
Summary
In this study, we compared the cellular uptake, intracellular localization and cytotoxicity of two groups of anthracycline derivatives in cultured H9c2(2-1) rat cardiomyoblasts. The first group consisted of doxorubicin (DOX) and two of its derivatives containing a formamidino group (–N = CH–N<) at the C-3′ position with a morpholine (DOXM) or a hexamethyleneimine (DOXH) ring. The second group consisted of daunorubicin (DRB) and its derivatives containing a morpholine (DRBM) or a hexamethyleneimine (DRBH) ring. DOXH and DRBH were taken up by cardiomyoblasts more efficiently than estimated for other tested anthracyclines. The cellular uptakes of DOXM and DRBM were reduced compared to those of the parent compounds. Applied structural modifications of DOX and DRB influenced the subcellular localization of the tested derivatives. DOX and DOXH were localized primarily in nuclei, whereas the other anthracyclines were found in the nuclei and cytoplasm. The percentages of the compounds that accumulated in the nuclei were 80.2 and 54.2 % for DOX and DOXH, respectively. The lowest nuclear accumulation values were observed for DRBM (19.9 %), DRBH (21.9 %) and DOXM (23.7 %). The ability of anthracyclines to accumulate in the nuclei correlated with their DNA binding constants (r = 0.858, P = 0.029). A correlation was found between the accumulation of the tested anthracyclines in the nuclei of cardiomyoblasts and their cardiotoxicity in vivo, which was observed in our previous study. We suggest that cytotoxicity and the anthracycline accumulation level in the nuclei of cultured cardiomyoblasts could be used for early prediction of their cardiotoxicity.
Similar content being viewed by others


Introduction
Anthracyclines have received significant attention due to their effectiveness and extensive use as anticancer agents. At present, the clinical use of these drugs is offset by drug resistance in tumors and cardiotoxicity [1, 2]. One of the many possible strategies for improving the therapeutic effectiveness of anthracyclines is the synthesis of new analogs with modified structures. Structural modifications of the anthracycline molecule may change many biological properties of these drugs, such as cellular uptake, subcellular localization, and cellular target affinity, which may affect the drugs’ selectivity toward cancer cells [3]. Modifications at the C-3′ position of the daunosamine moiety of anthracycline may result in improvement of some biological properties that are important for the anticancer action of these compounds [4–7].
Formamidinoanthracyclines are a new group of chemically modified anthracyclines. The most important feature of this novel class of anthracyclines is their lower cardiotoxicity compared to that of the parent compounds; this lower cardiotoxicity has been demonstrated in animal models [4, 8]. Although the low cardiotoxicity of these compounds seems to be a very promising feature, how modification of the daunosamine group of anthracycline at the C-3′ position produces this effect remains unclear. One possible method to reduce the cardiotoxicity of anthracyclines may be to reduce their accumulation in cardiac cells.
Heart tissue was demonstrated to have a small subpopulation of immature myocytes produced by stem-like cells [9, 10]. Such cycling cardiogenic precursors may be more sensitive to antiproliferative drugs than terminally differentiated cardiomyocytes, and their drug-induced death may play an important role in the cardiotoxic effects of anthracyclines [11]. Therefore, cardiac myoblasts in the form of rat embryonic H9c2(2-1) cells may be a good model for assessing the cardiotoxic properties of potential anticancer agents.
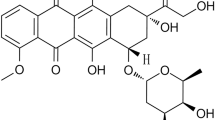
In this study, we attempted to determine whether the cytotoxicity of some doxorubicin (DOX) and daunorubicin (DRB) analogs, which were modified in the daunosamine moiety at the C-3′ position by the introduction of a formamidino group containing a morpholine or hexamethyleneimine ring (see Fig. 1 for structures), in cultured rat cardiomyoblasts is related to their cellular uptake and cellular localization.

Structures of the tested anthracyclines
Materials and methods
Drugs
DOX, DRB and their derivatives containing a formamidino group with a morpholine ring (DOXM and DRBM) or a hexamethyleneimine ring (DOXH and DRBH) were synthesized at the Institute of Biotechnology and Antibiotics in Warsaw [12]. Anthracyclines and their derivatives were dissolved in water and used immediately for each experiment.
Cell lines and cell culture
Rat DB1X cardiomyoblasts (H9c2(2-1) were purchased from the European Collection of Cell Cultures (ECACC). The cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10 % heat-inactivated fetal calf serum and 2 mM glutamine (all from Sigma-Aldrich) in a humidified 5 % CO2 atm at 37 °C. The cells were maintained as recommended by the supplier at a low density to avoid confluency and myoblast loss. All experiments were performed using cells at passages 4–27.
Cytotoxicity assay
The cytotoxic activity of the drugs was assayed by measuring their inhibitory effects on H9c2(2-1) cell proliferation. To measure the cytotoxicity of doxorubicin, daunorubicin, and their analogs, the tetrazolium dye method was used [13]. Cells were seeded at 1 × 104 cells/well in 48-well plates. After 24 h, the cells were incubated with different concentrations of anthracyclines for 72 h in triplicate in a final volume of 1 ml. Then, 50 μl of a sterile aqueous solution of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (5 mg/ml) (Sigma-Aldrich) was added to each well for an additional 3 h. The blue formazan precipitate was dissolved in DMSO (Sigma-Aldrich), and the absorbance of solutions was measured at 540 nm. For each experiment, determinations were performed in three replicates. The IC50 values (the anthracycline concentration effective at inhibiting cell growth after exposure to the drug for 72 h) were read from survival curves, which were fitted to an exponential equation using Xact v. 7.2, a statistical software tool by SciLab (Germany).
Cell cycle analysis
The cell cycle was analyzed by flow cytometry using a Becton-Dickinson FACSCanto II flow cytometer. H9c2(2-1) cells were seeded at 1 × 104 cells/cm2 in 10 cm2 culture flasks. After the cells were allowed to grow for 24 h, the medium was replaced and the cells were treated with an equitoxic anthracycline concentration corresponding to IC50. Treatment was performed for 24 h, and then the cells were washed in PBS and trypsinized. After the cells were washed in PBS again, they were fixed in 70 % ethanol and incubated for 1 h on ice. Fixed cells were washed twice in PBS, resuspended in commercially available propidium iodide staining solution and then subjected to flow cytometric assay. The percentages of H9c2(2-1) cells present in G1, S, and G2 phases of the cell cycle after drug treatment were analyzed using FACSDiva 6.1 software.
Cellular uptake
Anthracycline uptake was estimated by fluorimetric measurements after the drugs were extracted with an ethanol/HCl mixture as described by Riganti et al. [14]. H9c2(2-1) cells (2 × 105) were seeded onto 40 × 11 mm tissue culture dishes for 24 h. Then, the cells were treated in growth medium with 10 μM anthracycline solution at 37 °C for 1 h. After this time, the cells were gently scraped and suspended in 5 ml of ice-cold PBS. The cell suspension aliquots were centrifuged at 1500 rpm for 5 min and then washed twice in ice-cold PBS. Next, cell pellets were collected and resuspended in 2 ml of a 1:1 mixture of ethanol:0.3 M HCl, and the fluorescence intensities of these suspensions were measured at 20 °C using a Perkin-Elmer LS 55 spectrofluorimeter. The optimal excitation and emission wavelengths were 485 and 590 nm, respectively. To estimate the drug concentrations, calibration curves in the concentration range of 0.01–0.5 μM were prepared. Because the fluorescence was quenched slightly in the presence of cells, each point of the calibration curves was measured in the presence of 2 × 105 cells suspended in a mixture of ethanol/HCl.
Subcellular localization of anthracyclines
The subcellular localization of DOX, DRB and their analogs was examined using an Olympus BX-43 fluorescence microscope. H9c2(2-1) cells (2 × 104) were seeded onto 40 × 11 mm tissue culture dishes for 24 h. Then, the cells were incubated for 1 h at 37 °C in 2 ml of fresh growth medium supplemented with 10 μM drug. After incubation, the cells were washed in 4 ml of cold PBS, covered with a 24-mm cover glass, and observed at 400-fold magnification in visible light (dark field) and after excitation with a wide green filter. Cell images were acquired with an Olympus DP50 camera using the same exposure parameters for each image and saved in TIFF format as RGB images. Cell images were then processed by graphic software (Micrografx Picture Publisher v. 10) to prepare images suitable for densitometric measurements. First, the red channel for each image was separated and inverted to obtain images where areas corresponding to high fluorescence intensity became dark. Then, the part of the cell image corresponding to the nucleus was cut off and transferred above the part corresponding to the cytoplasm. This method allowed an image consisting of the separated nucleus and cytoplasm to be obtained. Such an image was then used as a path for densitometry. To estimate the percentages of drugs localized in the cytoplasm and in the nucleus, densitometric analysis was performed using GelScan v. 1.45 software (Kucharczyk T.E.).
Affinity of anthracyclines to DNA
The binding of the anthracyclines to double-stranded calf thymus DNA was tested by the ethidium bromide displacement assay [15]. This method is based on the competition between the tested compound and ethidium bromide for DNA intercalation sites. The fluorescence intensity of ethidium bromide increases upon DNA binding. Addition of a compound displacing intercalated ethidium bromide leads to quenching of the fluorescence of the DNA/ethidium bromide complex. The percentage of initial fluorescence was plotted against the concentration of the added compound, and the IC50 value was read. The IC50 value is defined as the concentration of added compound required to reduce the fluorescence of the DNA/ethidium bromide complex to 50 %. IC50 values were then used to calculate the apparent binding constants of the anthracyclines to DNA.
Statistical analysis
The data were analyzed using the SigmaPlot v. 12.5 statistics package (Systat Software, Inc. Chicago, USA). All the values in this study were expressed as the mean ± SD from at least three independent experiments. If no significant differences were detected between variances as assessed by the Snedecor–Fisher test, then the differences were compared by two-tailed Student’s t-test. The results were considered significant when P < 0.05. The relation between the DNA binding constants and nuclear localization of the anthracyclines was assessed by linear regression analysis.
Results
Cytotoxic activities of anthracyclines
The cytotoxicity potencies of DOX, DRB, and their analogs in H9c2(2-1) rat cardiomyoblasts exposed to the drugs for 72 h are presented in Table 1. All the tested anthracyclines were effective in inhibiting H9c2(2-1) cell proliferation. The IC50 values measured for doxorubicin and its derivatives were in the range of 35–138 nM. The most toxic compound in this group was the parent compound DOX, which exhibited approximately 4 times higher cytotoxic activity than DOXM and 2.5 times higher than DOXH. The IC50 values for daunorubicin derivatives were in the range of 75–626 nM. In this series of anthracyclines, the parent daunorubicin was the most cytotoxic drug, exhibiting 8 and 2 times higher activity compared to its analogs DRBM and DRBH, respectively.
Cell cycle analysis
Cell cycle analysis by flow cytometry was performed 24 h after cardiomyoblasts were treated with anthracyclines. The concentration of each drug that inhibited 50 % proliferation of cultured H9c2(2-1) cells was selected for these experiments. The results of the cell cycle analysis are shown in Fig. 2. The population of untreated control cells contained primarily 2 N content of DNA corresponding to G0/G1 phase (80.1 %). The S and G2/M fractions were 11 and 8.7 %, respectively. Of the H9c2(2-1) cells treated with DOX, 40.6 % showed increased accumulation in G2/M phase of the cell cycle and decreases in G0/G1 and S-phase populations compared with the untreated control. A different effect on the cycling of H9c2(2-1) cells was observed when cells were treated with DOXM and DOXH. These compounds induced increases in the S phase fraction (33.5 and 36.4 % for DOXM and DOXH, respectively). The accumulation of cells in S phase was accompanied by a decline in the G0/G1 population (53.5 and 50.1 % for DOXM and DOXH, respectively) and a slight increase in the percentage of cells in G2/M phase compared with untreated cells. Extremely similar results were obtained for DRB and its formamidine derivatives. DRB-induced cell cycle G2/M arrest resembled the arrest caused by DOX, whereas DRBM and DRBH produced extremely similar patterns of S phase arrest as observed for DOXH and DOXH (data not shown).

H9c2(2-1) cardiomyoblasts arrest in G2/M and S phases after anthracycline treatment. Cells were treated for 24 h with equitoxic concentrations of each drug that inhibited 50 % proliferation of cells and were subjected to flow cytometric analysis
Uptake of anthracyclines by rat cardiomyoblasts
The derivatives were taken up by H9c2(2-1) cells at different rates than those for their parent anthracyclines. Their uptake levels, expressed in nanomoles of compound per 1 × 106 cells, are shown in Table 1. Cellular accumulation of DOX was approximately 30 % higher than its derivative containing a morpholine ring (DOXM). DOXH was transported into the cells more efficiently than DOX and DOXM. The accumulation of DOXH in cardiac myoblasts was almost 2 times higher than that of DOXM and 1.5 times higher than that of the parent doxorubicin. The most “penetrating” compound from the entire group of examined analogs was the daunorubicin derivative with a hexamethyleneimine ring (DRBH); its accumulation in H9c2(2-1) cells was the highest among all tested drugs. After the cells were incubated with this compound for 1 h, the level of accumulation of this compound was more than 1.5 times higher than that of the parent drug (DRB) and at least 5–7 times higher than that of other tested compounds. Our experiments provided conclusive evidence that the hexamethyleneimine moiety enhances the uptake of anthracycline derivatives (DOXH and DRBH) by H9c2(2-1) cells. The lowest uptake values were found for both formamidino derivatives containing a morpholine ring (DOXM and DRBM). The lower cellular uptake values of these compounds translated into decreases in their cytotoxic potency in H9c2(2-1) cells (Table 1).
Subcellular localization of anthracyclines in rat cardiomyoblasts
The fluorescence microscopy images of H9c2(2-1) cells incubated with the compounds are presented in Fig. 3. The anthracyclines accumulated in the nuclei and in the cytoplasm, where they appeared in the form of small granules. As shown in Fig. 3, DOX and DOXH accumulated preferentially in cell nuclei. DOXM exhibited a different preference for subcellular compartments. The red fluorescence corresponding to DOXM was found primarily in the cytoplasm, whereas nuclear chromatin was stained much more weakly. DRB was present in both the nucleus and cytoplasm, whereas its formamidine derivatives DRBM and DRBH were localized primarily in the cytoplasm.

Images of H9c2(2-1) cells incubated with 10 μM anthracycline for 1 h at 37 °C. Cells were observed at 400-fold magnification after excitation with a wide green filter
To estimate the percentages of anthracyclines localized in the nucleus and in the cytoplasm of myocardial cells, densitometric quantification was performed. As shown in Table 1, the highest percentage of drug localized in cell nuclei, 80.2 %, was measured for DOX. DOXH exhibited a lower affinity to nuclear chromatin and to the cytoplasm compared to DOX (54.2 % of the DOXH drug was localized in nuclei). All other compounds exhibited the ability to accumulate in the cytoplasm rather than in nuclei. In particular, low affinity to nuclear chromatin was found for the morpholine derivatives DOXM and DRBM as well as for DRBH. All these compounds accumulated primarily in the cytoplasm.
Correlation between the affinity of anthracyclines to DNA and their localization in the nuclei of rat cardiomyoblasts
The DNA binding constant (Kapp) values estimated by the ethidium bromide displacement assay are shown in Table 1. The parent compounds DOX and DRB exhibited the highest affinities to double-stranded calf thymus DNA. Kapp values for their derivatives possessing a hexamethyleneimine ring (DOXH and DRBH) were 2.5–2.8 times lower. The lowest affinities to DNA were found for the morpholine derivatives DOXM and DRBM. When compared to the parent drugs, their Kapp values were approximately 20 times lower. When we plotted the percentage of anthracyclines localized in the nucleus against their Kapp values and performed a linear regression analysis, a significant correlation was found (r = 0.858, P = 0.029; Fig. 4). Thus, this result indicates that anthracyclines having high affinity to DNA are transported preferably into nuclei.

Correlation between the percentage of anthracyclines localized in the nuclei of H9c2(2-1) cells and their DNA binding constants (Kapp). Correlation coefficient r = 0.858, P = 0.029. Horizontal and vertical bars represent the standard deviation values
Discussion
The delivery and subcellular localization of drugs are important factors that influence their biological activity. Other authors have noted that the cytotoxic activity and clinical efficacy of anthracyclines may strongly correlate with their uptake potency and subcellular distribution [3, 16–19]. In this study, we used cultured rat cardiomyoblasts to compare the uptake, subcellular localization and cytotoxic activity of four anthracycline derivatives and two parent drugs. Our results show that the introduction of a formamidino group with a morpholine or hexamethyleneimine ring at C-3′ of daunosamine reduces the affinity of the tested anthracycline analogs to DNA as well as their cellular uptake and subcellular localization (Table 1). These results confirm the finding of Shaul et al. [3], who found that even small structural changes in the anthracycline molecule affects its cytotoxic activity and subcellular localization. The enhanced uptake of DOXH and DRBH by rat cardiomyoblasts can be explained by their lipophilicity, as the logP values of these derivatives are significantly higher than are those for DOX and DRB (Table 1). This observation is consistent with the findings of other authors who reported the strong impact of lipophilicity on the level of anthracycline accumulation in mammalian cells [16, 18, 19]. We assume that cell morphology is a very important factor that influences drug accumulation. In cardiomyoblasts that contain a large cytoplasm compartment (Fig. 3), the analogs accumulated in the cytoplasm, in contrast to the parent anthracyclines, which were transported preferentially to the nuclei. Our data suggest that cells containing large cytoplasm compartments and small nuclei accumulate hydrophobic anthracyclines preferentially, whereas the hydrophobicities of these drugs are less important for drug accumulation in cells with very large nuclei and small cytoplasmic compartments, e.g., leukemia cells [20].
Cell cycle analysis revealed that the compounds varied in their mode of cytotoxic action (Fig. 2). The growth inhibitory effect of DOX and its formamidine derivatives on cultured cardiomyoblasts appears to be due to blocks at G2/M phase (DOX) and G1/S phase (DOXM and DOXH) of the cell cycle. The same results were obtained for DRB, DRBM and DRBH (data not shown). This result is not surprising because our earlier studies demonstrated that formamidinoanthracyclines are not topoisomerase II inhibitors. These compounds produce different patterns of DNA damage than do their parent compounds, possibly mediated by a free radical mechanism [6, 7], whereas DOX and DRB are known to stabilize the “cleavable complex” of this enzyme with DNA [6, 7, 22].
The structures of the anthracyclines influenced their intracellular distribution. The morpholine as well as hexamethyleneimine substituents reduced the ability of the anthracyclines to bind to nuclear components (Fig. 3 and Table 1). This effect may be explained by the different affinities of the tested anthracyclines to DNA. When the percentages of anthracyclines localized in the nucleus was plotted against their DNA -binding constants, a significant correlation was found (r = 0.858, P = 0.029; Fig. 4). Derivatives exhibiting low affinity to DNA may accumulate with higher efficiency in the cytoplasm, as cytoplasmic components such as RNA molecules and proteins may compete with DNA for binding of these drugs.
Affinity to DNA seems to be a primary driving force directing anthracycline molecules into the cell nucleus, and this finding raises the question as to whether nuclear uptake of anthracyclines depends on the DNA content in cells. This possibility exists, as there are cells in different cycle phases and containing different DNA amounts in a population of cardiomyoblasts. Such cell populations should contain cells of different brightness after incubation with fluorescent compounds. However, we did not observe any differences in cell nuclear fluorescence when we viewed several cardiomyoblasts at once in the viewing field at low magnification. The brightness of the nuclei and cytoplasm was dependent on the type of anthracycline derivative, as shown in Fig. 3. This result can likely be explained by the fact that all experiments were performed 24 h after seeding the cells, when the majority of cells were at G0/G1 phase and contained 2 N DNA content (Fig. 2). Nonetheless, this issue is important; therefore, cells that are used to study the intracellular localization of fluorescent drugs should be subjected to cell cycle analysis to ensure that the majority of cells contain similar DNA content.
As mentioned in the Introduction, anthracycline analogs with a modified daunosamine moiety (DOXM, DOXH, DRBM and DRBH) were tested previously in animal models, and all of them exhibited lower cardiotoxicity than did DOX and DRB [4]. Interestingly, in this study, the same group of analogs exhibited decreased cytotoxicity in cultured cardiomyoblasts as well as lowered affinity to nuclear components, presumably primarily to DNA (Table 1 and Fig. 3), when compared to DOX and DRB. The correlation between cytotoxicity in vitro, accumulation of the tested anthracyclines in the nuclei of cardiomyoblasts, and their cardiotoxicity in vivo observed in our previous work [4] leads to the hypothesis that the cytotoxic potencies and the accumulation of anthracyclines in the nuclei of cultured cardiomyoblasts may be good and simple predictors of cardiotoxicity. This is an attractive possibility, as reliable methods that can be used for early prediction of anthracycline cardiotoxicity are needed [21].
The mechanism of anthracycline cardiotoxicity in vivo is very complex, and many cellular metabolic reactions may play a very important role in this process. Therefore, several possible mechanisms of anthracycline cytotoxicity are discussed in the literature [1, 2, 22]. The relatively new concept concerning anticancer drug cardiotoxicity is based on the finding that a small fraction of proliferating cells is present in cardiac tissue and may be a target for antiproliferative agents. [9–11]. The results of our work support this concept, as DOX and DRB, which exhibited the highest accumulation in cell nuclei and were the most potent inhibitors of cardiomyoblast proliferation among the tested compounds, were also found to be the most cardiotoxic in an in vivo animal model.
In conclusion, considering the complexity of the problem, we think that estimating cytotoxic activity and analyzing intracellular distribution of new potential anticancer anthracyclines in cultured cardiomyoblasts may be helpful in their early preclinical evaluation.
Abbreviations
- DOX:
-
doxorubicin
- DOXM:
-
doxorubicin containing a formamidino group with a morpholine ring
- DOXH:
-
doxorubicin containing a formamidino group with a hexamethyleneimine ring
- DRB:
-
daunorubicin
- DRBM:
-
daunorubicin containing a formamidino group with a morpholine ring
- DRBH:
-
daunorubicin containing a formamidino group with a hexamethyleneimine ring
References
Sheppard RJ, Jenna Berger J, Sebag IA (2013) Cardiotoxicity of cancer therapeutics: current issues in screening, prevention, and therapy. Front Pharmacol 4:19. doi:10.3389/fphar.2013.00019
Volkova M, Russell R III (2011) Anthracycline cardiotoxicity: prevalence, pathogenesis and treatment. Curr Cardiol Rev 7:214
Shaul P, Frenkel M, Goldstein EB, Mittelman L, Assaf Grunwald A, Ebenstein Y, Tsarfaty I, Fridman M (2013) The structure of anthracycline derivatives determines their subcellular localization and cytotoxic activity. ACS Med Chem Lett 4:323
Wasowska M, Oszczapowicz I, Wietrzyk J, Opolski A, Madej J, Dzimira S, Oszczapowicz J (2005) Influence of the structure of new anthracyclines antibiotics on their biological properties. Anticancer Res 25:2043
Wasowska M, Wietrzyk J, Opolski A, Oszczapowicz J, Oszczapowicz I (2006) Effect of structural modification of anthracyclines on the ability to overcome drug resistance of cancer cells. Anticancer Res 26:2009
Ciesielska E, Studzian K, Wasowska M, Oszczapowicz I, Szmigiero L (2005) Cytotoxicity, cellular uptake and DNA damage by daunorubicin and its new analogues with modified daunosamine moiety. Cell Biol Toxicol 21:139
Kik K, Wąsowska-Lukawska M, Oszczapowicz I, Szmigiero L (2009) Cytotoxicity and cellular uptake of doxorubicin and its formamidine derivatives in HL60 sensitive and resistant cells. Anticancer Res 29:1429
Lukawska M, Wietrzyk J, Opolski A, Oszczapowicz J, Oszczapowicz I (2010) Synthesis and biological properties of oxazolinodaunorubicin – a new derivative of daunorubicin with a modified daunosamine moiety. Investig New Drugs 28:600
Quaini F, Cigola E, Lagrasta C, Saccani G, Quaini E, Rossi C, Olivetti G, Anversa P (1994) End-stage cardiac failure in humans is coupled with the induction of proliferating cell nuclear antigen and nuclear mitotic division in ventricular myocytes. Circ Res 75:1050
Bearzi C, Rota M, Hosoda T, Tillmanns J, Nascimbene A, Angelis D et al (2007) Human cardiac stem cells. Proc Natl Acad Sci U S A 104:14068
Prezioso L, Tanzi S, Galaverna F, Frati C, Testa B, Savi M et al (2010) Cancer treatment-induced cardiotoxicity: a cardiac stem cell disease? Cardiovasc Hematol Agents Med Chem 8:55
Oszczapowicz I, Wasowska M, Oszczapowicz J, Owoc A, Dominiczak E, Wietrzyk J, Opolski A (2005) Nowe pochodne antybiotyków, sposób ich wytwarzania oraz zawierający je środek farmaceutyczny. Polish patent no 210494
Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB (1987) Evaluation of tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res 47:936
Riganti C, Miraglia E, Viarisio D, Costamagna C, Pescarmona G, Ghigo D, Bosia A (2005) Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res 65:516
Boger DL, Fink BE, Brunette SR, Tse WC, Hedrick MP (2001) A simple, high-resolution method for establishing DNA binding affinity and sequence selectivity. J Am Chem Soc 123:5878
Lotfi K, Zackrisson AL, Peterson C (2002) Comparison of idarubicin and daunorubicin regarding intracellular uptake, induction of apoptosis, and resistance. Cancer Lett 178:141
Mamidipudi V, Shi T, Brady H, Surapaneni S, Chopra R, Aukerman SL, Heise C, Sung V (2012) Increased cellular accumulation and distribution of amrubicin contribute to its activity in anthracycline-resistant cancer cells. Cancer Chemother Pharmacol 69:965
Tidefelt U, Prenkert M, Paul C (1996) Comparison of idarubicin and daunorubicin and their main metabolite regarding intracellular uptake and effect on sensitive and multidrug-resistant HL60 cells. Cancer Chemother Pharmacol 38:476
Garnier-Suillerot A, Marbeuf-Gueye C, Salerno M, Loetchutinat C, Fokt I, Krawczyk M et al (2001) Analysis of drug transport kinetics in multidrug-resistant cells: implications for drug actions. Curr Med Chem 8:51
European LeukemiaNet (2008) An atlas of cells with consensual nomenclature by the WP10 Morphology Faculty. http://www.leukemianet.org/content/diagnostics/-diagnostics/morphology/index_eng.html. Accessed 25 Oct 2008
Han Y, Zhang JP, Qian JQ, Hu CQ (2014) Cardiotoxicity evaluation of anthracyclines in zebrafish (Danio rerio). J Appl Toxicol 35:241
Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L (2004) Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 56:185
Acknowledgments
This work was supported by grant 503/2-159-01/503-01 from the Medical University of Lodz.
Compliance with Ethical Standards
This article does not contain any studies with human participants or animals performed by any of the authors. The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Studzian, K., Kik, K., Lukawska, M. et al. Subcellular localization of anthracyclines in cultured rat cardiomyoblasts as possible predictors of cardiotoxicity. Invest New Drugs 33, 1032–1039 (2015). https://doi.org/10.1007/s10637-015-0276-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0276-9