego, California, USA.
Author Contributions
ED, CH, JSM, HLH—study design; ED, CH, JA, LNW, MG, JCB, HR, NER, LMH, WHP—data collection and analysis; ED, CH—writing the first draft; ED, CH, JA, JSM, LNW, MG, JCB, HR, NER, WHP, AWF, NSA, HLH—criticial review of manuscript and approval of final manuscript version.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None of the authors have any conflict of interest to declare.
Ethical Approval
All procedures performed in studies involving human subjects were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants (parents) included in the study.
Rights and permissions
About this article

Cite this article
Danzer, E., Hoffman, C., D’Agostino, J.A. et al. Rate and Risk Factors Associated with Autism Spectrum Disorder in Congenital Diaphragmatic Hernia. J Autism Dev Disord 48, 2112–2121 (2018). https://doi.org/10.1007/s10803-018-3472-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10803-018-3472-6